
Abstract
Increased levels of oleic acid may enhance the nutritional and functional value of corn. Corn oil is primarily composed of palmitic, stearic, oleic, linoleic and linolenic fatty acids. Delta-12 desaturase in plants converts oleic acid (18:1) to linoleic acid (18:2) by inserting a double bond at the delta-12 position. Fatty acid desaturase-2 (fad2) encodes delta-12 desaturase that functions in the endoplasmic reticulum while fatty acid desaturase-6 (fad6) encodes delta-12 desaturase that functions in plastids. Complementary DNA (cDNA) clones from putative maize homologs for fad2 and fad6 were identified and the entire clones DNA sequenced. The maize fad2 cDNAs showed an amino-acid identity of 67–77% to fad2 of Glycine, Arabidopsis and Brassica species. Our cDNA sequence comparisons suggested that more than one fad2 gene is transcribed in maize embryos. Two different fad2 cDNAs from an embryo cDNA library map to separate chromosomal positions, providing evidence consistent with two different isoforms of fad2 expressed in the embryo. The fad2 cDNAs from multiple tissue sources clustered into three groups on a phenogram, and map to different positions on chromosomes 4, 5 and 10, which suggests at least three different isoforms of fad2 may be expressed in the maize plant. The two maize fad6 cDNAs share 81% amino-acid identity with the Arabidopsis fad6 and map to chromosome 1. Northern analysis revealed that fad2 is transcribed in embryos at 14, 21, 28 and 35 days after pollination, with the highest level observed at day 14. None of the fad2 or fad6 clones mapped to maize chromosome bins associated with QTLs for the ratio of oleic/linoleic acid, notably bin 6.04 which contains the linoleic1 locus and the largest reported QTL for the oleic/linoleic ratio. This suggests, but does not prove, that some of the QTLs for the oleic/linoleic acid ratio do not involve allelic variants of fad2 or fad6 but rather involve other genes that may influence flux through the enzymes encoded by fad2 or fad6.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The fatty acid composition of oil determines its use as a food source or for industrial applications (Harwood 1997). A large variety of fatty acids exist in nature but only five fatty acids account for about 90% of the commercial vegetable oil produced: palmitic (16:0), stearic (18:0), oleic (18:1), linoleic (18:2) and linolenic (18:3) (Browse 1991). Monounsaturated fatty acids (oleic acid) have many health and cooking benefits when compared to saturated and polyunsaturated fatty acids. (Mattson and Grundy 1985) Therefore, increasing the oleic acid content of the corn kernel could enhance the economic value and nutritional quality of corn oil. Corn oil is typically 25% oleic acid, but there have been reports of oleic acid levels varying up to 64% in maize germplasm (Jellum 1966).
Fatty acid biosynthesis is a complex process that is regulated at several points. In this pathway, saturated fatty acids, palmitic and stearic are synthesized and stearic acid is subsequently desaturated to oleic, linoleic and linolenic fatty acids (Harwood 1997). Biosynthesis of seed oils (triacylglycerols or TAGs) is extremely complex and has been reviewed in detail (Browse 1991; Ohlrogge and Jaworski 1997). Many of the genes for the enzymes that perform de novo fatty acid synthesis in plastids have been characterized in Arabidopsis thaliana and in other crop species. Several genes involved in the synthesis of TAGs in the endoplasmic reticulum have also been characterized (Browse 1991; Ohlrogge and Jaworski 1997; Ohlrogge et al. 1991, 1993).
The majority of polyunsaturated fatty acids are synthesized through the 18:1 desaturase in the endoplasmic reticulum (Browse 1991). The biochemical pathway for fatty acid biosynthesis in A. thaliana and Glycine max indicates that the enzyme delta-12 desaturase, encoded by the single gene fatty acid desaturase-2 (fad2) converts 18:1 to 18:2 by inserting a double bond at the 12th carbon in the fatty acid hydrocarbon chain (Okuley et al. 1994; Byrum et al. 1997). The plastidial fatty acid desaturase-6 (fad6) gene which encodes the enzyme delta-12 desaturase also converts oleic acid to linoleic acid in the chloroplast in the prokaryotic pathway (Somerville and Browse 1996).
In Brassica napus, desaturase multigene families have been shown to arise from genome duplication. Delta-12 desaturases have been shown to be active in the endoplasmic reticulum (4–6 gene copies per haploid genome) while the omega-6 desaturases have been shown to be active in a parallel pathway located in the plastids (6–8 copies per haploid genome) (Scheffler et al. 1997). In soybeans, two different genes have been reported to encode the delta-12 desaturases, 'FAD2-1' has been shown to be strongly expressed in developing seeds and 'FAD2-2' has been shown to be expressed in both vegetative tissues and developing seeds (Heppard et al. 1996).
Several classical mapping studies were conducted to identify loci that control or modify the conversion of oleic to linoleic acid in maize (Poneleit 1976) suggesting that a single gene locus linoleic1 (ln1) was involved in regulating the levels of oleic and linoleic acids in corn (Poneleit and Alexander 1965), resulting in higher levels of oleic acid and lower levels of linoleic acid. Subsequent studies showed a more complex mode of inheritance for levels of oleic and linoleic acid. In 1971 de la Roche et al. reported that two loci were involved in regulating levels of oleic acid. The linoleic (ln1) locus was mapped to the long arm of chromosome 6 (Poneleit 1976). Chromosome regions on 1S (Widstrom and Jellem 1984), chromosome 2 (Plewa and Plewa 1975), 4L (Widstrom and Jellum 1984) and 5L (Shadley and Shadley 1980; Widstrom and Jellem 1984) were also associated with control of ratios for oleic and linoleic acids. More recently, oleic acid 1 (olc1), which reduces further desaturation of oleic acid to linoleic acid, was identified and mapped to chromosome 1 (Wright 1995).
Quantitative trait loci (QTLs) in eight chromosome regions were associated with the concentration of oleic acid (Alrefai et al. 1995). The ratio of 18:1 to 18:2 was interrelated as the same QTLs were associated with concentrations of 18:1 and 18:2. A QTL that explained 63% of the phenotypic variation in the ratio of 18:1 to 18:2 was linked to umc65 on chromosome 6, bin 6.04, in the same region of the ln1 locus. This QTL study supported the previous report that the ln1 locus has a major effect on levels of oleic acid as well as other reports indicating that levels of oleic acid can be influenced by a number of loci.
Our objectives include characterizing the extent of DNA sequence similarities among a series of cDNAs encoding the delta-12 fatty acid desaturase of maize, the cluster analysis of the cDNAs and mapping the cDNAs to determine their genomic organization. Since fad2 and fad6 genes encode the delta-12 desaturase that is involved in the conversion of 18:1 to 18:2, they were logical candidates for one or more of the QTLs affecting the ratio of oleic and linoleic acid. Therefore a candidate gene approach was taken to determine if any putative fad2 or fad6 genes of maize mapped to chromosome regions associated with QTLs for the 18:1/18:2 ratio. Since a longer-term goal produces high oleic acid in corn oil by down-regulating expression of fad2 and fad6 genes through selection of QTLs and/or transgenic technologies, we performed Northern analysis with fad2 cDNAs to examine temporal transcription patterns in the developing seed.
Materials and methods
ESTs/cDNAs
ESTs generated from maize cDNA libraries of Pioneer Hi-Bred International by performing single 5′ end sequence runs were screened for similarity with fad2 sequences from A. thaliana, G. max and Brassica napus. Putative maize fad2 ESTs from embryo, seed and non-seed source tissues were selected in order to be comprehensive in our study. The longest EST clones of the strongest hits from different contigs were obtained. Double-stranded DNA sequencing of entire clones, which varied in size, was performed at the W.M. Keck Center for Comparative and Functional Genomics, University of Illinois at Urbana-Champaign. The source tissue, clone designations, size and GenBank number are: Embryo – cdpey46 (953 bp) BI683571, cdpec60 (970 bp) BI683570, cdpfe85 (1,807 bp) BI683572; Leaves – cfbbi58 (980 bp) BI683573 cqrak57 (1,523 bp) BI683575; Shoot culture – ccmaa92 (1,780 bp) BI683576; and Callus – ccrap39 (1,792 bp) BI683574.
DNA sequence analysis
The basic Alignment Search Tool (BLAST) was used to search nucleotide and protein sequence databases (Altschul et al. 1997). Optimal Local Sequence Alignment (LALIGN) was used to compare two nucleotide or protein sequences for their identities (Huang and Huang 1991). Multiple Sequence Alignment of the nucleotide and protein sequences was performed using the CLUSTALW software (Higgins et al. 1992; Thompson et al. 1994). The DRAWTREE program was used to construct the phylogenetic tree (Felsenstein 1989). All of the above mentioned programs were a part of the software package, Biology Workbench (http://workbench.sdsc.edu ).
Genetic materials
A series of different molecular-marker mapping populations were used to map the fad2 and fad6 cDNAs. The use of more than one population enabled identification of polymorphic fragments from all cDNA clones that could be mapped, and in some cases provided confirmation of mapping locations. The following populations were used for genetic mapping: Tx232 × Cm37 and Tx303 × CO159 recombinant inbred lines [Burr and Burr 1991; (http://burr.bio.bnl.gov/ acemaz.html )]; two-hundred F2S1 lines developed from a cross of Illinois High Oil (IHO) × Illinois Low Oil (ILO) described previously (Berke and Rocheford 1995); a population of 147 BC1S1 lines developed from a single plant of IHO cycle 90 crossed to B73; a single F1 plant backcrossed with B73 and plants from a single BC1 ear were self-pollinated. The BC1S1 lines were genotyped with 144 RFLP and SSR markers. A linkage map was created for the ten chromosomes of maize using the mapping program JoinMap (Stam 1993; Stam and van Ooigen 1995), a linkage analysis package was designed to help construct the primary linkage maps of markers segregating in experimental crosses. The final map contained 110 markers with a map length of 1,455.8 cM and an average distance of 14.6 cM between markers (King et al. 1997; manuscript in preparation); a set of 176 F2:3 lines was developed from a cross of inbred lines B73 and Tex6; one F1 plant was self-pollinated to produce F2 seed and those plants were self-pollinated. The F2:3 lines were genotyped with 135 SSR markers and the final map constructed with JoinMap had 113 SSR markers. The linkage map was 1,489 cM with an average interval length of 13.7 cM between markers (Paul et al. 2003).
DNA isolation and southern analysis
DNA was isolated from a bulk of 25–30 seedlings, with an approximately equal amount of tissue from each member of a family of the mapping populations. DNA was isolated using hot CTAB buffer according to a modified DNA isolation procedure (Mikkilineni 1997). Restriction endonuclease digestion, gel electrophoresis and hybridizations were performed as per the procedures outlined in Berke and Rocheford (1995).
Mapping analysis
Map positions of the cDNAs relative to previously mapped molecular markers were determined using the MAPMAKER 3.0 software employing a LOD score of 3.0 (Lander et al. 1987).
RNA materials
Immature ears were sampled 14, 21, 28 and 35 days after self-pollination from 90 Iillinois High Oil (IHO) cycle plants, grown in the field. Total RNA extraction was based on the "single-step method" of RNA isolation (Chomczynski and Chomezynski 1987). Northern transfer and hybridizations with the cDNAs were performed as per Maniatis (1985).
Nomenclature
The terms delta-12 and omega-6 refer to the same 12th carbon position on the fatty acid hydrocarbon chains that is desaturated by fad2.
Results and discussion
Sequence analysis of putative maize fad2 and fad6 cDNAs
BLASTN searches using known fad2 sequences from A. thaliana, G. max and B. napus identified the most highly similar cDNAs in maize cDNA libraries from different tissues. The entire length of the selected clones were double-strand sequenced. To further confirm that the selected cDNA clones encode for the enzyme delta-12 desaturase, a BLASTX search was performed on the Genbank database with a 600–750 nt sequence from the 3′ and 5′ ends of each clone. The best hits were delta-12 desaturases for both 3′ and 5′-end sequences (data not shown). The BLASTX searches supported the assumption that the selected clones were the maize homologs of fad2 orfad6.
Table 1 shows the predicted amino-acid sequence identities of the maize fad2 and fad6 desaturases together with the amino-acid sequences of fad2 and fad6 desaturases of other species. The cdpfe85 clone showed an amino-acid sequence identity with fad2 of A. thaliana, G. max and B. napus species, ranging from 67% to 77% over a stretch of 294 amino acids, in contrast to a very low identity of 27.5% over a stretch of 280 amino acids with the A. thaliana fad6 gene. This provides evidence that cdpfe85 is the maize homolog of fad2. The leaf cDNA library clone cqrak57 shares an 81% amino-acid identity over a stretch of 354 amino acids with A. thaliana fad6 delta-12 desaturase, in contrast to a very low identity at the amino-acid level with fad2 genes from other species. These associations indicate that cqrak57 is most likely the maize homolog of fad6.
Differences at the nucleotide sequence level among cDNAs provide evidence that at least two different genes, fad2 and fad6, encode the enzyme delta-12 desaturase in maize. After the cDNAs were placed into the fad2 and fad6 groups, the Optimal Local Nucleotide Sequence Alignment (LALIGN) program was used to determine the percent identity of the fad2 and fad6 clones with each other. The nucleotide sequence alignment was performed using the 3′-ends of the clones because for some clones the 5′-ends were truncated and comparisons of the variable 5′-ends would not yield meaningful results. Table 2 presents the nucleotide identities of the different maize fad2 and fad6 cDNA clones. cDNAs cdpey46, cdpec60, ccmaa92 and ccrap39 have the highest identities with each other (greater than 90%), while cdpfe85 has approximately 70% identity to the other fad2 clones (Table 2).
For the three embryo-derived fad2 clones: cdpey46, cdpec60 and cdpfe85, the 3′-end sequence analysis in Table 2 shows that cdpey46 and cdpec60 are about 99.2% identical over a stretch of 640 nucleotides whereas the clone cdpfe85 is only about 70% identical over a stretch of 587 nucleotides to either cdpey46 or cdpec60. The magnitude of the difference for the second cDNA comparison supports the concept that cdpfe85 and cdpey46/cdpec60 most likely encode different isoforms of fad2. Since these three cDNAs are derived from the same embryo cDNA library, this suggests that more than one isoform of fad2 is expressed in the embryo (Table 2).
The fad2 and fad6 groups of cDNAs differ considerably at the nucleotide sequence level (Table 2). The extent of the nucleotide overlap (using standard defaults of the program) of the fad6 clones to that of the rest of the clones only ranges between 26 nt to 41 nt, which is very minimal. The percent identities in Table 2 are somewhat misleading for this reason as comparisons take into account only a short stretch of nucleotide sequence overlap. The presence of more than one gene encoding a delta-12 desaturase in maize is consistent with studies in other species that identified a fad2 gene which encodes a delta-12 desaturase in the endoplasmic reticulum and a fad6 gene which encodes a delta-12 desaturase in plastids (Okuley et al. 1994; Somerville and Browse 1996; Byrum et al. 1997).
The fad2 and fad6 cDNAs cluster separately from each other (Fig. 1) which is in agreement with the sequence differences between these groups. Within the fad2 cDNAs, cdpfe85 clusters separately from the rest of the fad2 clones, consistent with cdpfe85, showing only 70% identity with the other fad2 cDNAs. The clones ccmaa92 and ccrap39 cluster together as do the clones cdpec60 and cdpey46. This is consistent with the nucleotide similarities of these two pairs of clones. Notably the cdpfe85 fad2 clone clusters with the fad6 clones before it clusters with the other fad6 clones. This suggests that the composition of the cdpfe85 clone may be somewhat unique in that it is relatively intermediate in sequence composition between the fad2 and fad6 clones.

Phenogram of 3′-ends of the fad2 and fad6 cDNAs
Chromosomal locations of maize fad2 and fad6 genes
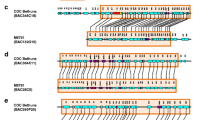
The fad2 and fad6 clones produced complex hybridization patterns in Southern analysis, making scoring of some autoradiograms difficult, with usually just a subset of the fragments visualized were segregating and scorable in each mapping population. The chromosome map positions of the fad2 and fad6 cDNAs determined in different populations is summarized in Fig. 2. In the Tex6 times B73 F2:3 population, hybridization fragments from the fad6 clones cqrak57 and cfbbi58 map to the same position in bin 1.07 on chromosome 1, flanked by markers php200855 and bngl615. Hybridization fragments from cqrak57 and cfbbi58 also map to bin 1.07 in the IHO × ILO F2:3 population, linked to marker npi236. Hybridization fragments from cqrak57 and from cfbbi58 appeared identical within each mapping population. The different mapping populations had different molecular markers on them, but all the markers listed as linked are in bin 1.07. This single locus identified by cqrak57 and cfbbi58 is designated uiu9.

Map positions of the fad2 and fad6 cDNAs in different genetic backgrounds
All of the fad2 clones produced hybridization fragments that mapped to bin 5.06 in three mapping populations. Hybridization fragments from cdpey46, cdpec60, cdpfe85, ccmaa92 and ccrap39 were flanked by markers umc68 and npi115 in IHO × ILO F2:3, php20531 and php10014 in IHO × B73 BC1S1, and umc51 and umc141 in Tex6 × B73 F2:3. This locus is designated uiu10 (Fig. 2).
In Burr's RI populations, which are genetically diverse from the above populations, additional map locations were detected for fad2 clones. Hybridization fragments from ccmaa92 mapped to: bin 4.06, flanked by markers uaz228 and npi584, designated as locus uiu11a; and to bin 10.03 linked to markers dpg5 and glu1, designated as locus uiu11b. Hybridization fragments from cdpfe85 mapped to bin 4.06, linked to uaz144a and dge18, and 2.3 cM from the position of ccmaa92 in bin 4.06, designated the locus of uiu12. Hybridization fragments from all the fad2 clones also mapped to bin 5.06, linked to markers umc108 and uaz204 (Fig. 2).
Hybridization fragments from the fad2 clones map to bin 5.06 in all of the mapping populations. Some of these may be due to cross hybridization of clones in genomic Southern analysis since fad2 in maize appears to be a small gene family with sequence similarities. Separate Southern analysis of all the fad2 and fad6 cDNA clones onto a filter created by blotting the cDNA inserts of the same clones, revealed that all fad2 clones cross hybridize to all of the other fad2 clones under normal hybridization conditions (data not shown). This experiment also revealed that the fad2 and fad6 clones did not cross-hybridize under these conditions.
Clone cdpfe85 has hybridization fragments that map to a unique location on chromosome 4, and cdpfe85 is the only member of a unique group in the phenogram (Fig. 1). The cdpfe85 clone is longer in nucleotide sequence length than the other fad2 clones. These observations strongly suggest that cdpfe85 represents a unique fad2 gene. Clone ccmaa92 has hybridization fragments that map to unique locations on chromosome 4 and 10, and ccmaa92 is in a distinct group with ccrap39 in the phenogram. Clone ccrap39 and ccmaa92 showed identical hybridization patterns, and thus ccrap39 was not mapped since all clones showing identical hybridization patterns in parental hybridizations were mapping to the same location. The ccmaa92/ccrap39 clones appear to represent a second unique fad2 gene.
Clones cdpec60 and cdpey46 map to bin 5.06 and are present in a same group in the phenogram. This suggests that cdpec60/cdpey46 represents a third unique fad2 gene. However all of the fad2 clones have hybridization fragments that map to bin 5.06, so we do not have a unique map location for hybridization fragments from the cdpec60/cdpey46 clones. There may be a unique locus for the cdpec60/cdpey46 clones in maize but the relevant hybridization fragment might have not been polymorphic in the three populations studied. It is possible that bin 5.06 represents the locus for cdpec60/cdpey46 and there are conserved domains in other fad2 genes that cross hybridize to the cdpec60/cdpey46 locus. Alternatively there may be more than one tightly linked fad2 gene or pseudogene present in the bin 5.06 region and fad2 clones may cross hybridize to both loci. The development and use of gene specific probes for each of the fad2 clones in conjunction with high resolution mapping populations in future studies would help resolve this question. Development and use of gene specific probes is beyond the scope of this publication.
Cluster analysis and mapping analysis support the separate identity and genome location of fad6 clones relative to the fad2 clones. The association of fad2 hybridization fragments with map locations on chromosomes 5 and 10, and two locations on chromosome 4 (Fig. 2), is in general consistent with the cluster analysis (Fig. 1) and supports the concept that we have identified at least three different fad2 genes of maize. When the cluster and mapping results are considered together, it is difficult to determine precisely how many different fad2 genes are present in the maize genome. The cluster analysis produced three different fad2 groupings, yet there are still nucleotide differences among the clones within each group. The detection of four different map locations for fad2 cDNAs is supportive of the potential existence of four different fad2 genes.
The duplicated nature of the maize genome (Helentjaris 1995) may have contributed to the genome organization and evolution of the fad2 and fad6 genes. There are large regions on chromosomes 4 and 5 that are considered duplicated (Moore 1995; Moore et al. 1995). Thus the possibility exists that the chromosome 4 and five fad2 loci, uiu10, uiu11a and uiu12, represent some form of duplicated loci.
The region on chromosome 1 where fad6 clones map, and the region on chromosome 4 where two closely linked fad2 genes map also, may represent a small duplicated segment of the maize genome (Helentjaris 1995). Notably the cdpef85 fad2 clone clusters with the fad6 clones before clustering with the other fad2 clones (Fig. 1). The cdpef85 clone is closely linked to another fad2 clone, ccmaa92, on chromosome 4. The possibility that some form of genomic duplication between the regions with bins 1.07 and 4.06 exists, and a local duplication event in the bin 4.06 region may have contributed to the genome organization of these fad2 and fad6 genes.
The fad2 map location in bin 10.03 may also be in a small duplicated region of the maize genome with the general region of bin 4.06 (Helentjaris 1995). This may also represent some form of duplication of the fad2 genes that contributed to their organization. However, until more precise and comprehensive maize genome duplication maps are available, it is not possible to determine whether all the cDNAs come from duplicated regions of the maize genome or not, and how this may have contributed to fad2 and fad6 gene evolution within the maize genome.
Transcript analysis
The fad2 cDNA clones were hybridized to RNA from embryo and endosperm tissue at four developing stages: 14, 21, 28 and 35 days after pollination (DAP). Northern analysis revealed the transcript size for all the fad2 clones to be 1.8 kb (Fig. 3). Hence the transcript size of at least one form of fad2 gene expressed in the maize embryo is 1.8 kb. Since all the clones resulted in the same transcript size, this suggests that the fad2 genes encoding different isoforms may have the same transcript size. However, since the fad2 clones cross-hybridize with each other it is also possible that the Northern blots with different cDNAs as probes only detected a highly abundant transcript of just one isoform.

Northern blot of RNA from embryo and endosperm at 14, 21, 28, 35 DAPs; hybridized with cDNA probe cdpec60 (fad2)
The Northern blots revealed that the fad2 gene temporal-transcription pattern appears to be relatively steady at 14, 21, 28 and 35 DAP in the embryo, with a higher level of transcription at day 14. In the endosperm, the expression is also relatively constant at 14, 21 and 28 DAP and then decreases by 35 DAP. However, without gene specific probes, it is difficult to determine the precise temporal patterns of individual clones, as some genes may exhibit different expression patterns that are masked by cross-hybridization. None of the fad6 cDNAs hybridized to the RNA from the embryo and endosperm. These findings suggest that fad6 is not transcribed in the embryo or endosperm.
Comparison of the fad2/fad6 candidate gene with QTLs for oleic acid levels
The map positions of fad2 and fad6 cDNAs were compared with the chromosome locations of QTLs for the ratio of oleic acid/linoleic acid. Most noteworthy no fad2 hybridization fragments were mapped in any of the populations to bin 6.04, which has the ln1 locus and the largest reported QTL for the ratio of oleic to linoleic fatty acid. Examination of the rest of the maize genome indicated that none of the fad2/fad6 genes appear to map precisely to any of the QTL positions previously reported for the ratio of oleic/linoleic acid. This strongly suggests, but does not prove, that the previously detected QTLs for the ratio of oleic/linoleic acid are not due to allelic variation in the structural gene fad2 orfad6. Furthermore the fad6 clones assayed do not appear to be transcribed in the embryo, which greatly reduces the likelihood that this gene is involved in affecting fatty acid levels in the seed.
Although we were comprehensive in our use of multiple mapping populations with genetically diverse parents, we cannot eliminate the possibility that there is a copy of a fad2 or fad6 gene in bin 6.04 with the ln1 locus, or in any of the previously mapped QTLs. A clone may map to one of these regions, but we may have not identified a polymorphic fragment from this locus and thus were not able to reveal it in mapping analyses.
The mapping results lead to considering the possibility that transcriptional, post-transcriptional and/or post-translational modifications of fad2 gene expression may be the basis of QTLs for the ratio of oleic/linoleic acid. Co-factors of fad2 might be involved in controlling relative levels of fad2 gene expression. Other genes in the fatty acid biosynthetic pathway, either upstream or downstream, might also influence delta-12 desaturase efficiency. Thus, although conversion of oleic to linoleic acid needs to go through the delta-12 desaturase, apparently there are a number of other loci throughout the genome that can modify this process.
References
Alrefai R, Berke TG, Rocheford TR (1995) Quantitative trait locus analysis of fatty acid concentrations in maize. Genome 38:894–901
Altschul SF, Thomas L, Madden, Alejandro A, Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Berke TG, Rocheford TR (1995) Quantitative trait loci for flowering, plant and ear height and kernel traits in maize. Crop Sci 35:1542–1549
Browse JaCS (1991) Glycerolipid metabolism: biochemistry and regulation. Annu Rev Plant Physiol Plant Mol Biol 42:467–506
Burr B, Burr FA (1991) Recombinant inbreds for molecular mapping in maize – theoretical and practical considerations. Trends Genet 7:55–60
Byrum JR, Kinney AJ, Stecca KL, Grace DJ, Diers BW (1997) Alteration of the omega-3 fatty acid desaturase gene is associated with reduced linolenic acid in the A5 soybean genotype. Theor Appl Genet 94:356–359
Chomczynski PaS, Chomczynski N (1987) Single-step Method of RNA isolation by acid guanidium thicyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Felsenstein J (1989) PHYLIP – Phylogeny Inference Package (Version 3.2). Cladistics 5:164–166
Harwood JL (1997) Plant lipid metabolism. In: Dey PM, Harborne JB (eds) Plant biochemistry. Academic Press, pp 237–272
Helentjaris T (1995) Atlas of duplicated sequences. Maize Genet Coop Newslett 69:67–81
Heppard EP, Kinney AJ, Stecca KL, Miao G-H (1996) Developmental and growth temperature regulation of two different microsomal omega-6 desaturase genes in soybeans. Plant Physiol 110:311–319
Higgins DG, Bleasby AJ, Fuchs R (1992) CLUSTAL V: improved software for multiple sequence alignment. CABIOS 8:189–191
Huang XM, Huang W (1991) A time-efficient, linear-space local similarity algorithm. Adv Appl Math 12:337–357
Jellum MD (1966) Fatty acid composition of corn oil of parental inbreds and reciprocal crosses. J Hered 57:243–244
King JJ, Hotchkiss JR, Rocheford TR (1997) Proc 33rd Annual Illinois Corn Breeders School. Champaign Itllinois March 3–4, pp 24–55
Lander ES, Green P, Abrahamson J, Barlow A, Daley MJ, Lincoln SE, Newburg L (1987) MAPMAKER: an interacative computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–183
Mattson FR, Grundy SM (1985) J Lipid Res 26:194
Maniatis T, Fritsch EF, Sambrook J (1985) Molecular cloning, a laboratory manual. Cold Spring Harbour Laboratory. Cold Spring Harbour, New York
Mikkilineni V (1997) Restriction fragment length polymorphism analysis of the Illinois Long-term selection chemical strains, crop sciences. University of Illinois, Urbana, pp 107–113
Moore G (1995) Cereal genome evolution: pastoral pursuits with lego genomes. Curr Opin Genet Dev 5:717–724
Moore G, Foote T, Helentjaris T, Devos K, Kurata N, Gale M (1995) Was there a single ancestral cereal chromosome? Trends Genet 11:81–82
Ohlrogge JB, Jaworski JG (1997) Regulation of fatty acid synthesis. Ann Rev Plant Physiol Plant Mol Biol 48:109–136
Ohlrogge JB, Browse J, Somerville CR (1991) The genetics of plant lipids. Biochim Biophys Acta 1082
Ohlrogge JB, Jaworski JB, Post-Beittenmiller D (1993) De novo fatty acid biosynthesis. In Lipid metablolism in plants. CRC Press, Boca Raton, Florida pp 3–32
Okuley J, Lightner J, Feldmann K, Yadav N, Lark E, Browse J (1994) Arabidopsis FAD2 gene encodes the enzyme that is essential for polyunsaturated lipid synthesis. Plant Cell 6:147–158
Paul C, Naidoo G, Forbes A, Mikkilinenini V, White D, Rocheford T (2003) Quantitative trait loci for low aflatoxin production in two related maize populations. Theor Appl Genet (in press)
Plewa MJ, Plewa DFW (1975) Monosomic analysis of fatty acid composition in embryo lipids of Zea mays L. Genetics 277–286
Poneleit CG (1976) Chromosome location for control of oleic and linoleic acids in corn oil. Agron Abs 8:59
Poneleit CG, Alexander DE (1965) Inheritance of linoleic and oleic acids in maize. Science 1585–1586
Roche IA de la, Alexander DE, Weber EJ (1971) Inheritance of Oleic and Linoleic acids in Zea mays L. Crop Sci 11:856–859
Scheffler JA, Schimdt H, Sperling P, Parkin IAP, Luhs W, Lydiate DJ, Heinz E (1997) Desaturase multigene families of Brassica napus arose through genome duplication. Theor Appl Genet 94:583–591
Shadley JD, Shadley DFW (1980) Identification of a factor in maize that increase embryo fatty acid unsaturation by trisomic and B-A translocation analysis. Can J Genet Cytol 11–19
Somerville C, Browse J (1996) Dissecting desaturation: plants prove advantageous. Trends Cell Biol 6:148–153
Stam P (1993) Construction of integrated genetic linkage maps by means of a new computer package – JoinMap. Plant J 3:739–744
Stam P, Van Ooijen JW (1995) JoinMap (tm) version 2.0: software for the calculation of genetic linkage maps. Centre for Plant Breeding and Reproduction Research, Wageningen, The Netherlands
Thompson JD, Thompson HDG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Widstrom NW, Jellum MD (1984) Chromosomal location of genes controlling oleic and linoleic acid composition in the germ oil of maize inbreds. Crop Sci 24:1113–1115
Wright A (1995) A gene conditioning high oleic maize oil, OLC 1. Maydica 40:85–88
Acknowledgements
We acknowledge Dr. Tim Helentjaris for assistance and advice in the identification and use of fad2 and fad6 cDNAs from Pioneer Hi-Bred International cDNA libraries. The cDNAs evaluated in this study were provided by Pioneer Hi-Bred International. Some of this work was performed while T.R.R was on sabbatical in the Cereals Department, John Innes Centre, Norwich, U.K. We acknowledge the assistance of Gordon Huestis in mapping some cDNAs while working with T.R.R at the John Innes Centre. This research was supported by a grant from the Illinois-Missouri Biotechnology Alliance Program (USDA), and facilitated by gift support from Monsanto and Pioneer Hi-Bred International.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Salamini
Rights and permissions
About this article
Cite this article
Mikkilineni, V., Rocheford, T.R. Sequence variation and genomic organization of fatty acid desaturase-2 (fad2) and fatty acid desaturase-6 (fad6) cDNAs in maize. Theor Appl Genet 106, 1326–1332 (2003). https://doi.org/10.1007/s00122-003-1190-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-003-1190-7