
Abstract
This study aims to explore the mechanism underlying miR-142-3p regulating myocardial injury induced by coronary microembolization (CME) through ATXN1L. miR-142-3p overexpression or ATXN1L knockout adenovirus vectors were injected into rats before CME treatment. Cardiac functions were examined by echocardiography, and pathologies of myocardial tissues were assessed. Then, serum cTnI and IL-1β contents and concentrations of IL-1β and IL-18 in cell supernatant were measured. Immunofluorescence determined the localization of histone deacetylase 3 (HDAC3). The interaction between miR-142-3p and ATXN1L as well as the binding between HDAC3 and histone 3 (H3) was identified. The binding of ATXN1L and HDAC3 to NOL3 promoter was verified using ChIP. The levels of ATXN1L, NOL3, and miR-142-3p as well as apoptosis- and pyroptosis-related proteins and acetyl-histone 3 (ac-H3) were evaluated. CME treatment impaired the cardiac functions in rats and increased cTnI content. CME rats showed microinfarction foci in myocardial tissues. After CME treatment, miR-142-3p and NOL3 were modestly expressed while ATXN1L content was elevated, in addition to increases in apoptosis and pyroptosis. miR-142-3p overexpression or ATXN1L knockout alleviated CME-induced myocardial injury, cardiomyocyte apoptosis, and pyroptosis in myocardial tissues. miR-142-3p regulated ATXN1L expression in a targeted manner. In the cellular context, miR-142-3p overexpression attenuated apoptosis and pyroptosis in cardiomyocytes, which was partly counteracted by ATXN1L overexpression. ATXN1L functioned on cardiomyocytes by promoting deacetylation of H3 through HDAC3 and thus inhibited NOL3 expression. Inhibition of HDAC3 or overexpression of NOL3 ameliorated the promotive effects of ATXN1L on cardiomyocyte apoptosis and pyroptosis. In vivo and in vitro evidence in this study supported that miR-142-3p could attenuate CME-induced myocardial injury via ATXN1L/HDAC3/NOL3.
Highlights
-
CME model witnessed aberrant expression of miR-142-3p, ATXN1L, and NOL3;
-
miR-142-3p negatively regulated ATXN1L;
-
miR-142-3p mediated CME-induced myocardial injury through ATXN1L;
-
ATXN1L promoted deacetylation of H3 through HDAC3 and thus inhibited NOL3 expression;
-
ATXN1L acted on cardiomyocyte apoptosis and pyroptosis through HDAC3/NOL3 axis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coronary microembolization (CME), caused by the spontaneous rupture of a susceptible coronary atherosclerotic plaque or by percutaneous coronary intervention (PCI) [1], could induce slow flow or no-reflow phenomenon, leading to myocardial infarction, contractile dysfunction, malignant arrhythmias, and even sudden cardiac death [2]. Growing evidence corroborated that inflammatory response and apoptosis are two pathological events involved in CME-induced myocardial injury and progressive cardiac dysfunction [3,4,5]. Apart from apoptosis, pyroptosis, an important type of programmed cell death accompanied by inflammatory responses, also occurs in the pathogenesis of cardiovascular diseases, such as myocardial infarction and atherosclerosis [6]. It is reasonable to believe that interventions which attenuate myocardial apoptosis and pyroptosis could be cardioprotective for CME-related cardiac dysfunction.
MicroRNAs (miRNAs) are noncoding RNAs with a length of 19 to 25 nucleotides that hold great significance for translational inhibition or the degradation of messenger RNA (mRNA) at the posttranscriptional level and are capable of affecting a variety of pathophysiological processes [7]. Previously, we identified the differentially expressed miRNAs in myocardial tissues of CME pigs by using miRNA chip analysis and found the expression of miR-142-3p and miR-136-5p was downregulated most significantly [8]. miR-142-3p has been demonstrated to serve, in part, as an anti-apoptotic gene in hypoxia/reoxygenation-induced cardiomyocytes [9]. More importantly, our preliminary work has demonstrated that overexpression of miR-142-3p mitigated CME-induced myocardial injury and inflammation response [10], yet the mechanisms behind miR-142-3p alleviating CME-induced myocardial injury remain obscure. ATXN1L is a member of the Ataxin protein family and shares an ATXM1 and HMG-box protein 1 (AXH) domain required for interaction with Capicua [11], and ATXN1L has been regarded as a regulator of hematopoietic stem cell function [12]. However, whether miR-142-3p mediates ATXN1L in CME is still unavailable.
Herein, this study used a CME rat model to explore the potential action mechanism of miR-142-3p in myocardial injury in vivo. LPS and ATP have been commonly used to induce inflammatory responses and pyroptosis in cells [13,14,15], and this study thereby established oxygen–glucose deprivation (OGD) and lipopolysaccharide (LPS) models in primary cardiomyocytes to identify the role of miR-142-3p in apoptosis and pyroptosis. More importantly, this study manifested the interactions between miR-142-3p and ATXN1L, providing an insight for treating CME-induced myocardial injury.
Materials and methods
A CME rat model
Healthy Sprague–Dawley (SD) rats (200~250 g) provided by SPF (Beijing) Biotechnology Co. were anesthetized with 50 mg/kg ketamine and 10 mg/kg xylazine by intraperitoneal injection. Then, the animals were subjected to tracheotomy after skin antisepsis and ventilated with an animal ventilator with a respiratory rate of 60~70/min and a tidal volume of 50 ml. The thoracotomy was performed to expose the heart, and the fat pad was separated to isolate the ascending aorta. A total of 0.1 ml microembolization spheres (3 × 104/ml, approximate 3000 microspheres) were suctioned and mixed before being infused into the left ventricle at the cardiac apex, and the aortic root was ligated for 15 s. The sternum was closed after the recovery of heart rate and respiration, and the ventilator was removed following the recovery of autonomous respiration. All experiments were ratified by Guilin Medical University and conducted per the guidance for the care and use of laboratory animals issued by the National Institutes of Health. All efforts had been made to minimize pain in animals.
The rats were randomly divided into sham, CME, CME + AAV-miR-NC, CME + AAV-miR-142-3p or CME + ATXN1L KO groups (n = 10 per group). Rats in the CME group were treated as described above. The sham-operated rats were administrated with an equal dose of normal saline. In the CME + AAV-miR-NC group or CME + AAV-miR-142-3p group, rats were intravenously injected in tails with AAV-miR-NC or AAV-miR-142-3p (1 × 1011 vector genomes; Hanbio Biotechnology, Shanghai, China) four weeks before the establishment of the CME rat model. Rats in the CME + ATXN1L KO group were intravenously injected with ATXN1L knockout adenovirus vectors in tails four weeks before the establishment of the CME rat model.
Evaluation of cardiac functions by echocardiography
An ultrasonic cardiograph (Philips sonos 7500) equipped with a 10-MHz transducer was used to detect left ventricular ejection fraction (LVEF), left ventricular end diastolic diameter (LVEDd), left ventricular fraction shortening (LVFS), and cardiac output (CO) at 0, 3, 6, 12, and 24 h after modeling. After anesthesia, echocardiography with the modalities of two-dimensional and M-mode imaging was conducted from parasternal long-axis view and apical four chamber and two chamber views to assess end-diastolic volume (EDV) and end-systolic volume (ESV) of left ventricle as well as LVEDd and left ventricular end systolic diameter (LVEDs). LVEF and FS were calculated as follows: LVEF (%) = (EDV – ESV)/EDV × 100; FS (%) = (LVEDd – LVEDs)/ LVEDd × 100. All values were obtained by averaging the measurements from three cardiac cycles. All echocardiographic detections were performed by experienced specialist doctors who were blind to the grouping of this study. After the last time of the echocardiography, the rats were anesthetized and euthanized, and immediately, blood samples were collected and the heart was obtained for following experiments.
Quantification of white blood cells in blood samples
The number of white blood cells in the blood samples was evaluated by a Beckman Coulter LH 750 hematology analyzer (Beckman Coulter, Miami, FL, USA).
ELISA
ELISA kits (R&D, USA) were used to evaluate cardiac troponin I (cTnI) and IL-1β levels in rat serum and IL-1β and IL-18 contents in the supernatant of cardiomyocytes.
Hematoxylin–eosin staining
The heart was isolated and sectioned parallel to the atrioventricular groove into 7 to 9 pieces (5 to 8 mm thick). Five pieces of the ventricular anterior wall (50~100 mg) were frozen in liquid nitrogen and retained in a refrigerator at – 80 °C to detect gene and protein expression levels. The rest of the myocardial tissues were used to prepare paraffin sections and the sections were fixed by 4% paraformaldehyde for 12 h prior to paraffin embedding. The tissues were further sliced along the left ventricular long-axis into 4-μm-thick sections. After that, the sections were dewaxed with alcohol into distilled water and subjected to hematoxylin staining for 9 min. Then, the sections were differentiated by 0.7% hydrochloric acid alcohol for 15 s and washed three times with flowing water. The sections were immersed in distilled water for 15 s until the nuclei turned blue and washed by flowing water. Alcoholic eosin staining was performed for 1 min before the sections were washed by tap water. After dehydration in absolute alcohol and sealing by neutral balsam, the sections were viewed under a microscope to assess the morphology of the myocardial tissues.
Masson staining
The paraffin sections of the myocardial tissues were dewaxed before Weigert’s iron hematoxylin staining for 5 min. Then, the sections were differentiated in hydrochloric acid alcohol, followed by washing in running water. After that, the sections were treated by acid fuchsin for 5 min before further treatment with phosphomolybdic acid hydrate for 3 min, counterstaining with toluidine blue for 5 min, and glacial acetic acid treatment for 1 min. The sections were conventionally dehydrated, transparent and sealed for observation under a microscope.
TUNEL staining
The paraffin sections of the myocardial tissues from the myocardial infarction border zone were washed with xylene for 5 min and rinsed successively in 100%, 95%, 90%, 80%, and 70% alcohol for 3 min each, followed by washing twice in PBS. The myocardial tissues were cultured with proteinase K for 30 min and permeabilized at 21–37 °C for 8 min. Cardiomyocytes were fixed with methyl aldehyde for 15 min and centrifuged at 4 °C and 1500 rpm for 5 min, after which the cells were washed twice with PBS. The supernatant was abandoned, and the cell pellet was fixed with 70% ice-cold alcohol for 15 min and centrifuged at 4 °C and 1500 rpm for 5 min. After washing twice in PBS, the supernatant was discarded, and the cells were incubated with 2 μl TdT and 48 μl biotin-labeled or Cy3 fluorescence probe-labeled dUTP (Beyotime, Shanghai, China) in a dark and wet box for 1 h. After washing three times with PBS, the sections were sealed with anti-fade mounting medium and observed under a fluorescence or an optical microscope. The nuclei were dyed by DAPI. TUNEL-positive cells (%) = the number of brown or red cells/the number of total cells × 100%.
Observation of myocardial ultrastructure by transmission electron microscope
The ventricular myocardium was collected from the left ventricular free wall by a double-edged blade (the tissues of each group were collected from the myocardial infarction border zone) and then sliced into 1-mm3 pieces. Then, the pieces were fixed with glutaraldehyde phosphate buffer at 4 °C for 24 h and subjected to dehydration, embedding, and staining. Afterwards, the tissues were prepared into 50- to 70-nm-thick sections before the observation of the myocardial ultrastructure by a transmission electron microscope (TEM).
Immunohistochemistry
The prepared sections were immersed in EDTA buffer (pH = 9.0) for antigen retrieval and heated in a boiling water bath for 10 min. After natural cooling, the sections were rinsed three times with PBS and incubated with 3% hydrogen peroxide solution for 10 min to inhibit the generation of endogenous peroxidases. The sections were blocked with serum for 30 min and incubated with 50 μl blocking solution-configured primary antibodies against caspase-1 (ab74279, 1:50, Abcam, CA, USA) and IL-1β (ab2105, 1:100, Abcam, CA, USA) at 4 °C overnight. After rinsing three times in PBS, the sections were incubated with secondary antibody at room temperature for 0.5 h, after which the sections were rinsed three times with PBS and developed in DAB. After being stained with hematoxylin for 3 min and differentiated with 1% hydrochloric acid alcohol for 1~3 s, the sections were washed with flowing water and treated with 0.6% ammonium hydroxide. The sections were subjected to dehydration in gradient alcohol, permeabilization by xylene, and sealing by neutral balsam. An optical microscope was used to observe and analyze the expression of caspase-1 and IL-1β in the rat myocardial tissues.
Primary rat cardiomyocytes
Primary cardiomyocytes were isolated from neonatal SD rats within 24 h of their birth. The neonatal rats were anesthetized and sacrificed by cervical dislocation. After the chest was sterilized, the hearts were isolated and placed in PBS to remove atrial tissues. The retained ventricular tissues were cut into 1-mm3 pieces and preserved in a dried vial containing penicillin and then disassociated using a water bath with collagenase and 0.08% neutral proteinase (Roche, Penzberg, Upper Bavaria, Germany) at 37 °C for 5 min, after which the cell supernatant was removed. The cell suspension was filtered twice (for 3~5 min each) to avoid biggish fragments of tissues. The digestion was terminated by addition of 5 ml DMEM (Thermo, Waltham, MA, USA) containing 10% fetal bovine serum (FBS). The isolated cardiomyocytes were centrifuged at 1500 rpm for 5 min and the supernatant was discarded. After that, the isolated cardiomyocytes were resuspended in 10% FBS-supplemented DMEM and centrifuged at 1500 rpm for 5 min, and the supernatant was then removed, after which the aforesaid procedures were repeated once. The cells were pre-incubated for 2 h and purified by differential adhesion method to remove fibroblasts and endothelial cells. Non-adherent cells were collected and seeded in 6-well plates containing 10% FBS-supplemented DMEM. Thereafter, the cells were incubated at 37 °C with 5% CO2 and the medium was replaced after 24 h, during which the cell growth was assessed to avoid contamination. After that, the medium was replaced in 1~2 days according to the cell growth. For 3~4 days, if spontaneously beating cells were observed, the cells could be digested and seeded into culture plates for later use. The morphology of the cardiomyocytes was visualized under an inverted microscope.
Detection of the purity of cardiomyocytes
The purity of cardiomyocytes was detected by immunofluorescence. After cell culture for 48 h, the slides of cardiomyocytes were washed slightly with PBS, fixed with ice-cold 75% alcohol for 15 min, and washed three times with PBS, followed by incubation with 0.3% Triton X-100 for 30 min. The cardiomyocytes were washed three times with PBS and sealed with goat serum at room temperature for 60 min. Afterwards, the cardiomyocytes were incubated with rabbit anti-rat cTnI polyclonal antibody (ab155047, 1:200, Abcam, Cambridge, MA, USA) or PBS (as the negative control) at 4 °C overnight and supplemented with Alexa Fluor 488-conjugated goat anti-rabbit IgG (ab150077, 1:100, Abcam) for 1-h incubation after being washed three times with PBS. DAPI was used to stain the nuclei for 10 min before washing three times with PBS. After sealing with anti-fade mounting medium, the purity of the cardiomyocytes was evaluated by an inverted microscope.
Cell transfection and grouping
Primary cardiomyocytes were incubated in vitro and randomly grouped into Blank, OGD, LPS (inflammation model), OGD + ATXN1L KO, LPS + ATXN1L KO, OGD + miR-142-3p, LPS + miR-142-3p, OGD + ATXN1L-OV + miR-142-3p, LPS + ATXN1L-OV + miR-142-3p, OGD + ATXN1L-OV, LPS + ATXN1L-OV, OGD + ATXN1L-OV + histone deacetylase (HDAC3)-in, LPS + ATXN1L-OV + HDAC3-in, OGD + NOL3-OV, LPS + NOL3-OV, OGD + ATXN1L-OV + NOL3-OV, and LPS + ATXN1L-OV + NOL3-OV groups.
Blank group was used as blank control. Cardiomyocytes in the OGD group were cultured in low glucose medium under a hypoxic condition (2% O2, 5% CO2, and 93% N2) for 12 h and then maintained in normal medium and normoxic atmosphere (5% O2, 20% CO2, and 75% N2) for 12 h. Cardiomyocytes in the LPS group were treated with LPS (1 μg/ml) for 4 h and ATP (5 mM) for 30 min. Cells in the OGD + ATXN1L KO, LPS + ATXN1L KO, OGD + ATXN1L-OV, and LPS + ATXN1L-OV groups were transfected with ATXN1L knockout plasmid (ATXN1L KO) or ATXN1L overexpression plasmid (ATXN1L OV) (Shanghai GenePharma Co., Ltd., Shanghai, China) and then treated with OGD or LPS plus ATP. Cells in the OGD + miR-142-3p and LPS + miR-142-3p groups were transfected with miR-142-3p mimic (Shanghai GenePharma Co., Ltd., Shanghai, China) before OGD or LPS plus ATP treatment. Cells in the OGD + NOL3-OV and LPS + NOL3-OV groups were transfected with NOL3 overexpression plasmid (NOL3-OV) (Shanghai GenePharma Co., Ltd., Shanghai, China) prior to OGD or LPS plus ATP treatment. The transfections were performed using Lipofectamine 2000 (Thermo Fisher Scientific, MA, USA) in line with the manufacturer’s protocols.
Dual luciferase reporter assay
The potential binding site between miR-142-3p and ATXN1L was predicted by DIANA and Starbase. The mutant-type and wild-type sequences of the binding site were designed and then inserted into luciferase reporter vectors (pGL3-Promoter). Accordingly, the vectors were separately named as MT-ATXN1L and WT-ATXN1L. Then, the MT-ATXN1L or WT-ATXN1L was transfected with miR-142-3p mimic or miR-142-3p inhibitor into cardiomyocytes. After 48 h, Firefly and Renilla luciferase activities were examined by dual luciferase reporter assay kit (Promega, Madison, USA). Renilla luciferase activity was reviewed as the internal control, and relative activity was defined as the ratio between Renilla luciferase activity and Firefly luciferase activity.
RNA binding protein immunoprecipitation
The binding between miR-142-3p and ATXN1L was determined using an RNA binding protein immunoprecipitation (RIP) kit (Merck Millipore, Billerica, MA, USA). In short, the cells were lysed with an equal volume of lysis buffer on ice for 5 min and centrifuged at 14,000 rpm and 4 °C for 10 min to collect the supernatant. A part of the cell lysate was used as an input, and the other part was co-immunoprecipitated with anti-AGO2 antibody (ab186733, 1:30; Abcam, CA, USA). Next, 50-μL magnetic beads were resuspended in RIP Wash Buffer (100 μL), and mixed with 5 μg antibody to obtain the magnetic bead-antibody complex. The magnetic bead-antibody complex was resuspended in RIP Wash Buffer (900 μL), and incubated with the cell lysate (100 μL) at 4 °C overnight. The eluted sample and input were detached by proteinase K, from which the RNA was extracted. The expression of miR-142-3 and ATXN1L was examined by reverse transcription quantitative polymerase chain reaction (RT-qPCR). IgG (ab205718, Abcam, CA, USA) was regarded as the negative control.
Flow cytometry
The cardiomyocytes were washed twice with PBS before trypsinization and centrifuged at 1000 rpm and room temperature for 5 min, after which the supernatant was removed. Following re-suspension with PBS, the cells were centrifuged at 1000 rpm and room temperature for 5 min, and the supernatant was removed. Ice-cold 1 × binding buffer (490 μl) at a density of 105~106/ml was applied to resuspend the cells, and 5 μl Annexin V-FITC and 5 μl PI were added and cultured with the cardiomyocytes on ice in the dark for 10 min before detection of cardiomyocyte apoptosis by flow cytometry.
Localization of HDAC3 by immunofluoresence
The density of the cardiomyocytes was adjusted to 1 × 104/ml before the preparation of cell slides. ATXN1L overexpression lentiviral vector was transfected into the cardiomyocyte. When the confluence reached 60~80%, the cells were washed three times with PBS for 5 min each and fixed by 4% paraformaldehyde for 15 min, followed by washing with PBS. Then, the cardiomyocytes were permeabilized with 1% Triton X-100 dissolved by PBS for 2 min and then rinsed three times with PBS for 5 min each, after which the cells were sealed with 5% serum for 1 h and washed with PBS. Next, the cardiomyocytes were incubated with primary antibody against HDAC3 (3949S, 1:100, Cell signaling technology, MA, USA) at 4 °C overnight and Alexa Fluor 647-labeled secondary antibody (1:100) in the dark for 2 h. The nuclei were stained by DAPI (1:150), and the cells were visualized under a fluorescent microscope.
Co-immunoprecipitation
The supernatant of cardiomyocytes (400 μl) was incubated with ATXN1L antibody or IgG antibody (the negative control) at 4 °C overnight, and incubated with Protein G/A beads at 4 °C for 3~5 h on a rotator. After the incubation, the mixture was centrifuged at 4 °C and 1000 g for 5 min to collect the bead-antibody-protein complexes. The complexes were washed three times with washing buffer (50 mM Tris–HCL/pH 7.4, 100 Mm NaCL, 5 mM CaCL2, 5 mM MgCL2, 0.1% Nonidet P-40) before resuspension with 1 × SDS-PAGE loading buffer. The prepared protein samples were heated in a metal bath at 100 °C for 5 min and separated by 10% SDS-PAGE. Next, the protein was blotted onto PVDF membranes, and the membranes were incubated with primary antibodies for ATXN1L (ab241958, 1:100, Abcam, CA, USA), HDAC3 (3949S, 1:1000, Abcam, CA, USA), and histone 3 (H3, ab1791, 1:1000, Abcam, CA, USA) and then goat anti-rabbit IgG (1:5000, Beijing ComWin Biotech Co., Ltd., Beijing, China) before western blot analysis.
Chromatin immunoprecipitation
The binding of ATXN1L and HDAC3 with NOL3 promoter was verified using a chromatin immunoprecipitation (ChIP) kit (Millipore, Billerica, MA, USA). The crosslink between DNA and protein was fixed for 30 min using formaldehyde, after which the DNA was isolated and sonicated into 200–1000 bp fragments. The fragment was incubated with ATXN1L, HDAC3, or IgG antibody. The DNA fragment pulled down was evaluated using qPCR.
RT-qPCR
After corresponding treatment, the cardiomyocytes were dissolved in 1 ml Trizol (Thermo Fisher Scientific, MA, USA), and total RNA was isolated in line with the based on the user manual. Total RNA was quantified and reverse transcribed. PCR reaction was conducted based on the protocols of fluorescent quantitative PCR kit (Takara, Dalian, China) on the fluorescent quantitative PCR instrument ABI7500 (Applied Biosystems, Inc., Foster City, San Francisco, CA, USA). The PCR reaction was initiated with pre-denaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 20 min, and extension at 72 °C for 34 s. RT-qPCR analysis quantified the expression of miR-142-3p, ATXN1L, and NOL3. All primer sequences (Table 1) were synthesized by GENEWIZ, Inc. GAPDH and U6 were separately used to normalize mRNA and miRNA expression levels. Data analysis was performed using 2−ΔΔCt method [16], and the formula presented as ΔΔCt = [Ct(target gene)-Ct(internal gene)]experimental group-[Ct(target gene)-Ct(internal gene)]control group.
Western blot
The cardiomyocytes were washed three times in ice-cold PBS before being lysed. After being placed on ice for 30 min, the lysate was centrifuged at 4 °C and 12,000 rpm for 10 min. The supernatant was aliquoted into centrifuge tubes (0.5 mL) and preserved at – 20 °C or quantified by a BCA kit (Beyotime, Shanghai, China). The protein was denatured with 6 × SDS loading buffer at 100 °C and separated using SDS-PAGE. Ice-cold (4 °C) transfer buffer was applied for membrane transfer for 1.5 h, and then the membranes were sealed with TBST-configured 5% non-fat milk powder for 1 h. TBST-configured primary antibodies for NOL3 (ab126238, 1:1000), caspase-3 (ab13847, 1:500), cleaved-caspase-3 (ab2302, 1:500), GSDMD-N (ab215203, 1:1000), caspase-1 (ab179515, 1:1000), acetyl-histone 3 (ac-H3, ab4729, 1:1000) (Abcam, CA, USA), β-actin (4970S, 1:1000), caspase-8 (4790S, 1:1000), cleaved-caspase-8 (8592S, 1:1000), NLRP3 (13158S, 1:1000), and ASC (67824 T, 1:1000) (Cell Signaling Technology, Boston, USA) were incubated with the membranes at 4 °C overnight. After washing with TBST, the membranes were incubated with goat anti-rabbit IgG or goat anti-mouse IgG (1:5000, Beijing ComWin Biotech Co., Ltd., Beijing, China) at room temperature for 2 h and washed with TBST before color development and detection.
Statistical analysis
The data analysis was conducted using SPSS 18.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism 6.0 (GraphPad Software Inc.). All data were displayed as mean ± standard deviation. T-test was applied for comparison between two groups, and one-way analysis of variance was applied to assess the differences among multiple groups. Linear relationship between two variables was produced by Pearson correlation coefficient. A P value less than 0.5 was considered statistically significant.
Results
miR-142-3p overexpression alleviates CME-induced myocardial injury
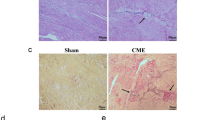
To disclose the action mechanism of miR-142-3p in CME-induced myocardial injury, a CME rat model was established. Based on the results of echocardiography, hematoxylin–eosin (H&E) staining, and Masson staining, the CME-induced rats had decreased LVEF, LVFS, and CO, in addition to increased LVEDd, than sham-operated rats; CME-induced rats had disordered myofiber, indistinct cellular structure, a fraction of swollen cells, and more microinfarction foci; relative to the sham-operated rats, the CME-induced rats had elevated serum cTnI expression and decreased miR-142-3p expression (Fig. 1A-I). These findings indicated the successful establishment of the CME rat model and that miR-142-3p expressed at a low level in the model rats. Moreover, cardiomyocyte apoptosis was enhanced in the model rats, concomitant with disordered myocardial fiber structure, raptured cardiomyocytes, and increased caspase-1 and IL-1β levels in myocardial tissues, compared with the sham-operated rats (Fig. 1J–P). In addition, the number of white blood cells and IL-1β expression in the blood of the model rats elevated significantly than those in the sham-operated rats (Fig. 1Q–R). The abovementioned results suggested CME-induced cardiomyocyte apoptosis and pyroptosis in rats.

miR-142-3p represses myocardial injury induced by CME. miR-142-3p expression in myocardial tissues was assessed by RT-qPCR (A); Echocardiography results for LVEF (B), CO (C), LVEDd (D), and LVFS (E); H&E staining measured the pathologies of rat myocardial tissues (F); Masson staining showed microinfarction foci and myocardial injury in myocardial tissues (G); Quantitative analysis for microinfarction size (H); Serum cTnI content was determined by ELISA (I); Apoptosis of cardiomyocytes in myocardial tissues as measured by TUNEL (J, K); Western blot analysis evaluated the levels of cleaved caspase-3/8 (L); Ultrastructure of myocardial tissues by TEM (M); Contents of caspase-1 (N) and IL-1β (O) in myocardial tissues were detected by immunohistochemistry (P). The number of white blood cells (Q) and IL-1β level in rat blood (R) were assessed. N = 10. *P < 0.05, **P < 0.01, ***P < 0.001. CME, coronary microembolization; LVEF, left ventricular ejection fraction; LVEDd, left ventricular end diastolic diameter, LVFS, left ventricular fraction shortening; CO, cardiac output; H&E, hematoxylin–eosin; TEM, transmission electron microscope
To probe the impact of miR-142-3p on CME-induced myocardial injury, the rats were intravenously injected with miR-142-3p overexpression adenovirus vectors or the negative control in tails before modeling, after which RT-qPCR was performed to examine miR-142-3p expression. As expected, miR-142-3p expression was increased in CME + AAV-miR-142-3p group relative to CME + AAV-miR-NC group (Fig. 1A). Echocardiography results revealed that CME + AAV-miR-142-3p group had substantial increases in LVEF, LVFS, and CO and a decrease in LVEDd than those in CME + AAV-miR-NC group (Fig. 1B–E). Compared with CME + AAV-miR-NC group, CME + AAV-miR-142-3p group showed alleviated myocardial injury, less microinfarction foci, and decreased cTnI content, apoptosis rate, cleaved caspase-3/8, and caspase-1 and IL-1β levels (Fig. 1F–P). Moreover, the number of white blood cells and IL-1β expression were declined in the rat blood of the CME + AAV-miR-142-3p group than those in CME + AAV-miR-NC group (Fig. 1Q–R). Taken together, CME could elicit cardiomyocyte apoptosis and pyroptosis in rats, which was suppressed by miR-142-3p overexpression.
ATXN1L is a direct target of miR-142-3p
To determine the mechanism of miR-142-3p in CME-induced myocardial injury, we screened 10 potential target genes of both miR-142-3p and miR-136-5p, including ankyrin repeat domain 11 (ANKRD11), purine rich element binding protein B (PURB), C5orf24, zinc finger protein 827 (ZNF827), ten-eleven translocation 3 (TET3), rich interactive domain 5B (ARID5B), ATXN1L, basic nucleoprotein 2 (BNC2), hedgehog interaction protein (HHIP), and ZBTB20 using miRBase, TargetScan, StarBase, and miRanda. TET3, ARID5B, ATXN1L, BNC2, and HHIP were screened out owing to their regulatory effect on gene expression. Among these five genes, ARID5B had been reported in a previous research, and miR-142-3p failed to target human HHIP, while TET3 and BNC2 were downregulated in myocardial ischemia models. Therefore, ATXN1L was chosen for subsequent experiments.
As shown by RT-qPCR and western blot, the mRNA and protein expression levels of ATXN1L were strengthened in CME group relative to sham group, and ATXN1L expression was suppressed in CME + AAV-miR-142-3p group than in CME + AAV-miR-NC group (Fig. 2A, B). Moreover, Pearson correlation coefficient revealed that miR-142-3p expression was negatively correlated with ATXN1L in the CME rat model (Fig. 2C).

miR-142-3p targets ATXN1L. RT-qPCR and western blot examined the mRNA (A) and protein (B) expression levels of ATXN1L, N = 10; Linear correlation between miR-142-3p and ATXN1L was assessed by Pearson (C), N = 10; Morphology of primary rat cardiomyocytes after cell culture for 48 h (D), N = 3; cTnI content in cardiomyocytes as measured by immunofluorescence (E), N = 3; In OGD- and LPS-treated cardiomyocytes, miR-142-3p (F) was assessed by RT-qPCR, and the mRNA (G) and protein (H) expression levels of ATXN1L were measured by RT-qPCR and western blot, N = 3; The interaction between miR-142-3p and ATXN1L was identified by dual luciferase reporter gene assay (I) and RIP (J), N = 3. *P < 0.05, **P < 0.01, ***P < 0.001. OGD, oxygen–glucose deprivation; LPS, lipopolysaccharide; RIP, RNA binding protein immunoprecipitation
Rat primary cardiomyocytes were isolated. After cell cultures for 48 h, most of the isolated cells were adherent and in round, fusiform or triangle shape, with spontaneous pulsation and obvious three-dimensionality (Fig. 2D). After cell culture for three days, immunofluorescence revealed that over 97% of the cardiomyocytes expressed cTnI (Fig. 2E). Next, we established an OGD model and an inflammation model in cardiomyocytes. In OGD and LPS groups, miR-142-3p expression was inhibited (Fig. 2F), and the mRNA and protein levels of ATXN1L were increased (Fig. 2G-H), compared to blank group. Dual luciferase reporter assay and RIP test showed that miR-142-3p could bind to the 3'UTR region of ATXN1L (Fig. 2I–J). These results indicated the negative correlation between miR-142-3p and ATXN1L and that ATXN1L was a target gene of miR-142-3p.
ATXN1L knockout exhibits an ameliorative effect on CME-induced myocardial injury
Our previous results demonstrated that ATXN1L expressed at a high level in the myocardial tissues of CME-induced rats, and therefore the study focused on the effects of ATXN1L in CME-induced myocardial injury. ATXN1L was knocked out in the rats using adenovirus vectors before the CME rat model was established. As shown by RT-qPCR and western blot, the mRNA and protein levels of ATXN1L were significantly suppressed in the rat myocardial tissues of CME + ATXN1L KO group than those in CME group (Fig. 3A, B).

ATXN1L knockout alleviated CME-induced myocardial injury. The mRNA (A) and protein (B) levels of ATXN1L in myocardial tissues were detected by RT-qPCR and western blot; LVEF (C), CO (D), LVEDd (E); LVFS (F) were assessed by echocardiography; H&E staining (G) and Masson (H) staining examined the pathologies of myocardial tissues; Infarction size of the myocardial tissue was calculated (I); Serum cTnI expression was measured by ELISA (J); Detection of cardiomyocyte apoptosis in myocardial tissues by TUNEL (K); Western blot validated the expression of cleaved caspase-3/8 (L); TEM determined the ultrastructure of myocardial tissues (M); Caspase-1 (N) and IL-1β (O) expression levels in myocardial tissues were detected by immunohistochemistry (P); Western blot evaluated the levels of NLRP3, ASC, cleaved-caspase-1 and GSDMD-N (Q); The number of white blood cells (R) and IL-1β level in rat blood (S) were assessed. N = 10. *P < 0.05, **P < 0.01. CME, coronary microembolization; LVEF, left ventricular ejection fraction; LVEDd, left ventricular end diastolic diameter, LVFS, left ventricular fraction shortening; CO, cardiac output; H&E, hematoxylin–eosin; TEM, transmission electron microscope
Echocardiography revealed increases in LVEF, LVFS, and CO and a decrease in LVEDd in CME + ATXN1L KO group relative to CME group (Fig. 3C–F). As shown by H&E and Masson staining, in model rats, myofiber arranged disorderedly, and a fraction of cells were swollen, concurrent with uneven staining in the cytoplasm, strengthened local eosinophilic red staining, and microinfarction foci; while CME + ATXN1L KO group showed attenuated myocardial injury and less microinfarction foci (Fig. 3G, H). In addition, the size of microinfarction foci was markedly reduced in CME + ATXN1L KO group (Fig. 3I). Moreover, CME + ATXN1L KO group showed decreased cTnI content, declined cardiomyocyte apoptosis rate, less ruptured cells, and reduced caspase-1 and IL-1β levels (Fig. 3J-P). The detection of NLRP3 inflammasome by western blot revealed downregulated NLRP3, ASC, cleaved caspase-1, and GSDMD-N expression levels in CME + ATXN1L KO group than those in CME group (Fig. 3Q). In addition, the number of white blood cells and IL-1β expression were reduced than those in the CME group (Fig. 3R-S). These results confirmed that ATXN1L knockout significantly repressed cardiomyocyte apoptosis and pyroptosis in CME-induced rats.
ATXN1L exacerbates CME-induced cardiomyocyte apoptosis and pyroptosis
We next examined the effect of ATXN1L on cardiomyocyte apoptosis and pyroptosis in vitro. RT-qPCR and western blot displayed higher ATXN1L expression in ATXN1L-OV group and lower ATXN1L expression in ATXN1L-KO group, compared with blank group (Fig. 4A-B).

ATXN1L facilitates cardiomyocyte apoptosis and pyroptosis. The mRNA (A) and protein (B) expression levels of ATXN1L in cardiomyocytes were assessed by RT-qPCR and western blot; Apoptosis of cardiomyocytes was measured by TUNEL (C, D) and flow cytometry (E, F); Western blot detected cleaved caspase-3/8 expression (G, H); Detection of IL-1β and IL-18 levels by ELISA (I); Expression of NLRP3, ASC, cleaved-caspase-1 and GSDMD-N as detected by Western blot (J, K). N = 3. *P < 0.05, **P < 0.01, ***P < 0.001
In ATXN1L-OV group, the apoptosis rate of cardiomyocytes and the number of TUNEL positive cells were elevated and the levels of cleaved caspase-3/8 were enhanced than those in blank group; the apoptosis rate and the number of TUNEL positive cells were declined and cleaved caspase-3/8 were downregulated in OGD + ATXN1L KO and LPS + ATXN1L KO groups relative to OGD and LPS groups (Fig. 4C–H). These results demonstrated the inhibition of ATXN1L knockout on cardiomyocyte apoptosis.
Compared with blank group, IL-1β and IL-18 expression levels in the supernatant of cardiomyocytes were increased and the expression of NLRP3, ASC, cleaved-caspase-1, and GSDMD-N was elevated in ATXN1L-OV group (Fig. 4I–K). The expression of IL-1β and IL-18 was weakened and NLRP3, ASC, cleaved-caspase-1, and GSDMD-N expressed at a low level in OGD + ATXN1L KO group than those in OGD group; the expression of these pyroptosis-related proteins was downregulated in LPS + ATXN1L KO group than those in LPS group (Fig. 4I). These results indicated the promotive effect of ATXN1L on cardiomyocyte pyroptosis. Overall, ATXN1L facilitated cardiomyocyte apoptosis and pyroptosis, and ATXN1L knockout could impede apoptosis and pyroptosis induced by OGD and LPS in cardiomyocytes.
miR-142-3p implicates in cardiomyocyte apoptosis and pyroptosis through ATXN1L
The present study then intended to identify whether miR-142-3p could regulate cardiomyocyte apoptosis and pyroptosis through ATXN1L, we performed RT-qPCR analysis and found that miR-142-3p expression was elevated after miR-142-3p mimic transfection (Fig. 5A).

miR-142-3p reduces cardiomyocyte apoptosis and pyroptosis by ATXN1L. Transfection efficiency of miR-142-3p overexpression as measured by RT-qPCR (A); cardiomyocyte apoptosis was identified by TUNEL (B, C) and flow cytometry (D, E); Western blot determined the expression levels of cleaved caspase-3/8 (F, G); IL-1β and IL-18 expression in cardiomyocytes as measured by ELISA (H); Alterations of pyroptosis-related proteins were evaluated by western blot (I, J). N = 3. *P < 0.05, **P < 0.01, ***P < 0.001
In OGD + miR-142-3p and LPS + miR-142-3p groups, the apoptosis rate and the number of TUNEL positive cells were reduced, compared with OGD group and LPS group (Fig. 5B–E). Importantly, ATXN1L overexpression reversed the inhibitory effect of miR-142-3p overexpression on the apoptosis rate and the number of TUNEL positive cells in OGD + miR-142-3p + ATXN1L-OV and LPS + miR-142-3p + ATXN1L-OV groups when compared with OGD + miR-142-3p group and LPS + miR-142-3p group (Fig. 5B–E). Meanwhile, decreases in cleaved caspase-3/8 were found in OGD + miR-142-3p and LPS + miR-142-3p groups compared with OGD group and LPS group; in OGD + miR-142-3p + ATXN1L-OV and LPS + miR-142-3p + ATXN1L-OV groups, cleaved caspase-3/8 expression levels were increased when compared with OGD + miR-142-3p group and LPS + miR-142-3p group (Fig. 5F, G). These results demonstrated that ATXN1L overexpression counteracted the anti-apoptotic effect of miR-142-3p overexpression in cardiomyocytes.
In OGD + miR-142-3p and LPS + miR-142-3p groups, IL-1β and IL-18 in cardiomyocytes were downregulated than those in OGD and LPS groups; the contents of IL-1β and IL-18 both in OGD + miR-142-3p + ATXN1L-OV and LPS + miR-142-3p + ATXN1L-OV groups were promoted compared with OGD + miR-142-3p group and LPS + miR-142-3p group (Fig. 5H). Western blot of the pyroptosis-related protein levels demonstrated that the contents of NLRP3, ASC, caspase-1, and GSDMD-N were suppressed in OGD + miR-142-3p and LPS + miR-142-3p groups relative to OGD and LPS groups; while simultaneous overexpression of miR-142-3p and ATXN1L enhanced the expression of NLRP3, ASC, cleaved-caspase-1, and GSDMD-N more than miR-142-3p overexpression alone in cardiomyocytes treated with LPS or OGD (Fig. 5I–J). These results confirmed that the inhibition of miR-142-3p on cardiomyocyte pyroptosis was inhibited by ATXN1L overexpression. Taken together, while miR-142-3p overexpression could inhibit cardiomyocyte apoptosis and pyroptosis induced by OGD and LPS treatment, ATXN1L overexpression could counteract the inhibiting effect of miR-142-3p on cardiomyocyte apoptosis and pyroptosis.
ATXN1L regulates H3 deacetylation and NOL3 expression through HDAC3
A previous study presented that ATXN1L could interact with HDAC3 and thus epigenetically regulate gene expression [17]. Also, ATXN1L could bind to HDAC3 and promote nuclear translocation of HDAC3 and deacetylation of histone, thus inhibiting gene expression [18]. UCSC bioinformatics analysis demonstrated a histone acetylation peak in NOL3 promoter (Fig. 6A), suggesting the regulation of NOL3 expression by deacetylation and acetylation of histone 3. In diabetic cardiomyopathy, downregulation of NOL3 (ARC) facilitates caspase-1/IL-1β/IL-18 signaling and induces cardiomyocyte pyroptosis [19]. Hence, we speculated that ATXN1L elicited apoptosis and pyroptosis in CME-induced myocardial injury by regulating HDAC3 and suppressing NOL3 expression.

miR-142-3p suppresses cardiomyocyte apoptosis and pyroptosis by ATXN1L. NOL3 expression exhibited a peak of acetylation of H3 (red area) (A); RT-qPCR and western blot detected the mRNA (B) and protein (C) expression of NOL3 in rat myocardial tissues; Western blot was used to detect the expression of HDAC3 and NOL3 in rat myocardial tissues (D); NOL3 expression was assessed by RT-qPCR and western blot in OGD- or LPS-treated cardiomyocytes (E, F) and in cardiomyocytes transfected with ATXN1L overexpression or knockout plasmid (G, H); Content and localization of HDAC3 in the cytoplasm and nucleus were revealed by immunofluorescence (I); Correlation between ATXN1L and HDAC3 was examined by Co-IP (J); Acetylated H3 expression was reduced by ATXN1L overexpression and was enhanced by ATXN1L knockout (K). The binding of ATXN1L and HDAC3 with NOL3 promoter was verified by ChIP (L). N = 3. *P < 0.05, **P < 0.01, ***P < 0.001. H3, histone 3; OGD, oxygen–glucose deprivation; LPS, lipopolysaccharide; HDAC3, histone deacetylase 3; Co-IP, Co-immunoprecipitation
First, the detection of NOL3 expression in myocardial tissues showed downregulated expression of NOL3 mRNA and protein in the CME rats than in sham-operated rats (Fig. 6B, C). In CME + AAV-miR-142-3p group, HDAC3 expression was suppressed and NOL3 increasingly expressed, compared with CME + AVV-miR-NC group (Fig. 6D). Consistently, NOL3 expression in cardiomyocytes expressed at a low level in OGD and LPS groups than in blank group (Fig. 6E, F). Primary cardiomyocytes were transfected with ATXN1L overexpression. Compared with blank group, NOL3 expressed modestly in ATXN1L-OV group and was upregulated in ATXN1L KO group (Fig. 6G, H), indicating that ATXN1L negatively regulated NOL3. Immunofluorescence results presented that overexpression of ATXN1L accelerated HDAC3 translocation from the cytoplasm to the nucleus of cardiomyoctes, and nuclear HDAC3 content was decreased in ATXN1L KO group (Fig. 6I), implying that ATXN1L played a role in the deacetylation of histone. The interaction between ATXN1L and HDAC3 was identified by co-immunoprecipitation (Co-IP) assay (Fig. 6J). Moreover, the acetylation of H3 was repressed in ATXN1L-OV group and elevated in ATXN1L KO group, compared with blank group (Fig. 6K). ChIP assay demonstrated the enrichment of NOL3 promoter by ATXN1L and HDAC3 antibodies (Fig. 6L). Therefore, we concluded that ATXN1L facilitated the nuclear translocation of HDAC3, thus promoting H3 deacetylation and suppressing NOL3.
NOL3 inhibits the apoptosis and pyroptosis of cardiomyocytes
Our previous results identified downregulated NOL3 both in CME-induced myocardial tissues and OGD- and LPS-treated cardiomyocytes, and then we explored the implication of NOL3 in cardiomyocyte apoptosis and pyroptosis. RT-qPCR and western blot results manifested increases in the mRNA and protein levels of NOL3 in NOL3-OV group than in blank group (Fig. 7A, B).

NOL3 inhibits cardiomyocyte apoptosis and pyroptosis. The mRNA (A) and protein (B) expression levels of NOL3 were measured by RT-qPCR and western blot; Cardiomyocyte apoptosis as measured by flow cytometry (C, D) and TUNEL (E, F); Detections of apoptosis-related proteins (caspase-3/8) by western blot (G, H); IL-1β and IL-18 levels in cardiomyocytes were determined by ELISA (I); Levels of pyroptosis-related proteins (NLRP3, ASC, cleaved-caspase-1, and GSDMD-N) were examined by western blot (J, K). N = 3. *P < 0.05, **P < 0.01
Moreover, OGD + NOL3-OV and LPS + NOL3-OV groups had reduced apoptosis rate and less TUNEL positive cells compared with OGD and LPS groups (Fig. 7C–F). Also, the levels of cleaved caspase-3/8 were deficient in OGD + NOL3-OV group and LPS + NOL3-OV group than those in OGD group and LPS group (Fig. 7G, H). These finding suggested that overexpression of NOL3 could suppress cardiomyocyte apoptosis.
In OGD + NOL3-OV group, the expression of IL-1β, IL-18 and pyroptosis-related proteins was downregulated than in OGD group, and the same pattern was found in LPS + NOL3-OV group relative to LPS group (Fig. 7I–K). Together, these results confirmed the anti-apoptotic and anti-pyroptotic effects of NOL3 overexpression in OGD- and LPS-treated cardiomyocytes.
ATXN1L mediates apoptosis and pyroptosis through the HDAC3/NOL3 axis
In OGD + ATXN1L-OV group, the apoptosis rate of cardiomyocytes and the number of TUNEL positive cells were increased, compared with OGD group; the apoptosis rate and the number of TUNEL positive cells were reduced in both OGD + ATXN1L-OV + HDAC3-in and OGD + ATXN1L-OV + NOL3-OV groups, compared with OGD + ATXN1L-OV group (Fig. 8A–D). Compared with LPS group, the apoptosis rate and the number of TUNEL positive cells were elevated in LPS + ATXN1L-OV group; the apoptosis rate and the number of TUNEL positive cells were declined in LPS + ATXN1L-OV + HDAC3-in and LPS + ATXN1L-OV + NOL3-OV groups than in LPS + ATXN1L-OV group (Fig. 8A–D). In OGD + ATXN1L-OV and LPS + ATXN1L-OV groups, the levels of cleaved caspase-3/8 were substantially higher than those in OGD and LPS groups (Fig. 8E, F). Compared with OGD + ATXN1L-OV group, OGD + ATXN1L-OV + HDAC3-in and OGD + ATXN1L-OV + NOL3-OV groups had suppressed levels of cleaved caspase-3/8, and similar expression trends of caspase-3/8 were found in LPS + ATXN1L-OV + HDAC3-in group and LPS + ATXN1L-OV + NOL3-OV group (Fig. 8E, F). These results revealed that HDAC3 inhibition or NOL3 overexpression attenuated the promoting effect of ATXN1L on cardiomyocyte apoptosis.

ATXN1L facilitates apoptosis and pyroptosis in cardiomyocytes by the HDAC3/NOL3 axis. Cardiomyocyte apoptosis was presented using flow cytometry (A, B) and TUNEL (C, D); Cleaved caspase-3/8 expression in cardiomyocytes as measured by western blot (E, F); IL-1β and IL-18 levels in the supernatant of the cells as detected by ELISA (G); Western blot displayed the expression of pyroptosis-related proteins (H, I). N = 3. *P < 0.05, **P < 0.01
In OGD + ATXN1L-OV group and LPS + ATXN1L-OV group, IL-1β, IL-18, and pyroptosis-related proteins (NLRP3, ASC, cleaved-caspase-1, and GSDMD-N) increasingly expressed, compared with OGD group and LPS group (Fig. 8G–I). The levels of IL-1β, IL-18, and pyroptosis-related proteins were inhibited in OGD + ATXN1L-OV + HDAC3-in and OGD + ATXN1L-OV + NOL3-OV groups than those in OGD + ATXN1L-OV group, and the same expression patterns of these proteins were shown in LPS + ATXN1L-OV + HDAC3-in group and LPS + ATXN1L-OV + NOL3-OV group (Fig. 8G–I). The aforementioned results revealed HDAC3 inhibition and NOL3 overexpression restrained cardiomyocyte pyroptosis exacerbated by ATXN1L overexpression. In summary, ATXN1L regulates cardiomyocyte apoptosis and pyroptosis by the HDAC1/NOL3 axis.
Discussion
CME is the main cause of no-reflow or slow-flow phenomena after PCI, leading to cardiomyocyte necrosis and apoptosis, ventricular remodeling, malignant arrhythmia, and heart failure [20, 21]. Experimentally induced CME caused by injection of embolic particles could preferably imitate the distal occlusion of small coronary arteries in patients who experience CME from a ruptured coronary atherosclerotic plaque [22]. Moreover, myocardial ischemia reperfusion (I/R) injury results in substantial tissue alterations such as cell death, inflammation, and oxidative stress, and the mechanism of myocardial I/R injury to coronary circulation is of great importance for cardioprotection [23]. Therefore, a better understanding about CME-induced myocardial injury may contribute to the preservation of cardiac function and therapeutic efficacy after heart failure. In this study, we made efforts to promote the existing understanding of the mechanism underlying CME-induced myocardial injury. First, we generated morphological and molecular evidence of myocardial injury in CME-treated rats and OGD/LPS-stimulated cardiomyocytes. We then obtained both in vivo and in vitro evidence that miR-142-3p mediated CME-induced myocardial injury by targeting ATXN1L. Moreover, our findings implied that the impediment of ATXN1L on myocardial function was involved with the HDAC3/NOL3 axis. These data highlight a novel strategy for improving cardiac function after CME by inhibiting apoptosis and pyroptosis in cardiomyocytes.
Dysregulation of miRNAs has been reported to elicit the aberrant upregulation or downregulation of their target genes and thus participate in CME pathogenesis [7, 24, 25]. In the CME rat model, downregulated miR-142-3p expression in myocardial tissues was observed, suggesting that miR-142-3p may hold a regulatory function in CME. Forced expression of miR-142-3p inhibits neonatal rat cardiomyocyte apoptosis by reducing caspase-3 and Bax expression levels [26, 27]. Consistently, our results manifested that miR-142-3p overexpression restored cardiac function and alleviated myocardial injury, supported by the recovery of coronary flow and the reduction of microinfarction foci after CME. Currently, ATXN1L was proven as a target gene of miR-142-3p and was upregulated in myocardial tissues after CME. ATXN1L knockout was also demonstrated to attenuate cardiac function and myocardial infarction induced by CME.
Inhibition of cardiomyocyte apoptosis has been highlighted as a potent therapeutic strategy for CME-induced myocardial injury [28]. Also, pyroptosis is an important contributor for the impairment of cardiac functions [29,30,31]. Hence, we examined apoptosis- and pyroptosis-related proteins to identify their correlation in CME pathology using OGD- and LPS-induced cellular models. The present study suggested that CME treatment induced the expression of cleaved caspase-3/8, caspase-1, and IL-1β, indicative of the occurrence of apoptosis and pyroptosis in cardiomyocytes after CME. The formation of NLRP3 inflammasome which is composed of NLR3, the adaptor protein ASC, and the cysteine protease caspase-1 results in the activation of caspase-1 [32]. The activation of caspase-1 is involved in pyroptosis, which could process and mature IL-1β and IL-18, but also cleave GSDMD and oligomerize GSDMD-N domain that mediates the formation of membrane pores, resulting the release of inflammatory factors, cell swelling, and pyroptosis [33]. Importantly, miR-142-3p overexpression or ATXN1L knockout was verified to inhibit apoptosis and pyroptosis both in vivo and in vitro by reducing the expressions of apoptosis- and pyroptosis-related proteins. Moreover, ATXN1L overexpression could counteract the cardioprotective effects of miR-142-3p, which confirmed that miR-142-3p affected myocardial injury via ATXN1L. In addition to cardiomyocytes, immune cells are also significant for cardiac remodeling after myocardial infarction by regulating inflammation and cardiac repair [34]. Inhibition of immune lineage cells during cardiac lineage development is required for cardiac reprogramming, but this study mentioned few about the role of immune cells in CME-induced myocardial injury. Our future study would concentrate on the identified transcriptional regulation, epigenetic modification, signaling pathways from the cellular microenvironment, and cell cycling regulation to improve cardiac reprogramming efficacy [35].
In heart, DNA methylation and histone modification regulate cardiomyocyte development [36]. Ischemic or hypoxia stress alters the mechanisms responsible for epigenetic modifications, such as inducing the activities of histone deacetylases (HDACs), a group of enzymes capable of deacetylating histone proteins [37]. HDAC3 is a major member of HDACs, whose enzymatic activity is targeted by small molecule inhibitors for treating a variety of conditions [38]. ATXN1 has been proven to bind HDAC3, which is required for ATXN1-induced transcriptional repression [38]. Also, ATXN1L could bind to HDAC3 and promote the nuclear translocation of HDAC3 and the deacetylation of histone [18]. In the current study, ATXN1L exhibited the promotive effect on the translocation of HDAC3 from the cytoplasm to the nucleus of cardiomyocytes. Our bioinformatics analysis indicated the correlation between NOL3 and H3, and Co-IP result verified the interaction between ATXN1L/HDAC3 and H3. ATXN1L was disclosed to negatively regulate H3 deacetylation. The anti-apoptotic role of NOL3 (ARC) has been mentioned in cardiomyocytes [39, 40]. In ischemia/reperfusion (I/R)-induced acute kidney injury, ARC significantly restrained the expression of IL-1β and caspase-3 [41]. Consistent observation was obtained from the present study that NOL3 overexpression showed a suppressive effect on cardiomyocyte apoptosis and pyroptosis by inhibiting the expression of caspase-3/8, IL-1β, IL-18, NLRP3, ASC, caspase-1, and GSDMD-N. In cardiomyocytes, the enhancement of ATXN1L on apoptosis and pyroptosis was mitigated by HDAC3 inhibition or NOL3 upregulation, which suggested that ATXN1L played its role through the HDAC3/NOL3 axis.
In conclusion, the current study showed that miR-142-3p was markedly decreased in response to CME-induced myocardial injury. Overexpression of miR-142-3p alleviated apoptosis and pyroptosis in OGD- and LPS-treated cardiomyocytes. ATXN1L was a downstream target of miR-142-3p and participated in the pathogenesis of myocardial injury after CME. Our findings revealed that miR-142-3p might play a promising role in ameliorating myocardial injury after CME.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Zhou Y, Li T, Chen Z, Huang J, Qin Z, Li L (2021) Overexpression of lncRNA TUG1 alleviates NLRP3 inflammasome-mediated cardiomyocyte pyroptosis through targeting the miR-186-5p/XIAP axis in coronary microembolization-induced myocardial damage. Front Immunol 12:637598. https://doi.org/10.3389/fimmu.2021.637598
Wang XT, Wu XD, Lu YX, Sun YH, Zhu HH, Liang JB, He WK, Zeng ZY, Li L (2017) Potential involvement of MiR-30e-3p in myocardial injury induced by coronary microembolization via autophagy activation. Cell Physiol Biochem 44:1995–2004. https://doi.org/10.1159/000485905
Liu T, Zhou Y, Liu YC, Wang JY, Su Q, Tang ZL, Li L (2015) Coronary microembolization induces cardiomyocyte apoptosis through the LOX-1-dependent endoplasmic reticulum stress pathway involving JNK/P38 MAPK. Can J Cardiol 31:1272–1281. https://doi.org/10.1016/j.cjca.2015.01.013
Liu Y, Liu Y, Huang X, Zhang J, Yang L (2019) Protective effects and mechanism of curcumin on myocardial injury induced by coronary microembolization. J Cell Biochem 120:5695–5703. https://doi.org/10.1002/jcb.27854
Wang JY, Chen H, Su X, Zhou Y, Li L (2017) Atorvastatin pretreatment inhibits myocardial inflammation and apoptosis in swine after coronary microembolization. J Cardiovasc Pharmacol Ther 22:189–195. https://doi.org/10.1177/1074248416662348
Zhaolin Z, Guohua L, Shiyuan W, Zuo W (2019) Role of pyroptosis in cardiovascular disease. Cell Prolif 52:e12563. https://doi.org/10.1111/cpr.12563
Chen ZQ, Zhou Y, Chen F, Huang JW, Li HL, Li T, Li L (2021) miR-200a-3p attenuates coronary microembolization-induced myocardial injury in rats by inhibiting TXNIP/NLRP3-mediated cardiomyocyte pyroptosis. Front Cardiovasc Med 8:693257. https://doi.org/10.3389/fcvm.2021.693257
Su Q, Li L, Zhao J, Sun Y, Yang H (2017) MiRNA expression profile of the myocardial tissue of pigs with coronary microembolization. Cell Physiol Biochem 43:1012–1024. https://doi.org/10.1159/000481699
Wang Y, Ouyang M, Wang Q, Jian Z (2016) MicroRNA-142-3p inhibits hypoxia/reoxygenation induced apoptosis and fibrosis of cardiomyocytes by targeting high mobility group box 1. Int J Mol Med 38:1377–1386. https://doi.org/10.3892/ijmm.2016.2756
Su Q, Lv X, Ye Z, Sun Y, Kong B, Qin Z, Li L (2019) The mechanism of miR-142-3p in coronary microembolization-induced myocardiac injury via regulating target gene IRAK-1. Cell Death Dis 10:61. https://doi.org/10.1038/s41419-019-1341-7
Wong D, Lounsbury K, Lum A, Song J, Chan S, LeBlanc V, Chittaranjan S, Marra M, Yip S (2019) Transcriptomic analysis of CIC and ATXN1L reveal a functional relationship exploited by cancer. Oncogene 38:273–290. https://doi.org/10.1038/s41388-018-0427-5
Kahle JJ, Souroullas GP, Yu P, Zohren F, Lee Y, Shaw CA, Zoghbi HY, Goodell MA (2013) Ataxin1L is a regulator of HSC function highlighting the utility of cross-tissue comparisons for gene discovery. PLoS Genet 9:e1003359. https://doi.org/10.1371/journal.pgen.1003359
Wang H, Zhou X, Li H, Qian X, Wang Y, Ma L (2017) Transient receptor potential melastatin 2 negatively regulates LPS-ATP-induced caspase-1-dependent pyroptosis of bone marrow-derived macrophage by modulating ROS production. Biomed Res Int 2017:2975648. https://doi.org/10.1155/2017/2975648
Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G (2015) Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 43:923–932. https://doi.org/10.1016/j.immuni.2015.10.009
Zhao LR, Xing RL, Wang PM, Zhang NS, Yin SJ, Li XC, Zhang L (2018) NLRP1 and NLRP3 inflammasomes mediate LPS/ATP induced pyroptosis in knee osteoarthritis. Mol Med Rep 17:5463–5469. https://doi.org/10.3892/mmr.2018.8520
Burja B, Kuret T, Janko T, Topalovic D, Zivkovic L, Mrak-Poljsak K, Spremo-Potparevic B, Zigon P, Distler O, Cucnik S et al (2019) Olive leaf extract attenuates inflammatory activation and DNA damage in human arterial endothelial cells. Front Cardiovasc Med 6:56. https://doi.org/10.3389/fcvm.2019.00056
Venkatraman A, Hu YS, Didonna A, Cvetanovic M, Krbanjevic A, Bilesimo P, Opal P (2014) The histone deacetylase HDAC3 is essential for Purkinje cell function, potentially complicating the use of HDAC inhibitors in SCA1. Hum Mol Genet 23:3733–3745. https://doi.org/10.1093/hmg/ddu081
Karagianni P, Wong J (2007) HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene 26:5439–5449. https://doi.org/10.1038/sj.onc.1210612
Li X, Du N, Zhang Q, Li J, Chen X, Liu X, Hu Y, Qin W, Shen N, Xu C et al (2014) MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis 5:e1479. https://doi.org/10.1038/cddis.2014.430
Ma J, Qian J, Chang S, Chen Z, Jin H, Zeng M, Zou Y, Ge J (2014) Left ventricular remodeling with preserved function after coronary microembolization: the effect of methylprednisolone. Eur J Med Res 19:7. https://doi.org/10.1186/2047-783X-19-7
Zhang Y, Zhang L, Zheng H, Chen H (2018) Effects of atrial fibrillation on complications and prognosis of patients receiving emergency PCI after acute myocardial infarction. Exp Ther Med 16:3574–3578. https://doi.org/10.3892/etm.2018.6640
Wang W, Ye S, Zhang L, Jiang Q, Chen J, Chen X, Zhang F, Wu H (2020) Granulocyte colony-stimulating factor attenuates myocardial remodeling and ventricular arrhythmia susceptibility via the JAK2-STAT3 pathway in a rabbit model of coronary microembolization. BMC Cardiovasc Disord 20:85. https://doi.org/10.1186/s12872-020-01385-5
Huang C, Qu Y, Feng F, Zhang H, Shu L, Zhu X, Huang G, Xu J (2022) Cardioprotective effect of circ_SMG6 Knockdown against myocardial ischemia/reperfusion injury correlates with miR-138-5p-mediated EGR1/TLR4/TRIF inactivation. Oxid Med Cell Longev 2022:1927260. https://doi.org/10.1155/2022/1927260
Kong B, Qin Z, Ye Z, Yang X, Li L, Su Q (2019) microRNA-26a-5p affects myocardial injury induced by coronary microembolization by modulating HMGA1. J Cell Biochem 120:10756–10766. https://doi.org/10.1002/jcb.28367
Zhu HH, Wang XT, Sun YH, He WK, Liang JB, Mo BH, Li L (2019) MicroRNA-486-5p targeting PTEN protects against coronary microembolization-induced cardiomyocyte apoptosis in rats by activating the PI3K/AKT pathway. Eur J Pharmacol 855:244–251. https://doi.org/10.1016/j.ejphar.2019.03.045
Sharma S, Liu J, Wei J, Yuan H, Zhang T, Bishopric NH (2012) Repression of miR-142 by p300 and MAPK is required for survival signalling via gp130 during adaptive hypertrophy. EMBO Mol Med 4:617–632. https://doi.org/10.1002/emmm.201200234
Zhao Z, Qu F, Liu R, Xia Y (2019) Differential expression of miR-142-3p protects cardiomyocytes from myocardial ischemia-reperfusion via TLR4/NFkB axis. J Cell Biochem. https://doi.org/10.1002/jcb.29506
Su Q, Lv X, Ye Z (2019) Ligustrazine attenuates myocardial injury induced by coronary microembolization in rats by activating the PI3K/Akt pathway. Oxid Med Cell Longev 2019:6791457. https://doi.org/10.1155/2019/6791457
Yang F, Qin Y, Wang Y, Li A, Lv J, Sun X, Che H, Han T, Meng S, Bai Y et al (2018) LncRNA KCNQ1OT1 mediates pyroptosis in diabetic cardiomyopathy. Cell Physiol Biochem 50:1230–1244. https://doi.org/10.1159/000494576
Ye B, Chen X, Dai S, Han J, Liang X, Lin S, Cai X, Huang Z, Huang W (2019) Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des Devel Ther 13:975–990. https://doi.org/10.2147/DDDT.S195412
Zheng X, Zhong T, Ma Y, Wan X, Qin A, Yao B, Zou H, Song Y, Yin D (2020) Bnip3 mediates doxorubicin-induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci 242:117186. https://doi.org/10.1016/j.lfs.2019.117186
Mishra PK, Adameova A, Hill JA, Baines CP, Kang PM, Downey JM, Narula J, Takahashi M, Abbate A, Piristine HC et al (2019) Guidelines for evaluating myocardial cell death. Am J Physiol Heart Circ Physiol 317:H891–H922. https://doi.org/10.1152/ajpheart.00259.2019
Jia C, Chen H, Zhang J, Zhou K, Zhuge Y, Niu C, Qiu J, Rong X, Shi Z, Xiao J et al (2019) Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol 67:311–318. https://doi.org/10.1016/j.intimp.2018.12.028
Kologrivova I, Shtatolkina M, Suslova T, Ryabov V (2021) Cells of the immune system in cardiac remodeling: main players in resolution of inflammation and repair after myocardial infarction. Front Immunol 12:664457. https://doi.org/10.3389/fimmu.2021.664457
Liu L, Guo Y, Li Z, Wang Z (2021) Improving cardiac reprogramming for heart regeneration in translational medicine. Cells. https://doi.org/10.3390/cells10123297
Arima Y, Fukuoka H (2020) Developmental origins of health and disease theory in cardiology. J Cardiol. https://doi.org/10.1016/j.jjcc.2020.02.003
Su Q, Liu Y, Lv XW, Dai RX, Yang XH, Kong BH (2020) LncRNA TUG1 mediates ischemic myocardial injury by targeting miR-132-3p/HDAC3 axis. Am J Physiol Heart Circ Physiol 318:H332–H344. https://doi.org/10.1152/ajpheart.00444.2019
Song S, Wen Y, Tong H, Loro E, Gong Y, Liu J, Hong S, Li L, Khurana TS, Chu M et al (2019) The HDAC3 enzymatic activity regulates skeletal muscle fuel metabolism. J Mol Cell Biol 11:133–143. https://doi.org/10.1093/jmcb/mjy066
Li Y, Liu X, Rong F (2011) PUMA mediates the apoptotic signal of hypoxia/reoxygenation in cardiomyocytes through mitochondrial pathway. Shock 35:579–584. https://doi.org/10.1097/SHK.0b013e318211601a
Wu L, Xi Z, Guo R, Liu S, Yang S, Liu D, Dong S, Guo D (2013) Exogenous ARC down-regulates caspase-3 expression and inhibits apoptosis of broiler chicken cardiomyocytes exposed to hydrogen peroxide. Avian Pathol 42:32–37. https://doi.org/10.1080/03079457.2012.757289
Ke Y, Yan H, Chen L, Zhong S, Dai Y, Cai S, Pan L, Wang Y, Zhou M (2019) Apoptosis repressor with caspase recruitment domain deficiency accelerates ischemia/reperfusion (I/R)-induced acute kidney injury by suppressing inflammation and apoptosis: The role of AKT/mTOR signaling. Biomed Pharmacother 112:108681. https://doi.org/10.1016/j.biopha.2019.108681
Acknowledgements
Thanks for all the support and contributions of the participators.
Funding
This research was funded by the grants from the National Natural Science Foundation of China (Grant No. 81960079); Guangxi Natural Science Foundation (Grant No. 2020GXNSFDA238007; Grant No. 2020GXNSFFA297002; Grant No. 2020GXNSFAA297009); The Key Research and Development Program of Guangxi (Grant No. AB20159005); Guangxi BaGui Scholars Special Project; and Guangxi Health Commission Key Laboratory of Disease Proteomics Research.
Author information
Authors and Affiliations
Contributions
Yuli Xu: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Writing–original draft. Xiangwei Lv: Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Writing–original draft. Ruping Cai: Data curation, Formal analysis, Investigation, Methodology. Yanling Ren: Methodology. Rixin Dai: Methodology. Shirong He: Resources. Wei Zhang: Supervision. Quanzhong Li: Methodology, Writing–original draft. Xiheng Yang: Data curation, Formal analysis. Qiang Su: Conceptualization, Formal analysis, Supervision, Funding acquisition, Validation, Project administration, Writing–review and editing. Riming Wei: Conceptualization, Supervision, Funding acquisition, Validation, Project administration, Writing–review and editing.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All experiments were approved by Guilin Medical University and produced in line with the guidance for the care and use of laboratory animals issued by National Institutes of Health (NIH). All efforts had been made to minimize pain in animals.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yuli Xu and Xiangwei Lv contributed equally to this research.
Rights and permissions
About this article

Cite this article
Xu, Y., Lv, X., Cai, R. et al. Possible implication of miR-142-3p in coronary microembolization induced myocardial injury via ATXN1L/HDAC3/NOL3 axis. J Mol Med 100, 763–780 (2022). https://doi.org/10.1007/s00109-022-02198-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-022-02198-z