
Abstract
Using comparative genetics, genes, repetitive DNA sequences and chromosomes were studied in the Oryzeae in order to more fully exploit the rice genome sequence data. Of particular focus was Zizania palustris L., n = 15, commonly known as American wildrice. Previous work has shown that rice chromosomes 1, 4 and 9 are duplicated in wildrice. The Adh1 and Adh2 genes were sequenced and, based on phylogenetic analyses, found to be duplicated in wildrice. The majority of the sequence diversity in the Adh sequences was in intron 3, in which were found several MITE insertions. Cytological and molecular approaches were used to analyze the evolution of rDNA and centromeric repetitive sequences in the Oryzeae. In wildrice, copies of the 5S rDNA monomer were found at two loci on two different chromosomes near the centromeres, as in rice. One nucleolar organizer region (NOR) locus was found adjacent to the telomere, as in rice. RCS1, a middle repetitive sequence in rice, was present in all of the centromeres of wildrice. RCS2/CentO, the highly repetitive component of Oryza sativa L. centromeres, was conserved in eight of the Oryza species examined, but was not found in wildrice. Three other middle repetitive centromeric sequences (RCH1, RCH2/CentO and RCH3) were also examined and found to have variable evolutionary patterns between species of Oryza and Zizania.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Poaceae has been divided by taxonomists into 43 tribes of grasses (Watson and Dallwitz 1992). These tribes include cereal grasses of economic importance such as the Triticeae (wheat), the Andropogoneae (corn and sorghum) and the Oryzeae (rice and wildrice), among others. Recent phylogenetic studies have shown that the majority of the tribes in the Poaceae fall into two major clades, termed the BOP and PACC clades; with the PACC clade, containing the Andropogoneae, diverging earlier than the BOP clade, which contains the Triticeae and the Oryzeae (Mathews et al. 2000).
The tribe Oryzeae consists of 12 genera and 71 species with a worldwide distribution (Vaughan 1994). The genus Oryza includes Oryza sativa L., the genetic model for cereal species (Goff 1999). Also included in the tribe is American cultivated wildrice (Zizania palustris L.), a crop undergoing continuing domestication, which is primarily grown in California and northern Minnesota. Breeding of wildrice is in its infancy as is the understanding of its genetics.
The Oryzeae is an excellent system in which to examine genome evolution, as the O. sativa genome is being sequenced by a public consortium, providing the basis for examining genome evolution at the DNA sequence level. The genus Oryza consists of cytogenetically characterized diploids and allotetraploids. Therefore the evolution of specific genomes and chromosomes within this set of species can be traced. Evolution of the genetic map can also be considered because genetic maps, consisting of common RFLP markers, exist for both O. sativa and Z. palustris (Kennard et al. 2000). Utilization of both DNA sequence and genetic linkage maps will allow for detailed comparative genome analyses of these species.
Both Z. palustris and O. sativa have been placed in the Oryzeae based on sequence comparisons of the phytochrome B gene in the Poaceae (Mathews et al. 2000). Recently, a genetic map of wildrice was published using a set of RFLP markers common to O. sativa (Kennard et al. 2000). Nearly 85% of the markers were colinear between the Z. palustris and O. sativa maps. Therefore, wildrice is poised to gain from the investment in rice genomics due to its close phylogenetic position and extensive genetic colinearity with O. sativa.
There are many reports of conserved linkage of RFLP loci in related grass species, referred to as synteny, resulting in the 'circle of genomes', with rice being the central genome to which others are compared (reviewed in Devos and Gale 2000). However, little is known about the physical basis of synteny other than a few small-scale sequencing comparisons (Chen et al. 1997; Feuillet and Keller 1999; Tikhonov et al. 1999). In contrast to the extensive genetic colinearity between rice and wildrice, there are extensive chromosomal differences between these two species. Z. palustris has a chromosome number of n = 15, three more chromosomes than rice (n = 12), and a DNA content approximately two times that of rice (Grombacher et al. 1997). All other members of the Oryzeae have basic chromosome numbers that are multiples of 12 (Vaughan 1994). Previous cytogenetic studies of wildrice have shown that there is considerably more centromeric heterochromatin than in rice and have indicated that this may account for a portion of the increased DNA content (Grombacher et al. 1997).
In this study, both cytological and molecular techniques were used to begin to examine the physical basis of genetic synteny between rice and wildrice. The alcohol dehydrogenase (Adh) genes were cloned and sequenced to examine the evolution of individual genes within the Oryzeae. Both Adh1 and Adh2 were found to be duplicated in wildrice with major structural changes in intron 3. The genomic architecture of wildrice was characterized using fluorescence in situ hybridization (FISH) of repetitive sequences previously mapped in rice. There are two 5S ribosomal DNA loci and one NOR locus in wildrice, all located on different chromosomes. Repetitive sequences native to rice centromeres are also present in the centromeres of wildrice except for the most prominent rice sequence, RCS2/CentO. PCR was used in combination with Southern analyses to determine the evolutionary relationships of five of these cytologically defined O. sativa sequences in the Oryzeae.
The objectives of this research were to elucidate the mode(s) of genome evolution within the Oryzeae at the level of chromosomes, repetitive DNA sequences and individual genes. We plan to further integrate physical mapping resources from the genus Oryza, such as BAC libraries, with genetic and molecular cytogenetic mapping approaches to further examine genome evolution in the Oryzeae. Z. palustris provides an unique opportunity to study genome evolution in a species undergoing continuing domestication, and will be a key to understanding genome evolution in the grasses.
Materials and methods
Probes and plant material
pTa71, the ribosomal DNA sequence located at the NOR and cloned from wheat (Gerlach and Dyer 1980), and the 5S coding region cloned from maize, were used as FISH probes to map the NOR and 5S ribosomal DNA arrays. Five rice centromeric sequences (RCS1 AF058901, RCS2/CentO AF058902, RCH1 AF058903, RCH2 AF058904 and RCH3 AF058905, Dong et al. 1998; Cheng et al. 2002) were used to examine centromere architecture in wildrice.
Genomic DNA was collected from O. sativa L. cv Nipponbare (AA), O. sativa cv IR36 (AA), O. sativa cv M202 (AA), Oryza rufipogon Griff. (PI 239671) (AA), Oryza officinalis Wall ex Watt (PI 590412) (CC), Oryza rhizomatis Vaughan (IRGC 105440) (CC), Oryza minuta Presl. et Presl. (IRGC 104674) (BBCC), Oryza alta Swallen (PI 590397) (CCDD), Oryza australiensis Domin (IRGC 101318) (EE), Oryza brachyantha Chev. et Roehr. (IRGC 105171) (FF), Oryza meyeriana (Zoll. et Mor. ex Steud.) Baill. (IRGC 106474) (GG), Z. palustris L. cv Johnson, and Zea mays L. (inbred B73) using standard CTAB-based procedures. IRGC accessions were provided by the International Rice Research Center-Genetic Resource Center, Los Banõs, Philippines and PI accessions by the USDA-ARS National Small Grains Research Facility, Aberdeen, Idaho.
Cloning, sequencing and analysis of the Adh genes of rice and wildrice
Adh genes of Z. palustris were PCR amplified and cloned using the AdhF1 and AdhR1 primers (Ge et al. 1999, supplied by T. Sang, Michigan State University). Thirty transformed colonies were picked, placed into liquid culture, grown overnight and then the DNA was extracted with a plasmid mini-prep kit (Qiagen). DNA was digested with EcoRI and HindIII and subjected to electrophoresis. Distinct restriction profiles were obtained, the clones were sorted into profile classes, and a representative clone from each class was selected for subsequent DNA sequence analysis.
Sequencing was conducted by end-sequencing the clones with T7 and SP6 sequencing primers in conjunction with internal primers designed with Oligo 6.1. Sequencing was performed using the BIG-DYE terminator (ABI) and analyzed on an ABI 377 automated sequencer. Sequence editing and alignment were done using Sequencer 3.1 (GeneCodes). Using the NCBI BLAST server, Z. palustris sequences were designated Adh1a and Adh1b or Adh2a and Adh2b based on their similarity to these O. sativa genes and deposited in GenBank (AF408764 to AF408767).
ClustalW sequence alignments were performed and the percent identities of Adh1 and Adh2 sequences in O. sativa and Z. palustris were determined using the "Multiple Sequence Alignment" portion of the Lasergene software package (DNASTAR, Inc.). Plot similarities were constructed using PlotSimilarity in the GCG software package (SeqWeb version 2.0.2; accelrys), using a comparison window of 10, a gap creation penalty of 5, a gap extension penalty of 1, a maximum input sequence range of 5,000 and a minimum number of gap characters of 2,000, scaling the continuous curve plot between the minimum and maximum values in the scoring matrix. The locations of putative MITEs in the O. sativa and Z. palustris sequences were determined by running BLASTN searches of The Institute for Genomic Research (TIGR) rice repeat databases (http://tigrblast.tigr.org/euk-blast/index.cgi?project = osal ).
PCR and primer design
Primers were developed for RCS1 (5′-GTTCGCTTGTTGTTGTTG-3′; 5′TCCTCATCATCCTCCCCATC-3′), RCS2/CentO (5′-GCATTCATGGCAAAAACTCAC-3′; 5′-AACGCACCCCAATACACT-3′), RCH1 (5′-ACCATTCCCCCTTTGTCACC-3′; 5′-CGCTCACTCTCACTTCACTC-3′), RCH2 (5′-GGCAGGATACAAGGCAACTC-3′; 5′-GTTTTCGATTTGCATCGGTTT-3′) and RCH3 (5′-GATAATGAGCCACCGGAAGA-3′; 5′-TGCTAGCCAAGTTGTTGCAG-3′) using the GCG package (Genetics Computing Group, Oxford Molecular Group). PCR conditions were 7 min at 95 °C followed by 25 cycles of 1 min at 95 °C, 2 min at 52 °C and 2 min at 72 °C with a final extension run of 72 °C for 10 min using Taq DNA Polymerase (Promega, USA). Primers for Adh1 (described below) were used as internal controls in PCR reactions, and reactions with no detectable product using the primers for a centromeric sequence were repeated at least twice.
Southern blotting and probe hybridization
Genomic DNA was digested to completion with restriction enzymes, separated on a 1% (w/v) agarose gel, stained and photographed before being transferred to Zeta-Probe GT membrane (BioRad) following the manufacturer's directions. The membranes were baked in a vacuum oven at 80 °C for 1 h. Probe DNA was isolated using Qiaprep Miniprep kits (Qiagen, USA) and radiolabeled using nick translation (Sambrook and Russel 2001). Blots were prehybridized for 24 h with excess sheared salmon sperm DNA before being hybridized with radiolabeled DNA probes. Blots were washed three times in 2 × SSC, 0.1% SDS (w/v) for 5 min at room temperature, once in 1 × SSC, 0.1% SDS (w/v) for 10 min at room temperature and finally in 0.5 × SSC, 0.1% SDS (w/v) for 30 min at 65 °C, then exposed to film.
Chromosome preparation and fluorescence in situ hybridization (FISH)
For chromosome preparations, growing plants were removed from paddies and actively growing root tips were excised and placed in 2-mM hydroxyquinoline for 2.5 h. The root tips were washed in ddH2O for 10 min after which they were fixed in methanol:glacial acetic acid (3:1, v/v) for at least 3 days. Chromosome preparations were screened with a phase-contrast microscope and those with good chromosome spreads were immediately placed in a –80 °C freezer until used for FISH. Preparation of the FISH probe, hybridization of the probe to chromosomes and detection of hybridization sites followed previously published protocols (Jiang et al. 1996; Jackson et al. 1998). The slides were viewed with an epifluorescence Zeiss microscope and grayscale images were captured with an Olympus MagnaFire CCD camera attached to a Macintosh computer. Images were analyzed and line measurements made with Image-Pro Express 4.0 (Media Cybernetics) software and final image adjustments made with Adobe Photoshop v7.0.1.
Results
Evolution of the Adh1 and Adh2 genes among species of Oryza and Zizania
During the PCR-based cloning of the Adh genes from Z. palustris, we unexpectedly found two distinct restriction digest profiles for each of the Adh genes (named Adh1a, Adh1b, Adh2a and Adh2b). Both variants for each Adh gene were sequenced and subjected to further computational analysis. A multiple sequence alignment of the O. sativa Adh1 gene (exons 3–7; AF14568) with Z. palustris sequences Adh1a and Adh1b (exons 3–7; AF408765 and AF408764, respectively) showed 86.1% identity between the two Z. palustris sequences, but only 67.5% identity between O. sativa and Z. palustris Adh1b and 64.2% identity between O. sativa and Z. palustris Adh1a (Fig. 1b). Similar results were obtained with the Adh2 sequence from O. sativa (AF148602) and with Z. palustris Adh2a and Adh2b (AF408766 and AF408767, respectively) (Fig. 1b). The lack of sequence conservation between Adh1a and Adh2a and Adh1b and Adh2b (Fig. 1b), respectively, indicates that these are duplications and not allelic variants. Using the Adh sequences from Z. palustris and several Oryza species, phylogenetic trees were built and the Adh duplicates were consistently placed in separate branches of the trees (data not shown).

Similarity among O. sativa and Z. palustris Adh1 and Adh2 sequences. a Similarity plot of the Adh1 sequence of O. sativa and the Adh1 sequences of Z. palustris showing that the greatest sequence divergence occurs in intron 3. Sequence data for O. sativa and both Z. palustris clones existed only for the region from 100 bases to 1,750 bases. Diagram (drawn to scale) of the Adh1 gene is shown above the plot. Black boxes represent exons 1–10; solid lines represent introns 1–9; arrowheads represent primers used to amplify the Adh1 sequences from Z. palustris; gray lines indicate regions in which putative MITEs are located in the indicated sequences. b Percent identity of Adh1 and Adh2 among O. sativa and Z. palustris species. c Diagram of the Adh2 gene. Black boxes represent exons 3–7; solid lines represent introns 3–6; dashed lines represent regions for which no O. sativa sequence was determined; gray lines indicate regions in which putative MITEs are located
Not surprisingly, alignment results combined with the similarity plot shown in Fig. 1a revealed the O. sativa and Z. palustris Adh1 sequences to be more divergent in introns than in exons, with the largest intron, intron 3, showing the greatest divergence. A similar result was obtained using the Adh2 sequences (Fig. 1b; similarity plot not shown). In order to determine if transposable element insertion events might have contributed to the changes leading to the observed sequence divergence, a BLASTN search of The Institute for Genomic Research (TIGR) rice repeat databases was performed on all six Adh sequences. Only hits to putative miniature inverted repeat transposable elements (MITEs) were obtained.
Regions with sequence similarity to putative MITEs were located in all three Adh1 sequences (Fig. 1a). Four putative MITEs were similar to overlapping regions of Z. palustris Adh1a, and two putative MITEs were similar to non-overlapping regions of Z. palustris Adh1b. All of the regions in the two Z. palustris sequences showing similarity to putative MITEs occur in intron 3. A single putative MITE with similarity to the O. sativa Adh1 sequence was also located in intron 3.
A BLASTN search of the TIGR rice repeat databases using the Adh2 sequences revealed hits to nine putative MITEs in the Z. palustris Adh2b sequence (no hits were found for the O. sativa Adh2 or Z. palustris Adh2a sequences). Six of these putative MITEs showed similarity to regions of intron 3, one to intron 4, and two putative MITEs hit regions of intron 7 (Fig. 1c).
Genetic colinearity between species in the Poaceae indicates conserved genetic relationships among these species (Devos and Gale 2000). To better understand the duplication of the Adh genes in Z. palustris we compared the homologous regions of rice, maize and wildrice. RFLP marker RZ53, closely linked to Adh2 in both rice and maize (Fig. 2), was determined to be single copy in Z. palustris (data not shown). This result is in contrast to the Adh genes that, based on sequence data, are both duplicated in Z. palustris.

Schematic of comparative mapping data of wildrice (Kennard et al. 2000), rice (http://ars-genome.cornell.edu/cgi-bin/WebAce/webace?db = ricegenes) and maize (http://www.agron.missouri.edu). Linkage groups are indicated above the vertical axis and colinearity is indicated by lines connecting linkage groups. The known positions of Adh1 and Adh2 are shown in parentheses next to the linkage groups. Based on sequence data from rice, RZ53 is approximately 34 kb from Adh2, and Adh1 and Adh2 are approximately 30 kb apart (Tarchini et al. 2000). In maize, RZ53 is duplicated but linked to Adh2 on chromosome 4
Conservation of centromeric sequences in the Oryzeae
A conserved centromeric sequence from Sorghum bicolor L., pSau 3A-9 (Jiang et al. 1996), was used to isolate a rice centromeric BAC (17p22) from which several sequences were subcloned (Dong et al. 1998). Five of these sequences, RCS1, RCS2/CentO, RCH1, RCH2 and RCH3, were used to examine centromeric architecture and DNA sequence conservation and evolution among species of Oryza and Zizania. Two approaches were taken with these sequences. First, the clones were used as probes in Southern analyses to determine their conservation, and secondly, primers were designed for use in PCR to more stringently ascertain the sequence conservation of these sequences at specific primer sites. The results of the Southern and PCR analyses are summarized in Table 1.
RCS1, a middle repetitive sequence, was shown to be conserved throughout the Poaceae (Dong et al. 1998). PCR products were found throughout the tested species (see Fig. 4a) confirming that RCS1 is highly conserved. Cytologically, RCS1 was located in the centromeres of Z. palustris (Fig. 3e). Although the FISH signal was distributed throughout the pericentromeric heterochromatin, the most intense signals were centromeric.

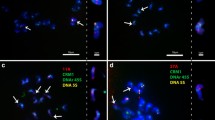
Fluorescence in situ hybridization of ribosomal DNA sequences to Z. palustris meiotic and mitotic chromosomes. a Biotin labeled 5S rDNA probe was hybridized to mitotic chromosomes and detected with FITC (green). Chromosomes are counterstained with Propidium Iodide (PI) (red). Arrows indicate larger 5S rDNA locus and arrowheads indicate a smaller locus; bar = 5 μm. b Biotin-labeled 18S–26S-5.8S rDNA probe hybridized to chromosomes from the first division of meiosis and detected with FITC (green). Chromosomes are counterstained with PI (red). One pair of distal hybridization signals is present. Arrows indicate three diminutive chromosome pairs; bar = 5 μm. c–d Biotin labeled 5S ribosomal DNA was hybridized to extended DNA fibers from Z. palustris and detected with FITC. Lengths ranged from 121.8 to 216.4 μm. A histogram of the raw data was used to determine that the average sizes of the two loci were 182.7 μm (panel c) and 136.3 μm (panel d). Bar = 20 μm for panels c–d. e FISH of RCS1 on metaphase chromosomes of Z. palustris. Left side of panel-chromosomes stained with PI (red), middle-RCS1 detected with FITC (green) and right-merged image of PI and FITC. Although RCS1 is found throughout the pericentromeric heterochromatin, the most intense signals appear to be centromeric; bar = 5 μm
RCS2/CentO is a high copy, tandemly repeated sequence found only in the Oryzeae (Dong et al. 1998; Cheng et al. 2002). Within the genus Oryza, PCR products were found in all AA-genome species, one CC genome (O. officinalis Wall ex Watt), and two allotetraploid species also containing the CC genome (O. minuta Presl. et Presl. BBCC and O. alta Swallen CCDD) (Fig. 4b). Minor products were also found in O. meyeriana (Zoll. Et Mor. Ex Steud.) Baill. (GG). This was concordant with Southern analysis except for O. australiensis Domin (EE), which had a faint signal, and O. meyeriana (GG), which had no hybridization signal (Table 1). This sequence was not found in Z. palustris by either PCR or Southern analysis.

Southern and PCR-based analysis of conservation of O. sativa centromeric repeats in the Oryzeae. a–e PCR was performed on genomic DNA with primers from a RCS1, b RCS2/CentO, c RCH1, d RCH2 and e RCH3. f–g Genomic DNA was digested with EcoRI, blotted and probed with f RCH2 and g RCH3. Lane 1 O. sativa ssp. indica, 2 O. sativa ssp. javonica, 3 O. sativa ssp. japonica, 4 O. rufipogon, 5 O. officinalis, 6 O. rhizomatis, 7 O. minuta, 8 O. alta, 9 O. australiensis, 10 O. brachyantha, 11 O. meyeriana, 12 Z. palustris, and 13 Z. mays
RCH1, RCH2 and RCH3 are all middle repetitive sequences conserved in the Poaceae (Dong et al. 1998). Based on Southern analysis, these three sequences were present in all Oryzeae species and maize, except for RCH3, which was not present in Z. palustris (Fig. 4f, g; RCH1 data not shown). The PCR-based approach had varied results. Primers designed for RCH1 had amplification products in the AA genome species, O. officinalis (CC), and one tetraploid, O. minuta (BBCC) (Fig. 4c). PCR analysis of RCH2 revealed products in the AA-genome species and one of the CC-genome species, O. officinalis (Fig. 4d). RCH3 had amplification products in the AA-genome species, O. officinalis (CC), both tetraploids (O. minuta, BBCC and O. alta CCDD) and minor amplification products in O. brachyantha Chev. Et Roehr. (FF) and Z. mays L. (Fig. 4e). RCH1 and RCH2 were present in Z. palustris based on Southern analysis but failed to give any amplification product with PCR.
Chromosomal structure and arrangement of ribosomal sequences in Z. palustris
Z. palustris has three more chromosome pairs than O. sativa. Interestingly, the chromosomes of Z. palustris are longer than rice chromosomes and contain more centromeric heterochromatin (Grombacher et al. 1997). However, at mitotic and meiotic metaphase, three chromosome pairs appear to be smaller than the rest of the chromosome complement (see Fig. 3b). Based on data presented above, all three of these diminutive chromosomes appear to have conserved certain O. sativa centromeric sequences present in the other 12 wildrice chromosome pairs.
The 5S rDNA sequences were mapped onto metaphase chromosomes using root-tip preparations. Four distinct loci were observed in nearly all nuclei and chromosome spreads indicating that there are two 5S rDNA loci in Z. palustris (Fig. 3a). Both loci are coincident with the centromeric heterochromatin, suggesting that the 5S rDNA sequences are either adjacent to or interspersed into pericentromeric sequences. The close juxtaposition of 5S rDNA with centromeric sequences has also been observed in rice (F. Dong and J. Jiang, personal communication).
The variable intensity of the fluorescence signals derived from the 5S rDNA probes indicates that the two loci in Z. palustris contain unequal copy numbers of the 5S rDNA monomer. To estimate the copy number of each 5S rDNA locus, 5S rDNA sequences were hybridized to genomic fibers of Z. palustris (fiber-FISH). Previous work with the fiber-FISH technique has shown that, in our hands, every micrometer of physical distance is equivalent to approximately 2.9 kb of DNA (Jackson et al. 1998). A total of 40 individual measurements were made and, based on histogram analysis, were distributed bimodally from which the approximate sizes of the two 5S rDNA loci were calculated to be 524.2 kb ± 152.1 (Fig. 3c) and 391.3 kb ± 113.3 (Fig. 3d) in size. Based on a 5S rDNA monomer of 324 bp (Kamisugi et al. 1994), the copy numbers were estimated to be 1,208 ± 350 and 1,618 ± 469 copies at each locus, respectively.
Using FISH, the nucleolar organizer region (NOR), including the 18S-26S-5.8S coding regions, was found on a single pair of chromosomes in Z. palustris (meiotic chromosomes, Fig. 3b). This is similar to O. sativa ssp. japonica, where the NOR is found only on chromosome 9 (Iijima et al. 1991). O. sativa ssp. indica has two NOR loci on the ends of chromosomes 9 and 10 (Islam-Faridi et al. 1990). During meiosis, the chromosomes of Z. palustris routinely form 8 rods and 7 rings (Grombacher et al. 1997). The NOR sequence was always located on a chromosome pair forming a rod configuration at the first meiotic division. Using fiber-FISH on Z. palustris, we were unable to measure a complete fiber corresponding to the NOR, but the data collected indicates that the locus is greater than 2 Mb in size (data not shown).
Discussion
Three approaches were used to characterize chromosome and DNA sequence evolution in the Oryza and Zizania species. The Adh genes of Z. palustris were cloned and sequenced to examine the evolution of individual genes; rDNA sequences were physically mapped to analyze the number, size and position of these chromosomal landmarks; and the evolution and conservation of centromeric sequences were evaluated using FISH, Southern analyses and the PCR of O. sativa centromeric sequences.
Based on Southern analysis, RFLP probe RZ53, which is closely linked to Adh2 in rice and maize, appears to be single copy in Z. palustris (data not shown). Duplications can complicate comparative genetic and physical mapping both within and across species. Using a comparative genetic mapping approach along with previously available data, the evolution of the Adh genes within a genomic context is not clear and may be independent of the closely linked marker, RZ53 (Fig. 2). Indeed, previous work with duplicated chromosomal segments in Arabidopsis has demonstrated that individual genes within a duplicated region can be lost creating a patchwork of synteny between the duplicated segments (Ku et al. 2000; Vision et al. 2000). Thus, it is possible that the chromosomal region containing the Adh genes was duplicated in Z. palustris but that the RZ53 sequence was lost in one of the two regions.
The majority of the sequence divergence among O. sativa and Z. palustris Adh genes occurs in the introns. This is not surprising, considering the evolutionary pressure to maintain conserved coding regions in order to maintain a functional Adh protein. Intron 3, which is approximately four times larger than the next largest intron (intron 4) in both Adh1 and Adh2, exhibited the greatest sequence diversity.
One factor contributing to at least some of the sequence diversity observed in the Adh genes could be the insertion and/or excision of transposable elements. A BLASTN search of the rice repeat database revealed similarities to a number of MITEs. No other type of transposon showed similarity to the Adh sequences in this search, although this does not rule out the possibility that other transposable elements have contributed to the sequence diversity over time. Genes of flowering plants have previously been shown to contain transposable elements, with MITEs being the most prevalent type of transposon found in rice genes (Bureau et al. 1996). No transposable elements were detected in the coding regions of the Adh sequences, but a number of putative MITEs were identified in intron 3 in both Adh1 and Adh2 sequences of Z. palustris.
It is possible that intron 3 contains sites targeting it for preferential MITE insertion, and that this has contributed to the increased size of intron 3 compared to the other introns of the Adh genes as well as to the sequence diversity of the region. MITEs have been shown to have target site preference (Bureau et al. 1996; Zhang et al. 2000), however the putative target sites of MITEs located in intron 3 have not been determined. A more likely explanation for the apparent preferential insertion of MITEs in intron 3 is that this region is already so much larger than the other introns, and therefore more able to tolerate and maintain MITE insertion without affecting gene splicing. This is supported by the fact that three MITEs showed similarity to regions of introns 4 and 7 of Z. palustris Adh2b, and that no MITEs were found in a BLASTN search using the other two Adh2 sequences. The differences in the locations of putative MITEs in Adh1, Adh1a and Adh1b, and the fact that nine putative MITEs were found in Adh1b but none were found in Adh2 (O. sativa) or Adh1a indicates that the insertions occurred after the duplication of the Adh genes in Z. palustris.
We attempted to use the Adh sequences to further resolve the phylogenetic position of Z. palustris within the Oryzeae. However, due to major structural changes in intron 3, it was difficult to align the duplicated Adh genes to the data matrix of Ge et al. (1999). This difficulty with intron 3 was also noted in the Adh2 gene of the sister taxon Rhynochoryza by Ge et al. (1999), and it was excluded from their analysis because its introns were difficult to align with those of the ingroup species. Using Adh1a sequence data, we determined that Z. palustris is either in a monophyletic branch with Rhynchoryza subulata (Nees.) Baill. or in a paraphyletic grade that includes Rhynchoryza, Zizaniopsis, Leersia and Oryza section Granulata – the most basal group of rice species (data not shown).
Recent work on grass centromeres has shown that there are basically two types of sequences present in grass centromeres. The first, represented by RCS2/CentO (Dong et al. 1998), is a tandemly repeated centromeric sequence about 155 bp in size, structurally similar to the pAL1 sequence of Arabidopsis (Martinez-Zapater et al. 1986) and the CentC sequence of maize (Ananiev et al. 1998). These tandemly repeated sequences evolve rapidly and tend to be conserved only in closely related species (Miller et al. 1998). A second type of sequence, represented by RCS1, RCH1, RCH2 and RCH3 is a low to middle-copy, interspersed sequence that may be conserved across many genera (Aragon-Alcaide et al. 1996; Jiang et al. 1996; Dong et al. 1998; Miller et al. 1998; Cheng et al. 2002). RCS1 was present at the centromeres of Z. palustris, whereas the highly repetitive RCS2/CentO sequence was not present in Z. palustris, even under low-stringency Southern conditions (data not shown). Thus, the tandemly repeated RCS2/CentO sequence in O. sativa is different from the major repeat in the centromeres of Z. palustris.
The conservation and evolution of the repetitive centromeric sequences cloned from O. sativa were also examined in the Oryzeae (Fig. 4, Table 1). Differences between Southern and PCR-based analyses can be attributed to the reliance of PCR on very high sequence identity at about 20 nt primer sites, whereas the DNA hybridization-based Southern can accommodate more sequence variation between related sequences. All of the sequences were present in the sampled AA genomes and one CC genome (O. officinalis). The shared PCR products between the CC genome species O. minuta (BBCC) and O. officinalis (CC) support the close phylogenetic relationship of these two species as shown by Adh1 and Adh2 gene phylogenies (Ge et al. 1999). It is of interest to note that RCS2, the major tandem repeat in O. sativa, is conserved in the AA, some CC, an EE and a GG genome Oryza species as shown by Southern and PCR-based analyses. Without testing the BB and DD progenitors of the tetraploids O. minuta and O. alta, respectively, it is impossible to know which or if both genomes contain the RCS2 repeat. The other interspersed repeats were shown to be conserved in almost all the species by either Southerns and/or PCR analyses.
Little is known about chromosome evolution within the Oryzeae. There is variability in genome size among the Oryza diploids most probably due to repetitive DNA sequences (Uozu et al. 1997). The genome of Z. palustris is two times the size of the O. sativa genome. Some of this increase in DNA content is due to the duplicated chromosomes; however, these may be the diminutive chromosomes and therefore do not contain much DNA. Expansion of 5S ribosomal arrays can be discounted, as the total amount of 5S rDNA in Z. palustris is only slightly larger than that of O. sativa. Accumulation of repetitive sequences in the pericentromeric regions most likely accounts for some or most of the increase in genome size, as these regions have been reported to be much larger than those observed in O. sativa (Grombacher et al. 1997). Indeed, in other Oryza species, the pericentromeric regions also appear, cytologically, to be larger than those of O. sativa and this has been correlated with genome size and the accumulation of repetitive sequences (Uozu et al. 1997). RCS2/CentO, the primary component of O. sativa centromeres, appears to be the most diverged centromeric repetitive sequence among the species in this tribe. In order to truly evaluate the evolution of this major centromere component in this tribe, these sequences should be cloned and sequenced from all the representative genomes and compared.
Two 5S ribosomal DNA loci were found in Z. palustris (Fig. 3a) of which one is present on one of the diminutive chromosome pairs. In O. sativa there is only one locus located on chromosome 11 in ssp. japonica (Kamisugi et al. 1994) and on chromosome 9 in ssp. indica (Song and Gustafson 1993). The sub-chromosomal location of the 5S rDNA on the chromosomes is similar between O. sativa and Z. palustris in that the 5S rDNA locus is intimately associated with the pericentromeric heterochromatin. Using fiber-FISH, the physical sizes of the two 5S rDNA loci in Z. palustris were estimated to be 391 and 524 kb (Fig. 3c, d). In O. sativa, the 5S rDNA locus was variable in size between the three cultivated subspecies, ssp. javonica, japonica and indica, ranging from 300 kb to 425 kb to 670 kb, respectively (Ohmido et al. 2000; S. Jackson and J. Jiang, unpublished results). Thus, the sizes of the two loci in Z. palustris are within the range observed in the O. sativa subspecies, although the total size of both loci is larger than the 670 kb observed in ssp. indica. However, the 5S rDNA repeats do not account for much of the increase in the genome size of Z. palustris.
A single NOR locus was detected in Z. palustris near the telomere of one chromosome pair. In O. sativa, there is one locus in ssp. japonica on chromosome 9 (Iijima et al. 1991) and two loci in ssp. indica on chromosomes 9 and 10 (Islam-Faridi et al. 1990). All of the NORs in the genus Oryza are located near the telomeres. The NORs in both Z. palustris and O. sativa are on the order of megabases in size using fiber-FISH (S. Jackson, unpublished results). In Arabidopsis thaliana (L.) Heynh., there are two NORs that abut the telomeres and each is three to four megabases in size (Copenhaver and Pikaard 1996a, b). The 5S rDNA loci of A. thaliana are variable in number between ecotypes and tend to be centromerically located (Fransz et al. 1998). Ribosomal DNA sequences are not conserved in physical location in other Oryza species (Shishido et al. 2000); therefore, these loci appear to be able to 'move' frequently although the tendency for the NORs to be telomeric and the 5S ribosomal loci to be centromeric in location remains conserved. The biological significance of the position and number of NOR and 5S rDNA loci is not yet understood.
Within the Oryzeae the basic chromosome number is n = 12; only the Zizania species deviate from the base number of 12. Based on the comparative genetic map constructed in Z. palustris, three of the Z. palustris chromosomes appear to be duplicated (Kennard et al. 2000). To test the extent of physical duplication on these chromosomes, O. sativa BACs can be mapped on Z. palustris chromosomes or a BAC-based physical map of Z. palustris can be developed and crosslinked to that of O. sativa.
One question that predicates the immediate utility of model genomes for related species is the mode of genome/chromosome evolution. The mode of genome evolution will delimit the phylogenetic distance over which comparisons are informative. Within the grass family there are several reports of comparative sequencing between rice, and sorghum, maize, oat and barley (SanMiguel et al. 1996; Chen et al. 1997; Feuillet and Keller 1999; Tikhonov et al. 1999). With few exceptions, it seems evident that there has been an accumulation of non-genic DNA between coding regions and that the number of these elements increases both with genome size and taxonomic distance from rice (Chen et al. 1997; Tikhonov et al. 1999). More recent work in rice and maize has shown that there are regions with gene densities of one gene for every 6.1 kb and 9.8 kb, respectively, much higher than predicted densities (Fu et al. 2001; Mayer et al. 2001). Thus, within cereal genomes there appear to be 'gene galaxies' (Walbot and Petrov 2001).
Although the comparative genetic map of rice and wildrice is about 85% colinear, the research presented here has shown that there appear to be substantial levels of chromosomal evolution between these two species, as indicated by the duplication of both chromosomes and genes and the evolution of repetitive DNA sequences. However, the relatedness of these two species means that wildrice improvement can be advanced via information on gene function as predicted by the rice genome sequencing project and rice physical mapping resources, such as BAC libraries, and can be used to construct a rice-based Z. palustris physical map. Additionally, due to the recent domestication of wildrice, we have the unique opportunity to examine genome evolution in the context of ongoing domestication.
References
Ananiev EV, Phillips RL, Rines HW (1998) Chromosome-specific molecular organization of maize (Zea mays L.) centromeric regions. Proc Natl Acad Sci USA 95:13,073–13,078
Aragon-Alcaide L, Miller T, Schwarzacher T, Reader S, Moore G (1996) A cereal centromeric sequence. Chromosoma 105:261–268
Bureau TE, Ronald PC, Wessler SR (1996) A computer-based systematic survey reveals the predominance of small inverted-repeat elements in wild-type rice genes. Proc Natl Acad Sci USA 93:8524–8529
Chen M, SanMiguel P de, Oliveira AC, Woo S-S, Zhang H, Wing RA, Bennetzen JL (1997) Microcolinearity in sh2-homologous regions of the maize, rice, and sorghum genomes. Proc Natl Acad Sci USA 94:3431–3435
Cheng Z, Dong F, Langdon T, Ouyang S, Buell CR, Gu M, Blattner FR, Jiang J (2002) Functional rice centromeres are marked by a satellite repeat and a centromere-specific retrotransposon. Plant Cell 14:1691–1704
Copenhaver GP, Pikaard CS (1996a) Two-dimensional RFLP analyses reveal megabase-sized clusters of rRNA gene variants in Arabidopsis thaliana, suggesting local spreading of variants as the mode for gene homogenization during concerted evolution. Plant J 9:273–282
Copenhaver GP, Pikaard CS (1996b) RFLP and physical mapping with an rDNA-specific endonuclease reveals that nucleolus organizer regions of Arabidopsis thaliana adjoin the telomeres on chromosomes 2 and 4. Plant J 9:259–272
Devos KM, Gale MD (2000) Genome relationships: the grass model in current research. Plant Cell 12:637–646
Dong F, Miller JT, Jackson SA, Wang G-L, Ronald PC, Jiang J (1998) Rice (Oryza sativa) centromeric regions consist of complex DNA. Proc Natl Acad Sci USA 95:8135–8140
Feuillet C, Keller B (1999) High gene density is conserved at syntenic loci of small and large grass genomes. Proc Natl Acad Sci USA 96:8265–8270
Fransz P, Armstrong S, Alonso-Blanco C, Fischer TC, Torres-Ruiz RA, Jones G (1998) Cytogenetics for the model system Arabidopsis thaliana. Plant J 13:867–876
Fu H, Wonkeun P, Yan X, Zheng Z, Shen B, Dooner H (2001) The highly recombinogenic bz locus lies in an unusually gene-rich region of the maize genome. Proc Natl Acad Sci USA 98:8903–8908
Ge S, Sang T, Lu B-R, Hong D-Y (1999) Phylogeny of rice genomes with emphasis on origins of allotetraploid species. Proc Natl Acad Sci USA 96:14,400–14,405
Gerlach WL, Dyer TA (1980) Sequence organization of the repeating units in the nucleus of wheat which contain 5S rRNA genes. Nucleic Acids Res 8:4851–4865
Goff S (1999) Rice as a model for cereal genomics. Curr Opin Plant Biol 2:86–89
Grombacher AW, Porter RA, Everett LA (1997) Breeding wildrice. Plant Breed Rev 14:237–255
Iijima K, Kakeda K, Fukui K (1991) Identification and characterization of somatic rice chromsomes by imaging methods. Theor Appl Genet 81:576–605
Islam-Faridi MN, Ishii T, Kumar V, Sitch LA, Brar DS (1990) Chromosomal location of ribosomal RNA genes in rice by in situ hybridization. Rice Genet Newslett l7:143–144
Jackson SA, Wang M-L, Goodman HM, Jiang J (1998) Application of fiber-FISH in physical mapping of Arabidopsis thaliana. Genome 41:566–572
Jiang J, Nasuda S, Dong F, Scherrer CW, Woo S-S, Wing RA, Gill BS, Ward DC (1996) A conserved repetitive DNA element located in the centromeres of cereal chromosomes. Proc Natl Acad Sci USA 93:14,210–14,213
Kamisugi Y, Nakayama S, Nakajima R, Ohtsubo H, Ohtsubo I, Fukui K (1994) Physical mapping of the 5S ribosomal RNA genes on rice chromosome 11. Mol Gen Genet 245:133–138
Kennard W, Porter R, Grombacher A, Phillips RL (2000) A comparative map of wildrice (Zizania palustris L. 2n = 2x = 30). Theor Appl Genet 99:793–799
Ku H-M, Vision T, Liu J, Tanksley SD (2000) Comparing sequenced segments of the tomato and Arabidopsis genomes: large-scale duplication followed by selective gene loss creates a network of synteny. Proc Natl Acad Sci USA 97:9121–9126
Martinez-Zapater JM, Estelle MA, Somerville CR (1986) A higly repeated DNA sequence in Arabidopsis thaliana. Mol Gen Genet 204:417–423
Mathews S, Tsai RC, Kellog EA (2000) Phylogenetic structure in the grass family (Poaceae): evidence from the nuclear gene phytochrome B. Am J Bot 87:96–107
Mayer K, Murphy G, Tarchini R, Wambutt R, Vockaert G, et al (2001) Conservation of microstructure between a sequenced region of the genome of rice and multiple segments of the genome of Arabidopsis thaliana. Genome Res 11:1167–1174
Miller JT, Jackson SA, Nasuda S, Gill BS, Wing RA, Jiang R (1998) Cloning and characterization of a centromere-specific repetitive DNA element from Sorghum bicolor. Theor Appl Genet 96:832–839
Ohmido N, Kijima K, Akiyama Y, de Jong JH, Fukui K (2000) Quantification of total genomic DNA and selected repetitive sequences reveals concurrent changes in different DNA families in indica and japonica rice. Mol Gen Genet 263:388–394
Sambrook J, Russel D (2001) Molecular cloning: a laboratory manual, edn 3. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
SanMiguel P, Tikhonov A, Jin Y-K, Motchoulskaia N, Zakharov D, Melake-Berhan A, Springer PS, Edwards KJ, Lee M, Avramova Z, Bennetzen JL (1996) Nested retrotransposons in the intergenic regions of the maize genome. Science 274:765–768
Shishido R, Sano Y, Fukui K (2000) Ribosomal DNAs: an exception to the conservation of gene order in rice genomes. Mol Gen Genet 263:586–591
Song YC, Gustafson JP (1993) Physical mapping of the 5S rDNA gene complex in rice (Oryza sativa). Genome 36:658–661
Tarchini R, Biddle P, Wineland R, Tingey S, Rafalski A (2000) The complete sequence of 340 kb of DNA around the rice Adh1–Adh2 regions reveals interrupted colinearity with maize chromosome 4. Plant Cell 12:381–391
Tikhonov AP, SanMiguel PJ, Nakajima Y, Gorenstein NM, Bennetzen JL, Avramova Z (1999) Colinearity and its exceptions in orthologous Adh regions of maize and sorghum. Proc Natl Acad Sci USA 96:7409–7414
Uozu S, Ikehashi J, Ohmido N, Ohtsubo H, Ohtsubo E, Fukui K (1997) Repetitive sequences: cause for variation in genome size and chromosome morphology in the genus Oryza. Plant Mol Biol 35:791–799
Vaughan DA (1994) The relationship between the genus Oryza and other grasses. In: Vaughan D (ed) The wild relatives of rice: a genetic resources handbook. International Rice Research Institute, Manila, Philipppines, pp 3–5
Vision TJ, Brown DG, Tanksley SD (2000) The origins of genomic duplications in Arabidopsis. Science 290:2114–2117
Walbot V, Petrov DA (2001) Gene galaxies in the maize genome. Proc Natl Acad Sci USA 98:8163–8164
Watson L, Dallwitz MJ (1992) The grass genera of the world. CAB International, Wallingford, Oxon, UK, pp 48–54
Zhang Q, Arbuckle J, Wessler SR (2000) Recent, extensive, and preferential insertion of members of the miniature inverted-repeat transposable element family Heartbreaker into genic regions of maize. Proc Natl Acad Sci USA 97:1160–1165
Acknowledgements
We would like to thank J. Lilly for assistance with DNA sequencing. This research was supported by a grant from the United States Department of Agriculture National Research Initiative Competitive Grants Program 00-35300-9214. This is journal paper number 16813 of the Purdue University Agricultural Experimental Station.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by B. Friebe
Rights and permissions
About this article
Cite this article
Hass, B.L., Pires, J.C., Porter, R. et al. Comparative genetics at the gene and chromosome levels between rice (Oryza sativa) and wildrice (Zizania palustris). Theor Appl Genet 107, 773–782 (2003). https://doi.org/10.1007/s00122-003-1273-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-003-1273-5