
Abstract
The SGLT2 inhibitor empagliflozin improved cardiovascular outcomes in patients with diabetes. As the cardiac mechanisms remain elusive, we investigated the long-term effects (up to 2 months) of empagliflozin on excitation-contraction (EC)-coupling in human cardiomyocytes derived from induced pluripotent stem cells (iPSC-CM) in a blinded manner. IPSC from 3 donors, differentiated into pure iPSC-CM (4 differentiations), were treated with a clinically relevant concentration of empagliflozin (0.5 μmol/l) or vehicle control. Treatment, data acquisition, and analysis were conducted externally blinded. Epifluorescence microscopy measurements in iPSC-CM showed that empagliflozin has neutral effects on Ca2+ transient amplitude, diastolic Ca2+ levels, Ca2+ transient kinetics, or sarcoplasmic Ca2+ load after 2 weeks or 8 weeks of treatment. Confocal microscopy determining possible effects on proarrhythmogenic diastolic Ca2+ release events showed that in iPSC-CM, Ca2+ spark frequency and leak was not altered after chronic treatment with empagliflozin. Finally, in patch-clamp experiments, empagliflozin did not change action potential duration, amplitude, or resting membrane potential compared with vehicle control after long-term treatment. Next-generation RNA sequencing (NGS) and mapped transcriptome profiles of iPSC-CMs untreated and treated with empagliflozin for 8 weeks showed no differentially expressed EC-coupling genes. In line with NGS data, Western blots indicate that empagliflozin has negligible effects on key EC-coupling proteins. In this blinded study, direct treatment of iPSC-CM with empagliflozin for a clinically relevant duration of 2 months did not influence cardiomyocyte EC-coupling and electrophysiology. Therefore, it is likely that other mechanisms independent of cardiomyocyte EC-coupling are responsible for the beneficial treatment effect of empagliflozin.
Key messages
-
This blinded study investigated the clinically relevant long-term effects (up to 2 months) of empagliflozin on cardiomyocyte excitation-contraction (EC)-coupling.
-
Human cardiomyocytes derived from induced pluripotent stem cells (iPSC-CM) were used to study a human model including a high repetition number of experiments.
-
Empagliflozin has neutral effects on cardiomyocyte Ca2+ transients, sarcoplasmic Ca2+ load, and diastolic sarcoplasmic Ca2+ leak.
-
In patch-clamp experiments, empagliflozin did not change the action potential.
-
Next-generation RNA sequencing, mapped transcriptome profiles, and Western blots of iPSC-CM untreated and treated with empagliflozin showed no differentially expressed EC-coupling candidates.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The antidiabetic drug empagliflozin, which inhibits the sodium-dependent glucose cotransporter 2 (SGLT2) in the kidney, has been shown to reduce cardiovascular mortality, all-cause mortality, and hospitalization rates for heart failure (HF) in patients at high cardiovascular risk and type 2 diabetes [1]. These beneficial effects have been also confirmed for other SGLT2 inhibitors [2, 3] and, importantly, also in a diabetes-independent manner in patients with HF [4]. Moreover, recent clinical data indicate that this improved cardiovascular outcome might not be dependent on modulation of cardiovascular risk factors as the effects occur already after a few months [5]. Therefore, the question arises whether empagliflozin has direct cardiac effects. Despite a growing number of studies investigating the cardiovascular effects of empagliflozin, the underlying mechanisms are still under debate and remain controversial. Importantly, direct myocardial effects of empagliflozin on cardiac metabolism [6], contractility [7, 8], and cardiomyocyte Ca2+ and Na+ homeostasis [9, 10] have been proposed. One potential mechanisms might be an inhibitory effect of empagliflozin on the Na+/H+ exchanger (NHE) causing a reduction in myocardial cytoplasmic Na+ and Ca2+ and increased mitochondrial Ca2+ [10, 11]. As Ca2+ cycling fundamentally determines cardiac excitation-contraction (EC)-coupling and thereby cardiac function, changes in Ca2+ homeostasis are associated with pathological cardiac remodeling and HF [12]. Therefore, the evaluation whether empagliflozin may affect cardiomyocyte EC-coupling is of importance to understand its potential cardiac effects and for further translational investigation. However, the role of empagliflozin for cardiomyocyte EC-coupling is not fully elucidated, and previous reports are limited by small n-numbers. As the systemic effects of empagliflozin may indirectly affect the myocardium, the investigation of the direct cardiac effects of empagliflozin is thus only possible in in vitro models. However, isolated adult cardiomyocytes (CM), as investigated previously [7, 9, 10], are not suitable for chronic culture and treatment protocols according to the clinical setting, where the beneficial effects of empagliflozin became apparent after ~ 2 months of treatment [1].
The aim of this study was to fundamentally investigate the clinically relevant direct long-term effects of empagliflozin on human cardiomyocyte EC-coupling and electrophysiology in a standardized and blinded manner by using human-induced pluripotent stem cell cardiomyocytes (iPSC-CM). Based on the clinical data, iPSC-CM were chronically treated for 2 and 8 weeks with empagliflozin (0.5 μmol/l), and EC-coupling and electrophysiology were studied by blinded investigators.
Methods
IPSC-CM and treatment
All procedures were performed according to the Declaration of Helsinki and were approved by the local ethics committee. Informed consent was obtained from all tissue donors. IPSCs were cultured feeder-free and adherent by cultivating on Geltrex-coated cell culture dishes in the presence of chemically defined medium E8 (Life Technologies). Cardiac differentiation of iPSCs was performed by sequential targeting of the WNT pathway as described previously [13]. Briefly, undifferentiated iPSCs were cultured as a monolayer on Geltrex-coated 12-well dishes to a confluence of 85–95%. Medium was changed to cardio differentiation medium composed of RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 0.02% L-ascorbic acid 2-phosphate (Sigma Aldrich) and 0.05% albumin (Sigma Aldrich) including the GSK3 inhibitor CHIR99021 (4 μmol/L, Millipore) (d0). After 48 h, medium was changed to fresh media supplemented with 5 μmol/L of the inhibitor of Wnt production-2 (IWP2, Millipore) for 2 days. From day 10 on, the cells were cultured in cardio culture medium (RPMI 1640 medium supplemented with 2 mmol/L l-glutamine and 2% B27 with insulin (Life Technologies)), with a medium change every 2–3 days. CM were purified using metabolic selection for 2–4 days by using 4 mmol/L lactate as carbon source after 20–40 days of differentiation [14] and studied on day 90 after initiation of differentiation except when mentioned otherwise. Following differentiation, purity of iPSC-CM was determined by flow analysis (> 90% cardiac TNT+) or by morphology. Four- to 5-week-aged iPSC-CM were treated with 0.5 μmol/l empagliflozin for 2 or 8 consecutive weeks, which corresponds to the clinical relevant plasma concentration [15, 16]. All investigators were blinded by an external independent instance during treatment, data acquisition, data analysis, and statistical testing. All analyses were performed in iPSC-CM with or without empagliflozin treatment. Four differentiation experiments of different iPSC lines from 3 healthy probands (female) were used.
Epifluorescence microscopy (Ca2+ transients and sarcoplasmic reticulum Ca2+ content)
IPSC-CMs were loaded for 15 min with the ratiometric Ca2+ dye Fura-2 AM (5 μmol/l). Solution was replaced by Tyrode’s solution (in mmol/l: KCl 4, NaCl 140, MgCl2 1, HEPES 10, glucose 10, CaCl2 1.25, pH 7.4, NaOH), and the respective agent (empagliflozin/vehicle control) and cells were left to incubate for another 15 min to ensure complete deesterification of intracellular Fura-2 and allow cellular rebalance of Ca2+ cycling properties. The Tyrode’s solution was again replaced before measurements were started. Fura-2 fluorescence ratio was calculated using alternating excitation at 340 nm and 380 nm. The emitted fluorescence was collected at 510 nm. Measurements were performed with a fluorescence detection system (ION OPTIX Corp.). Ca2+ transients were recorded at steady-state conditions under constant field stimulation (30 V, 10 ms) during increasing frequencies (0.25, 0.5, 1 Hz) at room temperature. Sarcoplasmic Ca2+ content was assessed by caffeine application (10 mmol/L) after stopping field stimulation (0.25 Hz) to induce a complete sarcoplasmic reticulum Ca2+ release. The recorded Ca2+ transients were analyzed with the software IONWizard (ION OPTIX Corp.).
Confocal microscopy (diastolic Ca2+ sparks)
IPSC-CM were incubated with Fluo-4 AM (10 μmol/l) for 15 min before cells were incubated with Tyrode’s solution for another 15 min as described above. Ca2+ spark measurements were performed with a laser scanning confocal microscope (LSM 7 Pascal, Zeiss) at room temperature. Fluo-4 was excited by an argon ion laser (488 nm), and emitted fluorescence was collected through a 505-nm long-pass emission filter. Fluorescence images were recorded in the line scan mode with 512 pixels per line (width of each scan line: 35.4 μm) and a pixel time of 0.64 μs. One image consists of 10,000 unidirectional line scans, equating to a measurement period of ~ 6.9 s. Experiments were conducted at resting conditions after loading the SR with Ca2+ by repetitive field stimulation for 10 s (at 0.5 Hz, 30 V, 5 ms). Diastolic Ca2+ sparks were analyzed with the program SparkMaster for ImageJ. The Ca2+ spark frequency of each cell resulted from the number of sparks normalized to cell width and scan rate (100 μm−1*s−1). The Ca2+ spark size was calculated as the product of spark amplitude, duration, and width. To compute the full Ca2+ leak per cell, Ca2+ spark size was multiplied with Ca2+ spark frequency [17].
Action potential measurements
IPSC-CM were incubated with Tyrode’s solution and the respective agent (empagliflozin/vehicle control) for 15 min before measurements were started. For action potential recordings, whole-cell patch-clamp technique was used (current clamp configuration, HEKA electronics) [17, 18]. Microelectrodes (2–3 MΩ) were filled with (in mmol/L) 122 K-aspartate, 8 KCl, 10 NaCl, 1 MgCl2, 10 HEPES, and 5 Mg-ATP (pH 7.2, KOH). Action potentials were continuously elicited by square current pulses of 0.5–1 nA amplitude and 1–5 ms duration at 0.25, 0.5, 1, and 2 Hz. Access resistance was < 15 MΩ after rupture. Fast capacitance was compensated in cell-attached configuration. Membrane capacitance and series resistance were compensated after patch rupture. Signals were filtered with 2.9 and 10 kHz Bessel filters and recorded with an EPC10 amplifier (HEKA Elektronik) [19]. All experiments were conducted at room temperature.
RNA sequencing (RNA-seq) and bioinformatics
Differential gene expression was obtained by use of RNA sequencing performed on an Illumina HighSeq-4000 platform and bioinformatics. Total RNA was isolated from 3-month-old iPSC-CM from 2 controls with 2 differentiation each (n = 4 samples) treated and non-treated with empagliflozin using standard protocols (Promega).
Quality and integrity of RNA were assessed with the Fragment Analyzer from Advanced Analytical by using the standard sensitivity RNA Analysis Kit (DNF-471). All samples selected for sequencing exhibited an RNA integrity number over 8. RNA-seq libraries were performed using 150 ng total RNA of a nonstranded RNA-seq, massively parallel mRNA sequencing approach from Illumina (TruSeq stranded total RNA Library Preparation, Illumina). Libraries were prepared on the automation (Beckman Coulter’s Biomek FXP workstation). For accurate quantitation of cDNA libraries, a fluorometric based system, the QuantiFluor™dsDNA System from Promega, was used. The size of final cDNA libraries was determined by using the dsDNA 905 Reagent Kit (Fragment Analyzer from Advanced Bioanalytical) exhibiting a sizing of 300 bp in average. Libraries were pooled and sequenced on the Illumina HiSeq 4000 (SE; 1 × 50 bp; 30–35 Mio reads/sample).
Sequence images were transformed with Illumina software BaseCaller to BCL files, which was demultiplexed to fastq files with bcl2fastq v2.17.1.14.
Raw read and quality check
Sequence images were transformed with Illumina software BaseCaller to BCL files, which was demultiplexed to fastq files with bcl2fastq v2.20.0.422. The sequencing quality was asserted using FastQC (Andrews, S. (2014). FastQC A Quality Control tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)(version 0.11.5).
Mapping and normalization
Sequences were aligned to the reference genome Homo sapiens (hg38 version 96, https://www.ensembl.org/Homo_sapiens/Info/Index) using the STAR aligner (Dobin, Alexander, et al. “STAR: ultrafast universal RNA- seq aligner.” Bioinformatics 29.1 (2013): 15–21.) (version 2.5.2a) allowing for 2 mismatches within 50 bases. Subsequently, read counting was performed using featureCounts (Liao, Yang, Gordon K. Smyth, and Wei Shi. “featureCounts: an efficient general purpose program for assigning sequence reads to genomic features.” Bioinformatics 30.7 (2013): 923–930.) (version 1.5.0-p1). Read counts were normalized in the R/Bioconductor environment (version 3.6.1, www.bioconductor.org) using the DESeq2 R package (Love, Michael I., Simon Anders, and Wolfgang Huber. “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.” Genome biology 15.12 (2014): 550.) version 1.24.0, where normalization was done by calculating the size factors for each sample and dividing by them.
Western blots
Pellets of iPSC-CMs were snap-frozen in liquid nitrogen and stored at − 80 °C. Protein lysates were prepared in cell lysis buffer containing (mmol/L) 20 Tris-HCl, 200 NaCl, 20 NaF, 1 Na3VO4, 1 DTT, 1% Triton X-100 (pH 7.4), and complete protease and phosphatase inhibitor cocktails (Roche Diagnostics, Germany). About 20–25 μg of protein was loaded with SDS sample buffer (30 min, 37 °C in 2% beta-mercaptoethanol). The protein samples were separated on 5/10/15% SDS-PAGE, transferred, and probed with the following primary antibodies: mouse monoclonal anti-GAPDH (1:30000, BTMC-A473-9, BIOTREND), rabbit polyclonal anti-RyR2 (1:15000, HPA020028, Sigma), mouse monoclonal anti-PLB (1:5000, MA3-922, Thermo Scientific), rabbit polyclonal anti-pCaMKII (1:1000, PA5–36838, Thermo Scientific), rabbit polyclonal anti-CaMKII (1:5000, PA5-22168, Thermo Scientific), mouse monoclonal anti-NCX (1:1000, R3F1, Swant), and mouse monoclonal anti-SERCA (1:20000, MA3–919, Thermo Scientific). Secondary antibodies included HRP-conjugated goat anti-rabbit and goat anti-mouse (1:30000, 111-035-144 and 115-035-062, respectively, Jackson Immunoresearch). ImmobilonTM Western Chemiluminescent HRP Substrate (Millipore) was used for the chemiluminescent detection. The intensity of individual bands from Western blots was quantified using Image Studio Lite software, normalized to GAPDH, and then shown relative to the control group value.
Statistics
All data are presented as mean values ± SEM. For statistical testing of longitudinal data including more than two groups, 2-way ANOVA corrected for multiple comparisons by the Sidak method was used. For comparison of two groups, Student’s t test was applied. Analysis was performed using GraphPad Prism 8. P values were two-sided and considered statistically significant if P < 0.05.
Results
Effects of empagliflozin on systolic Ca2+ transients and SR Ca2+ load
As empagliflozin was shown to (acutely) influence Ca2+ cycling [9, 10], we investigated the chronic effects (2 and 8 weeks) of empagliflozin on stimulated Ca2+ transients using epifluorescence microscopy (Fura-2). Caffeine application was used to assess the SR Ca2+ load. After 2 weeks of treatment, empagliflozin did not change systolic Ca2+ transient amplitude, diastolic Ca2+ levels, or the Ca2+ elimination kinetics during increasing stimulation frequencies (0.25, 0.5, 1 Hz) in iPSC-CM (n = 127, 116, 91 cells) compared with control (n = 124, 116, 88 cells, Fig. 1 a–b and e–g). Moreover, SR Ca2+ load (caffeine-transient amplitude) was not altered in the empagliflozin group (n = 14 cells) compared with the control group (n = 12 cells, Fig. 1c–d and h). After 8 weeks of treatment, which corresponds to the time of onset of the clinical effects of empagliflozin, Ca2+ transient amplitude in control iPSC-CM (F340/380: 0.54 ± 0.03, n = 136 cells) was not significantly different compared with the empagliflozin-treated group (0.61 ± 0.04, n = 125 cells) at 0.25 Hz and during increasing frequencies up to 1 Hz (Fig. 2a–b and e). Also, diastolic Ca2+ levels (F340/380 at 0.25 Hz: control: 0.99 ± 0.02, empagliflozin: 0.96 ± 0.03) and Ca2+ elimination (tau (s), 0.25 Hz; control: 1.53 ± 0.07 s; empagliflozin: 1.54 ± 0.07 s) were not changed (Fig. 2f–g). In line with that, SR Ca2+ load did not differ between control (n = 31 cells) and empagliflozin (n = 31 cells) after 8 weeks of treatment (Fig. 2c–d and h). Thus, chronic treatment with empagliflozin has neutral effects on cardiomyocyte systolic and diastolic Ca2+ cycling in iPSC-CM.

Systolic Ca2+ transients and SR Ca2+ load (epifluorescence microscopy, Fura-2 AM 5 μM) of human-induced pluripotent stem cell cardiomyocytes (iPSC-CM) after 2 weeks of treatment with either vehicle control (control) or 0.5 μmol/l empagliflozin (EMPA). (a–b) Original representative stimulated Ca2+-transient recordings at 0.25 Hz after 2 weeks of treatment and (c–d) Ca2+ transients recorded during application of 10 mM caffeine indicating SR Ca2+ load. (e) Mean data during increasing stimulation frequencies (0.25, 0.5, and 1 Hz) for systolic Ca2+-transient amplitude (f), diastolic Ca2+ levels, and (g) exponential decay time of Ca2+ transients (τ). (h) Mean data for caffeine-transient amplitude. The sample sizes of iPSC-CM are depicted below each column. Groups were statistically compared using two-way ANOVA with Sidak’s test for multiple comparisons or Student’s t test (h)

Systolic Ca2+ transients and SR Ca2+ load (epifluorescence microscopy, Fura-2 AM 5 μM) of human-induced pluripotent stem cell cardiomyocytes (iPSC-CM) after 8 weeks of treatment with either vehicle control (control) or 0.5 μmol/l empagliflozin (EMPA). (a–b) Original representative stimulated Ca2+-transient recordings at 0.25 Hz of 8 weeks treated iPSC-CMs and (c–d) caffeine-induced transients indicating SR Ca2+ content. (e) Mean data during increasing stimulation frequencies (0.25, 0.5, and 1 Hz) for systolic Ca2+-transient amplitude (f), diastolic Ca2+ levels, and (g) exponential decay time of Ca2+ transients (τ). (h) Mean data for caffeine-transient amplitude. The sample sizes of iPSC-CM are provided below the respective column. For statistical analysis, two-way ANOVA with Sidak’s test for multiple comparisons or Student’s t test (h) was used
Influence of chronic empagliflozin treatment on diastolic Ca2+ release in iPSC-CM
Diastolic Ca2+ release via leaky RyR2 is a key mechanism for contractile dysfunction and arrhythmias in HF and other cardiac diseases [12]. Previous investigation suggested that empagliflozin reduces diastolic Ca2+ release in HF CM [9]. We therefore investigated the direct long-term effects of empagliflozin on diastolic SR Ca2+ release using confocal microscopy (Fluo-4). After 2 weeks of treatment with either empagliflozin (n = 46 cells) or vehicle control (n = 45), empagliflozin showed no effects on diastolic Ca2+ spark frequency (Fig. 3a–b) and the total calculated diastolic Ca2+ leak (Fig. 3c) in iPSC-CM. After 8 weeks of treatment, which corresponds to the time where the clinical effects of empagliflozin became apparent, diastolic Ca2+ spark frequency in empagliflozin-treated iPSC-CM (n = 102 cells) was determined at 2.3 ± 0.4/100 μm/s, but was not changed compared with iPSC-CM treated with vehicle control (2.3 ± 0.3/100 μm/s, n = 108, Fig. 3d–e). Accordingly, the total calculated diastolic Ca2+ leak did not differ in iPSC-CM treated with either empagliflozin or vehicle control (Fig. 3f).

Diastolic sarcoplasmic reticulum Ca2+ release (confocal microscopy, Fluo-4 AM 10 μM) of human-induced pluripotent stem cell cardiomyocytes (iPSC-CM) after 2 and 8 weeks of treatment with either vehicle control (control) or 0.5 μmol/l empagliflozin (EMPA). (a) Representative original confocal line scans showing diastolic Ca2+ sparks in iPSC-CM after 2 and (d) after 8 weeks treatment with empagliflozin compared with vehicle control. (b) Mean data of diastolic Ca2+ spark frequency and (c) the total calculated diastolic Ca2+ leak (normalized to vehicle control) after 2 weeks and (e–f) 8 weeks of empagliflozin treatment. The sample sizes of iPSC-CM are presented below the respective column. Groups were statistically tested using Student’s t test
Effects of empagliflozin on cardiac action potentials
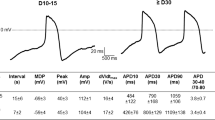
The action potential is a critical component of cardiomyocyte electrophysiology and adverse electric remodeling in HF. To study potential direct effects of chronic empagliflozin treatment on the action potential, we investigated stimulated action potentials in iPSC-CM using the patch-clamp technique. In iPSC-CM treated for 2 weeks with empagliflozin (n = 53 at 0.25 Hz), we observed no differences in action potential duration at 80% repolarization (APD80), resting membrane potential, or action potential amplitude compared with control iPSC-CM (n = 50, Fig. 4a–e). After 8 weeks of treatment, APD80 at 0.25 Hz was 344.4 ± 15.4 ms in control-treated cells (n = 45). APD80 (358.5 ± 18.5 ms) in empagliflozin-treated iPSC-CM (n = 43) was not significantly different compared to control (Fig. 4f–h). Also, resting membrane potential (control: − 62.4 ± 1.0 mV; empagliflozin: − 64.3 ± 1.1 mV) and action potential amplitude (control: 103.4 ± 1.7 mV; empagliflozin: 104.4 ± 1.8 mV) were not changed by empagliflozin after 8 weeks of treatment. In conclusion, empagliflozin showed neutral effects on cellular action potentials.

Action potentials (patch-clamp technique) of human-induced pluripotent stem cell cardiomyocytes (iPSC-CM) after treatment with either vehicle control (Control) or 0.5 μmol/l empagliflozin (EMPA). (a) Representative action potential recordings (0.25 Hz) of human iPSC-CM treated for 2 weeks with vehicle control or (b) empagliflozin. (c) Effects of 2 weeks treatment with control or empagliflozin on action potential duration at 80% (APD80), (d) resting membrane potential (RMP) and (e) action potential amplitude (APA). (f) Original action potential recordings (0.25 Hz) of 8 weeks treated human iPSC-CM with either control or (g) empagliflozin. (h) Mean data for APD80, (i) RMP, and (j) APA of iPSC-CM after 8 weeks of treatment with control or empagliflozin. The sample sizes of iPSC-CM are provided below the respective column. For statistical testing, two-way ANOVA with Sidak’s test for multiple comparisons (c and h) or Student’s t test was used
Effects of chronic empagliflozin treatment on gene expression profiles of human iPSC-CM
To further elucidate the molecular mechanisms underlying the pathogenesis of the empagliflozin-dependent phenotype, we performed RNA sequencing of iPSC-CM (4 differentiations from different cell lines from 2 donors), all with and without 0.5 μmol/l empagliflozin exposure for 8 weeks. After normalization for baseline expression, we did not identify differentially expressed genes (DEGs) between the untreated iPSC-CM population and the iPSC-CM after empagliflozin treatment while focusing on important EC-coupling associated genes (Fig. 5a–d). Normalized counts of empagliflozin-treated genes associated with systolic Ca2+ rise (RYR2), cytoplasmic Ca2+ extrusion (NCX, SERCA, PLN), cardiac remodeling (CAMKII), calcium binding (CALM1, CALM2, CASQ2), voltage-gated ion channels (SCN5A, CACNA1C, KCN5A), and Ca2+ transporting ATPase (ATP2B1) are shown in Fig. 5a–d.

Modulation of gene expression in human-induced pluripotent stem cell cardiomyocytes (iPSC-CM) after chronic (8 weeks) treatment with empagliflozin (0.5 μmol/l, n = 4 differentiations). (a) Empagliflozin-affected normalized counts of genes as ryanodine-receptor type 2 (RYR2), sodium-calcium exchanger (NCX1), sarcoplasmic reticulum Ca2+ ATPase (SERCA), Ca2+-/calmodulin-dependent protein kinase II (CaMKII), phospholamban (PLN), (b) calsequestrin 2 (CASQ2), calmodulin 1 (CALM1), calmodulin 2 (CALM2), (C) Na+ voltage-gated channel alpha subunit 5 (SCN5A), Ca2+ voltage-gated channel subunit alpha1 C (CACNA1C), K+ voltage-gated channel subfamily A member 5 (KCN5A), (d) ATPase plasma membrane Ca2+ transporting 1 (ATP2B1). For statistical analysis, Student’s t test was applied
Protein expression of EC-coupling proteins
To confirm our NGS findings on the protein level, we investigated the chronic effects of empagliflozin on regulatory EC-coupling proteins using Western blots analysis of treated iPSC-CM cultures (4 differentiations from different cell lines from 2 donors). Protein expression levels of EC-coupling proteins did not differ between 2 and 8 weeks. After 2 and 8 weeks of treatment, empagliflozin had no influence on protein expression of RyR2, which determines the systolic rise in [Ca2+]i (Fig. 6a, b). Furthermore, SERCA2a, PLB, and NCX, regulating cytosolic Ca2+ extrusion, were not affected in iPSC-CM after chronic empagliflozin treatment for 2 or 8 weeks (Fig. 6c, d, g). Also, CaMKII, which directly regulates cardiomyocyte Ca2+ cycling and which is strongly associated with adverse cardiac remodeling, was in terms of expression and autophosphorylation not affected in iPSC-CM after chronic in vitro treatment for 2 or 8 weeks (Fig. 6e, f).

Western blot of EC-coupling proteins in human-induced pluripotent stem cell cardiomyocytes (iPSC-CM). (a) Representative original Western blots after treatment with empagliflozin (EMPA) or vehicle control (control) for 2 or 8 weeks from 4 differentiation experiments (Diff.) from 2 healthy donors. GAPDH was used as loading control. (b) Mean protein expression levels (normalized to the respective control group at 2 or 8 weeks) in iPSC-CM (n = 4 differentiations, matched groups are displayed with matched individual symbols) and effects of 2 and 8 weeks treatment with empagliflozin (EMPA) on ryanodine-receptor type 2 (RyR2), (c) sodium-calcium exchanger (NCX), (d) sarcoplasmic reticulum Ca2+ ATPase (SERCA), (e) phosphorylated Ca2+-/calmodulin-dependent protein kinase II (pCaMKII), (f) Ca2+-/calmodulin-dependent protein kinase II (CaMKII), and (g) phospholamban (PLB). Groups were statistically analyzed using two-way ANOVA with Sidak’s test for multiple comparisons
Discussion
This study investigated the direct long-term and thereby clinically relevant effects of empagliflozin on cardiomyocyte EC-coupling and electrophysiology in iPSC-CM. As different mechanisms of SGLT2 inhibitors have been proposed, we aimed to perform blinded treatment, data acquisition, and analysis with a high amount of experimental repetitions. After 2 or 8 weeks of treatment, empagliflozin showed neutral effects on systolic Ca2+ transients and SR Ca2+ content as well as diastolic Ca2+ release. Action potential properties were unchanged upon chronic empagliflozin treatment. Finally, mRNA and protein expression levels of key genes and proteins involved in EC-coupling were not changed by empagliflozin.
Mechanistic evidence is still lacking to fully understand the beneficial clinical effects of SGLT2 inhibitors on cardiovascular and HF outcomes. In particular, the effects of empagliflozin on EC-coupling, which is centrally involved in pathological cardiac remodeling and HF [12], are not fully understood. In order to enhance the translational relevance, this study therefore used human iPSC-CM as a human cardiomyocyte model since the translational potential of animal models is limited. Moreover, utilizing iPSC-CM facilitates large numbers of standardized individual measurements. Of note, we could demonstrate that human iPSC-CM represent a valid translational system for cardiomyocyte function and electrophysiology [13, 20]. To investigate the direct cardiac effects of empagliflozin on cellular EC-coupling and to exclude systemic (secondary) effects on the myocardium (i.e., reduction in pre- and afterload), which could confound our observations, we directly treated iPSC-CM in vitro. Moreover, we performed a chronic treatment for up to 8 weeks in order to better mimic the situation of respective clinical trials, where the beneficial effects of empagliflozin became apparent after ~ 2 months of treatment [1]. Finally, iPSC-CM treatment, data acquisition, and data analysis were performed by externally blinded investigators.
Our data demonstrated that in human iPSC-CM, empagliflozin had neutral effects on systolic Ca2+ transients and SR Ca2+ content after 2 and 8 weeks of treatment. This result constitutes a safety signal as disturbed Ca2+ homeostasis can be associated with arrhythmias and negative inotropy. Moreover, long-term treatment with empagliflozin had neutral effects on action potential properties in human iPSC-CM, which is of relevance for cardiomyocyte electrophysiology as well as clinical safety. According to our findings on systolic Ca2+ transients, we previously did also not detect acute effects of empagliflozin on systolic Ca2+ transients in isolated CM from patients with HF [7]. However, data from animal cardiomyocyte studies showed an acute reduction of diastolic and systolic [Ca2+]i [10], while other experimental animal data showed an increase in Ca2+ transient amplitude and SR Ca2+ load upon exposure to empagliflozin [9]. Yet, adult CM from either animals or human are barely suitable for long-term treatment protocols, and methodological and species differences may limit the comparison of these studies. Nevertheless, while acute changes (up to 24 h) in Ca2+ cycling might be suggested by the aforementioned studies [9, 10], we could here demonstrate that after a treatment duration according to the clinical trial situation, empagliflozin has neutral effects on systolic Ca2+ cycling. Of note, it should also be considered that most already established prognostic relevant drugs for treating HF have neutral or even negative effects on systolic Ca2+ cycling and inotropy [21]. According to our functional findings, mRNA and protein expression of RyR2 and CaMKII mediating systolic Ca2+ release were not affected by chronic empagliflozin treatment. Furthermore, NCX, SERCA, and PLB involved in cytosolic Ca2+ extrusion and (for SERCA and PLB) SR Ca2+ load were not altered in iPSC-CM after chronic empagliflozin treatment. Thus, these first data on the long-term effects of empagliflozin indicated that, at least in CM, empagliflozin has neutral effects on EC-coupling gene as well as protein expression.
Furthermore, in human iPSC-CM, long-term treatment with empagliflozin did not affect diastolic Ca2+ release, which can contribute to cellular arrhythmogenesis and contractile dysfunction [22, 23]. In line with that, RyR2 and CaMKII protein and mRNA expression as well as pCaMKII, which have been shown to strongly regulate diastolic Ca2+ sparks [12, 24], were not altered after chronic treatment with empagliflozin in iPSC-CM. However, previous data reported a reduction of diastolic Ca2+ sparks as well as CaMKII activity in both human and murine failing CM after 24-h treatment with empagliflozin [9]. In these cells, based on the HF phenotype (while our iPSC-CMs are healthy), a distinct cellular remodeling is present including altered CaMKII activity. Considering the duration of the treatment, acute regulatory stress/oxidative processes may underlie the acute CaMKII-mediated reduction of Ca2+ sparks after exposure to empagliflozin in isolated human failing cells [9]. Nevertheless, we think that the treatment duration used in this study is according to the clinical trial situation, the more relevant approach to investigate the effects of empagliflozin.
In our study, we used iPSC-CM from healthy donors. Thus, the transfer of our results for particular cardiovascular diseases like diabetes or heart failure, where cardiac remodeling is present, might however be limited.
As another recently proposed mechanism, inhibitory effects of SGLT2 inhibitors on NHE1 have been discussed [10]. Since NHE1 inhibition has less effects in healthy models [25] and its activity is less in healthy hearts [26], it would be possible that in a healthy model, empagliflozin could have effects on the NHE1 and subsarcolemmal Na+/Ca2+ homeostasis without greatly changing bulk [Ca2+]i in the SR or the cytoplasm. Therefore, in our study, we cannot rule out effects on NHE1 activity. Finally, also other beneficial direct cardiac mechanisms of empagliflozin, i.e., on myofilament function [7] and on inflammatory and oxidative (paracrine) signaling cascades, have been proposed [8, 27, 28], which may serve as a rationale for further mechanistic studies.
In conclusion, we could demonstrate that after a clinically relevant treatment concentration and duration, empagliflozin has neutral effects on cardiomyocyte EC-coupling and electrophysiology in human iPSC-CM. Thus, at least in healthy cardiomyocytes, the beneficial clinical effects of empagliflozin seem to be not mediated by remodeling of cardiomyocyte EC-coupling, which has implications for further mechanistic investigation.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE (2015) Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 373:2117–2128
Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR (2017) Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 377:644–657
Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Ruff CT, Gause-Nilsson IAM, Fredriksson M, Johansson PA, Langkilde AM, Sabatine MS (2019) Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 380:347–357
McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Belohlavek J, Bohm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukat A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O'Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjostrand M, Langkilde AM, Committees D-HT, Investigators (2019) Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med 381:1995–2008 doi:
Fitchett D, Inzucchi SE, Cannon CP, McGuire DK, Scirica BM, Johansen OE, Sambevski S, Kaspers S, Pfarr E, George JT, Zinman B (2019) Empagliflozin reduced mortality and hospitalization for heart failure across the Spectrum of cardiovascular risk in the EMPA-REG OUTCOME trial. Circulation 139:1384–1395
Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, Flores E, Garcia-Ropero A, Sanz J, Hajjar RJ, Fuster V, Badimon JJ (2019) Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol 73:1931–1944
Pabel S, Wagner S, Bollenberg H, Bengel P, Kovacs A, Schach C, Tirilomis P, Mustroph J, Renner A, Gummert J, Fischer T, Van Linthout S, Tschope C, Streckfuss-Bomeke K, Hasenfuss G, Maier LS, Hamdani N, Sossalla S (2018) Empagliflozin directly improves diastolic function in human heart failure. Eur J Heart Fail 20:1690–1700
Kolijn D, Pabel S, Tian Y, Lodi M, Herwig M, Carrizzo A, Zhazykbayeva S, Kovacs A, Fulop GA, Falcao-Pires I, Reusch PH, Van Linthout S, Papp Z, van Heerebeek L, Vecchione C, Maier LS, Ciccarelli M, Tschope C, Mugge A, Bagi Z, Sossalla S, Hamdani N (2020) Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Galpha oxidation. Cardiovasc Res. https://doi.org/10.1093/cvr/cvaa123
Mustroph J, Wagemann O, Lucht CM, Trum M, Hammer KP, Sag CM, Lebek S, Tarnowski D, Reinders J, Perbellini F, Terracciano C, Schmid C, Schopka S, Hilker M, Zausig Y, Pabel S, Sossalla ST, Schweda F, Maier LS, Wagner S (2018) Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail 5:642–648
Baartscheer A, Schumacher CA, Wust RC, Fiolet JW, Stienen GJ, Coronel R, Zuurbier CJ (2017) Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia 60:568–573
Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, Jancev M, Hollmann MW, Weber NC, Coronel R, Zuurbier CJ (2017) Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia 61:722–726
Fischer TH, Maier LS, Sossalla S (2013) The ryanodine receptor leak: how a tattered receptor plunges the failing heart into crisis. Heart Fail Rev 18:475–483
Borchert T, Hubscher D, Guessoum CI, Lam TD, Ghadri JR, Schellinger IN, Tiburcy M, Liaw NY, Li Y, Haas J, Sossalla S, Huber MA, Cyganek L, Jacobshagen C, Dressel R, Raaz U, Nikolaev VO, Guan K, Thiele H, Meder B, Wollnik B, Zimmermann WH, Luscher TF, Hasenfuss G, Templin C, Streckfuss-Bomeke K (2017) Catecholamine-dependent beta-adrenergic signaling in a pluripotent stem cell model of Takotsubo cardiomyopathy. J Am Coll Cardiol 70:975–991
Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M, Fukuda K (2013) Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12:127–137
Heise T, Seewaldt-Becker E, Macha S, Hantel S, Pinnetti S, Seman L, Woerle HJ (2013) Safety, tolerability, pharmacokinetics and pharmacodynamics following 4 weeks' treatment with empagliflozin once daily in patients with type 2 diabetes. Diabetes Obes Metab 15:613–621
Scheen AJ (2014) Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet 53:213–225
Pabel S, Mustroph J, Stehle T, Lebek S, Dybkova N, Keyser A, Rupprecht L, Wagner S, Neef S, Maier LS, Sossalla S (2020) Dantrolene reduces CaMKIIδC-mediated atrial arrhythmias. Europace 22:1111–1118
Pabel S, Ahmad S, Tirilomis P, Stehle T, Mustroph J, Knierim M, Dybkova N, Bengel P, Holzamer A, Hilker M, Streckfuss-Bomeke K, Hasenfuss G, Maier LS, Sossalla S (2020) Inhibition of NaV1.8 prevents atrial arrhythmogenesis in human and mice. Basic Res Cardiol 115:20 doi:https://doi.org/10.1007/s00395-020-0780-8
Sossalla S, Maurer U, Schotola H, Hartmann N, Didie M, Zimmermann WH, Jacobshagen C, Wagner S, Maier LS (2011) Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na(+) current. Basic Res Cardiol 106:263–272
Streckfuss-Bomeke K, Wolf F, Azizian A, Stauske M, Tiburcy M, Wagner S, Hubscher D, Dressel R, Chen S, Jende J, Wulf G, Lorenz V, Schon MP, Maier LS, Zimmermann WH, Hasenfuss G, Guan K (2013) Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur Heart J 34:2618–2629
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González-Juanatey JR, Harjola V-P, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P (2016) 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failureThe task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the heart failure association (HFA) of the ESC. Eur Heart J 37:2129–2200
Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El-Armouche A, Hasenfuss G, Maier LS (2010) Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res 107:1150–1161
Hartmann N, Pabel S, Herting J, Schatter F, Renner A, Gummert J, Schotola H, Danner BC, Maier LS, Frey N, Hasenfuss G, Fischer TH, Sossalla S (2017) Antiarrhythmic effects of dantrolene in human diseased cardiomyocytes. Heart Rhythm 14:412–419
Fischer T, Herting J, Tirilomis T, Renner A, Neef S, Toischer K, Ellenberger D, Forster A, Schmitto J, Gummert J, Schondube F, Hasenfuss G, Maier L, Sossalla S (2013) CaMKII and PKA differentially regulate SR Ca2+−leak in human cardiac pathology. Circulation 128:970–981
Baartscheer A, Schumacher CA, van Borren MMGJ, Belterman CNW, Coronel R, Opthof T, Fiolet JWT (2005) Chronic inhibition of Na+/H+-exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc Res 65:83–92
Yokoyama H, Gunasegaram S, Harding SE, Avkiran M (2000) Sarcolemmal Na+/H+ exchanger activity and expression in human ventricular myocardium. J Am Coll Cardiol 36:534–540
Juni RP, Kuster DWD, Goebel M, Helmes M, Musters RJP, van der Velden J, Koolwijk P, Paulus WJ, van Hinsbergh VWM (2019) Cardiac microvascular endothelial enhancement of cardiomyocyte function is impaired by inflammation and restored by empagliflozin. JACC Basic Transl Sci 4:575–591
Zhazykbayeva S, Pabel S, Mügge A, Sossalla S, Hamdani N (2020) The molecular mechanisms associated with the physiological responses to inflammation and oxidative stress in cardiovascular diseases. Biophys Rev 12:947–968
Acknowledgments
We gratefully acknowledge the technical assistance of D. Riedl, J. Heine, S. Georgi-Beres, and Y. Metz.
Code availability
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL. S. Pabel, K. Streckfuss-Bömeke, and S. Sossalla are funded by the Else-Kröner-Fresenius Stiftung. S. Pabel is funded by the German Society of Internal Medicine. K. P. Hammer is funded by the DFG (HA 60107/3-1). L. Maier is funded by the DFG (MA 1982/5-1, 7-1).
Author information
Authors and Affiliations
Contributions
S.P., F.R., N.D., O.S., G.S., K.H., and K.S.B. performed experiments and analyzed experimental data; S.P.,G.S., K.S.B., and S.S. interpreted the results of experiments; S.P., K.S.B., and S.S. performed the conception and design of research; J.M., G.H., N.H., L.S.M., K.S.B., and S.S edited and revised the manuscript; S.P. drafted the manuscript; S.S. and K.S.B. approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
SS and LSM receive speaker’s/consultancy honoraria from Boehringer Ingelheim Pharma GmbH. SP, FR, ND, OS, GS, JM, KH, GH, NH, and KSB have nothing to declare.
Ethics approval
All procedures were performed according to the Declaration of Helsinki and were approved by the local ethics committee of the University Medical Center Göttingen.
Consent to participate
Informed consent was obtained from all tissue donors.
Consent for publication
The author transfers to Springer (respective to owner if other than Springer and for US government employees: to the extent transferable) the nonexclusive publication rights, and he/she warrants that his/her contribution is original and that he/she has full power to make this grant. The author signs for and accepts responsibility for releasing this material on behalf of any and all co-authors. This transfer of publication rights covers the nonexclusive right to reproduce and distribute the article, including reprints, translations, photographic reproductions, microform, electronic form (offline, online), or any other reproductions of similar nature.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pabel, S., Reetz, F., Dybkova, N. et al. Long-term effects of empagliflozin on excitation-contraction-coupling in human induced pluripotent stem cell cardiomyocytes. J Mol Med 98, 1689–1700 (2020). https://doi.org/10.1007/s00109-020-01989-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-020-01989-6