
Abstract
Transcription factors (TFs) participate in a wide range of cellular processes due to their inherent function as essential regulatory proteins. Their dysfunction has been linked to numerous human diseases. The forkhead box (FOX) family of TFs belongs to the “winged helix” superfamily, consisting of proteins sharing a related winged helix-turn-helix DNA-binding motif. FOX genes have been extensively present during vertebrates and invertebrates’ evolution, participating in numerous molecular cascades and biological functions, such as embryonic development and organogenesis, cell cycle regulation, metabolism control, stem cell niche maintenance, signal transduction, and many others. FOXD1, a forkhead TF, has been related to different key biological processes such as kidney and retina development and embryo implantation. FOXD1 dysfunction has been linked to different pathologies, thereby constituting a diagnostic biomarker and a promising target for future therapies. This paper aims to present, for the first time, a comprehensive review of FOXD1’s role in mouse development and human disease. Molecular, structural, and functional aspects of FOXD1 are presented in light of physiological and pathogenic conditions, including its role in human disease aetiology, such as cancer and recurrent pregnancy loss. Taken together, the information given here should enable a better understanding of FOXD1 function for basic science researchers and clinicians.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Transcription regulation depends on correct transcription factors (TFs) functioning and chromatin modulation. Two main categories (initiation and elongation) of TFs have been defined depending on their role during transcriptional regulation [1,2,3]. These proteins do not usually act alone as they bind cofactors, thereby forming protein complexes to activate or repress target gene transcription [4, 5]. Epigenetic modifications orchestrated by histone-modifying enzymes are also necessary to transform nucleosome dynamics and structure for producing interaction spaces for transcription complexes. Specific gene expression programmes are greatly related to particular sets of TFs’ temporal and spatial activity leading to cells’ inherent functional properties. Regulation in specific cells and during developmental stages appears to be particularly intricate in some cases, as different TF combinations can bind various genome sites to form regulatory networks [6,7,8,9,10,11].
TFs participate in a wide range of cellular processes due to their function as essential regulatory proteins. Numerous human diseases have been linked to their dysfunction (secondary to encoding or regulatory mutations). Greater than 15% of reported TFs have been related to Online Mendelian Inheritance in Man (OMIM-FOXD1: 601091) links. However, computational exploration of databases has predicted thousands of disease-associated TFs [12]. The amount of this kind of proteins has not yet been stated accurately, but it has been estimated that at least ~ 1400 have high confidence sequence-specific DNA binding domains (DBD) [13,14,15]. TFs are classified into families and subfamilies depending on their DBD sequence/structure and function [12, 16, 17].
The forkhead box (FOX) family of TFs belongs to the “winged helix” superfamily, consisting of proteins sharing a related winged helix-turn-helix DNA-binding motif. FOX genes have been extensively present during vertebrates and invertebrates’ evolution, participating in numerous physiological processes and biological functions, such as embryonic development and organogenesis, cell cycle regulation, metabolism control, stem cell niche maintenance, signal transduction, and many others [18, 19]. A wide range of phenotypes are associated with FOX genes’ dysregulation and mutations in human species [19,20,21,22,23,24,25,26,27,28,29].
Different key biological processes are related to FOXD1 function (also known as BF-2 such as kidney, retina, facial growth centres, hypothalamus, cardiomyocytes, and lung pericyte development, as well as embryo implantation. FOXD1 dysfunction, secondary to FOXD1 coding mutations and expression disturbances, has been linked to different diseases, suggesting it as a possible diagnostic biomarker and a promising target for future therapies.
This paper aims to present, for the first time, a comprehensive review of FOXD1’s role in mouse development (e.g., kidney and retina) and human disease. Molecular, structural, and functional aspects of FOXD1 are presented in the light of physiological and pathogenic conditions, including its role in human disease aetiology, such as cancer and recurrent pregnancy loss (RPL). Taken together, the information given here should enable a better understanding of FOXD1 function for basic science researchers and clinicians.
FOXD1 and the forkhead family of transcription factors
The first Forkhead (FKH) factor was described almost 30 years ago when a new mutant protein was shown to be involved in the formation of the spiked head phenotype in D. melanogaster [30]. This morphological modification resulted from a dysfunction resembling a “forked head” of animals’ foregut during their physiological development. Members of this family have been named forkhead (or fox) proteins since then. Their characteristic DBD (forkhead) is a ~ 100 amino acid long conserved region, defined because of its similarity with a hepatocyte nuclear factor 3 (HNF3) region [31]. Thereafter, Clark et al., used X-ray crystallography at 2.5 Å resolution to describe the HNF-3/forkhead DNA-recognition motif’s DBD three-dimensional structure [32]. FOX gene amount varies considerably in each species, although they are widely distributed amongst vertebrate and non-vertebrate organisms (> 2000 members throughout > 100 species) [19, 22]. At least 19 FOX protein subclasses (A through S) have been defined, depending on their conservation and DBD similarity [18, 33, 34]. FOX proteins’ DBD recognises and binds to the 5′-(G/A)(T/C)(A/C)AA(C/T)A-3′ core sequence to transactivate selected target gene promoters [22, 35]. However, the transactivation domain and accurate mechanisms for regulating target genes have not yet been completely elucidated for most of them.
Regarding human disease, numerous disorders are related to FOX gene dysfunction, such as cancer (e.g. FOXA1, FOXA2, FOXC2, FOXE1, FOXM1, FOXO1, FOXO3, FOXD1), vitiligo, lymphedema-distichiasis syndrome (FOXC2), thyroid dysgenesis, spiky hair and cleft palate (FOXE1, FOXF2, FOXP2, FOXC2 and FOXG1), eye abnormalities (FOXE3), Rett syndrome (FOXG1), heart disease (FOXH1), blepharophimosis, ptosis and epicanthus inversus syndrome (BPES) and non-syndromic primary ovarian insufficiency (FOXL2), intellectual disability (FOXP1), speech-language disorders (FOXP2) and RPL (FOXD1) [19, 36,37,38,39,40,41,42,43,44] (and references therein).
FOXD1 was identified and described for the first time within the forebrain neuroepithelium, and it has been suggested to be an important factor during retina development [45]; this study showed that the BF-2 DBD was closely related to those from other winged helix proteins, which were described thereafter as forkhead transcription factors (e.g. HFH-2 or FOXD3, FKH2 or FOXG1). At chromosome level, FOXD1 is an autosomal monoexonic gene located on chromosome 13 in mice and 5q12-q13 in humans. FOXD1 protein consists of 465 and 456 amino acids in mouse and human, respectively. They have similarity regarding their DBD (Supplementary Fig. S1). FOXD1 has various GC guanine-cytosine (GC)-rich regions encoding different lengths of homopolymeric polyamino acid stretches (e.g. Ala, Pro) having unknown function (Supplementary Fig. S1). These DNA regions are prone to forming hairpin DNA structures and suffering polymerase slippage, thereby hampering PCR, cloning and sequencing of coding regions. Apart from the forkhead domain, FOXD1 has a 122 amino acid-long hyperacidic NH2-terminal domain which might have transactivation properties [46]. Two polyproline runs (positions 241–312) are located within the COOH-terminal domain and described as potential effector sites. FOXD1 is expressed in different tissues and cells such as testes, kidneys, the central nervous system (CNS) (e.g. optic chiasm, retina, pituitary gland), the mesenchyme of facial growth centres, neuroepithelial cells of the prethalamus and hypothalamus, cadiomyocytes, lung pericytes and placenta, while overexpression has been observed in certain types of cancer (see below) [32, 41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. Due to its inherent nature as a transcription factor, FOXD1’s subtle expression is mandatory in regulating gene targets belonging to intricate molecular cascades. Several genes have been described as being direct or indirect targets for FOXD1, such as DCN, PGF, C3, DAX1, HMX1 (GH6), HMX4 (SOHo), EPHA3, EPHA6, LHB, PAI-1, PRKAR1A (Riα), p27, ALDH1A3, NKX2–2, AGT and REN [36, 49, 65, 66, 70, 72,73,74,75,76,77,78,79,80] (Fig. 1). In mice, Gli2 is an upstream direct regulator of Foxd1 expression during medulloblastoma pathogenesis [66]. Moreover, Shh transcriptionally regulates Foxd1 during mitogenic activity using Gli2−/−3−/− irradiated mouse embryonic fibroblast (iMEF) cells. In addition, Fgf, Wnt and TGFβ regulate Foxd1 expression in developing chick retina [60, 80, 81]. FOXD1 protein-protein interactions have not yet been validated experimentally.

FOXD1 direct and indirect regulatory proteins and target genes. a FOXD1 direct and indirect regulatory proteins. A1 Fgf8 and Wnt signalling repress FoxD1 expression in optic vesicles. A2 transcription factors such as Ets-1, Wt1 and Wt11, along with the p53 protein, are upstream regulators of Foxd1 gene expression during kidney development. A3 TGFβ and WNT5B factors involved in Hodgkin lymphoma have a transcriptional effect on FOXD1 expression. A4 Gli2 is an upstream direct regulator of Foxd1 expression during medulloblastoma pathogenesis. b FOXD1 direct and indirect target genes. B1 Retina development. B2 Kidney development. B3 Placenta and endometrium. B4 Testes and adipocytes. B5 Induced-pluripotent stem cells (iPSCs) b6 Different types of human cancer. *DEG genes, red: direct FOXD1 regulators and target genes
FOXD1 in kidney development
The kidneys develop during three stages in mammalian embryogenesis: pronephros, mesonephros and metanephros. Kidney development in mice begins in E8.5 and lasts to E11.5 whilst it becomes initiated during the 4th week to 35th week of gestation in humans. The metanephros develops during the last stage of renal embryogenesis, giving rise to the mature kidneys, which consist of the ureteric bud (UB) and the metanephric mesenchyme (MM) (Fig. 2). MM stroma mediates UB development, thereby enhancing its differentiation into ureters, pelvises, calyces and the collecting system. The branching UB induces the MM to form the nephrons’ epithelia and renal stroma [50, 82]. Foxd1 expression in mice is constrained to the MM cortical stroma in the developing kidneys (where nephrons differentiate), and it has lower expression in the medulla stroma around the collecting system (Fig. 2). Osr1 is an early marker of kidney development which acts as an upstream regulator of the expression of Six-2 and Pax-2 transcription factors. The Osr1+ cell population is divided into various groups of cells in developing kidneys (E11.5): Foxd1+ cells (which are stromal cell lineage progenitors), Pax-2+ epithelial cell progenitors and Six-2+ cell cap mesenchyme cell progenitors [57, 83,84,85,86]. Foxd1+ and Six-2+ coordinate cell differentiation into distinct kidney compartments [86].

WT and mutant forms of mouse Foxd1 affecting kidney development. a Foxd1 WT. Foxd1 during normal kidney development. Foxd1 expression in mice is constrained to the MM cortical stroma. Hox10, Tcf21, Ecm1, Fat4, Alcam and Rarb are implicated in UB branching. Ets-1, Wt1 and Wt11, along with the p53 protein regulates Foxd1 gene expression. Foxd1+ together with Six-2+ coordinate cell differentiation into distinct kidney compartments. Dcn becomes repressed by Foxd1 in cortical cells required for regulation of Bmp/Smad signalling and Cited1+. Foxd1-expressing population cells (Foxd1+ progenitors) can differentiate into mesangial cells, vascular smooth muscle cells, perivascular fibroblast, renin cell precursors and pericytes. b Foxd1 KO models. The lack of Foxd1 causes small kidneys and alterations in UB epithelium and the MM. Sfrp1 and Raldh2 gene expression is absent, and there is an increase in non-capsular (Bmp4+) and endothelial cells, accompanied by overexpression of Ret in the UB. Dcn is ectopically overexpressed altering Bmp/Smad signalling. Foxd1-null kidneys have shown decreased expression of Agt, Ren, Ace and At1r. MM: metanephric mesenchyme, UB: ureteric bud, red: red: direct FOXD1 regulators and target genes
Stroma cells play a pivotal role in signalling pathway activation and the expression of molecules enabling ureters and collecting system development, as well as epithelial and capsule differentiation regulation. Studies using reporter gene activity and a knockout murine model (C57Bl6 Wnt11−/−) have shown that transcription factors such as Ets-1, Wt1 and Wt11, along with the p53 protein, are upstream regulators of Foxd1 gene expression during kidney development [46, 71, 87] (Figs. 1 and 2). The Wnt11−/− murine model has revealed alterations in renal tubule morphology due to downregulation of Foxd1 [87].
Proteins such as Hox10, Tcf21, Ecm1, Fat4, Alcam and Rarb have also been implicated in stroma differentiation and UB branching [88,89,90]. Moreover, Hox10 (a homebox transcription factor) enables proper Foxd1+ cell location within the kidneys. Studies using Hox10-deficient mice models have shown that Foxd1+ cell expression was restricted to the kidney periphery, thereby impairing cortical stromal cell integration during renal embryogenesis (Fig. 2) [85, 89].
Studies of renal development using Foxd1 murine genetic modified models, during E14.5, have shown alterations in UB epithelium due to a small collecting system and inhibited nephron development in MM cortical stroma [50, 58, 85] (Table 1). Hatini et al. used KO mice to determine Foxd1 role during kidney development; they showed that Foxd1 was the earliest genetic marker of kidney stromal cell lineage and that its absence produced defects in UB epithelium and the MM [50]. These authors demonstrated inhibited UB growth and branching, as well as increased condensation in the mesenchyme with a decreased amount of nephrons. The KO mice had misplaced small kidneys which were separated from the adrenal glands.
Levinson et al. used in situ hybridisation and immunohistochemistry to show that Foxd1−/− animals’ kidneys were fused by defects in the renal capsule’s stromal cell population, thus inhibiting their detachment from the midline; they also showed that capsule development was impaired. The renal capsule normally expresses Sfrp1 and Raldh2 genes. However, these proteins were absent in Foxd1-deficient mice. Instead, there was expression of non-capsular (Bmp4+) and endothelial cells, accompanied by overexpression of Ret in the UB, thereby altering its branching. Bmp4 overexpression contributed to ectopic location of the glomeruli in the kidneys [58]. All these defects in kidney development might have been due to alterations in the signalling pathways required for renal capsule maturation [58, 85].
Other studies, using Foxd1 murine models (Foxd1lacZ/+ and Foxd1GFPCre), ChIP and reporter gene assays, have found that decorin (Dcn) became a repressed direct transcriptional target of Foxd1 in cortical cells [74] (Table 1; Fig. 1). Dcn in mice regulates the activation of Bmp/Smad signalling required for Cited1+ nephron progenitor cell differentiation to epithelial cells. Such mice’s differentiated kidneys retained cap mesenchyme cells within the Cited1+ compartment, thus inhibiting nephron progenitor differentiation. Furthermore, Dcn was ectopically overexpressed, thereby altering nephron differentiation by abnormal Bmp/Smad signalling (Bmp-4 and Bmp-7) and epithelial transition induction (Fig. 2) [74, 85, 95].
Hum et al. used a Foxd1-eGFP/Cre transgenic mouse line, immunohistochemistry and in situ hybridisation to determine the role of the renal stroma in patterning developing kidneys (Table 1). They found that Foxd1 played a key role in maintenance of renal stroma integrity [90]. Furthermore, they replicated previous studies’ findings regarding disorganised stromal pattern, loss of limits between nephrons, reduced ureteric branching, differentiation of nephrons from the medulla and abnormal vasculogenesis patterns [50, 58, 85, 90]. This suggests that Foxd1 might be used as an early biomarker of stromal cell lineage during kidney embryogenesis, enabling proper cell differentiation within the renal cortex and capsule.
Foxd1-expressing population cells (Foxd1+ progenitors) can differentiate into mesangial cells, vascular smooth muscle cells, perivascular fibroblast, renin cell precursors (expressing renin-Ren+) and pericytes once a stromal cell lineage has been developed (Fig. 2) [57, 96,97,98]. Foxd1+ cells in mice are endothelial (EPC) and mural renal arteriole progenitor cells, located in peritubular capillaries. Mukherjee et al. isolated Foxd1+ stroma from the Foxd1-eGFP/Cre transgenic mouse strain to grow them as organoids, thereby confirming that the endothelium was differentiated from Foxd1+ cell renal stroma [91, 99]. Moreover, these cells are angiogenic regulators which can synthesise erythropoietin and also play a key role during the arterial tree’s suitable expansion and orientation in the kidneys [90, 92, 94, 97, 100,101,102].
Foxd1+ cell differentiation into mesangial and vascular cells depends on the recombining binding protein suppressor (Rbpj) which is the downstream effector in the Notch signalling pathway [103, 104]. Sequeira-Lopez et al. have used Foxd1-GFP/Cre mice to study whether Foxd1+ stromal cells were renal artery-derived vascular progenitor precursors regulating kidney vascular morphogenesis and orientation. The authors showed that these cells differentiated into Ren+ cells, making cellular networks with capillary epithelium and thus regulating vascular resistance, blood pressure and fluid-electrolyte balance [92].
Moreover, studies using Foxd1-null kidneys have shown that a reduced amount of Ren+ cells can alter renal perfusion due to these cells’ detachment from the capillary walls, as well as decreased expression of Agt (angiotensinogen), Ren (renin), Ace (angiotensin I converting enzyme) and At1r (angiotensin II, type I receptor-associated protein). It is worth noting that Agt and Ren promoters have putative binding sites for Foxd1. Foxd1 therefore might act as an upstream regulator of the renin-angiotensin system [76] (Figs. 1 and 2). Renin-angiotensin system proteins are key molecules during UB branching morphogenesis. Their dysregulation due to the absence of Foxd1 might thus contribute toward abnormal UB development [85, 91, 92, 96].
Some studies have found that Foxd1+ cells, such as pericytes and mesangial cells, are involved in myofibroblast and endothelial cell development, causing kidney fibrotic diseases and a renal pro-inflammatory environment [93, 94, 96, 100, 105,106,107,108]. Humphreys et al. (2010) used a FoxD1/Cre knock-in mouse model for labelling the renal stroma and determining myofibroblast development during fibrosis (Table 1). It was found that Foxd1+-derived pericytes differentiated into smooth muscle actin-positive myofibroblast during fibrogenesis [93].
A recent study by Chang et al. used a Foxd1Cre/+ model and methylation techniques to demonstrate the molecular mechanism regulating Epo expression in fibrotic kidneys. They used Foxd1+ progenitor-derived Col1a1-GFP+Pdgfrβ+ pericytes to differentiate them into myofibroblasts and reported that pericytes expressed Epo, though its expression was inhibited by its promoter’s methylation when differentiating into myofibroblasts during kidney fibrogenesis (Table 1) [94]. Foxd1+ cell homeostatic functions have also been seen to become lost during kidney fibrinogenesis which involved upregulation of Adamts-2 and downregulation of Timp-3 [91, 108].
Foxd1+ cells have thus been defined as key players during proper kidney organogenesis. They contribute toward activating the signalling pathways necessary for the development, differentiation and compartmentalisation of renal cells belonging to the stroma, renal capsule, endothelium and vascular system. FOXD1 in humans may be a useful marker for cell fate maps from kidney embryogenesis to possible kidney diseases. However, how FOXD1 variants affect kidney development in humans or its pathogenesis still needs elucidating.
FOXD1 in retina development
The retina is the part of the eye in close contact with the CNS; its main function is to convert light into electrical signals. This organ consists of photoreceptors, rods and cones. These cells send a light signal to the bipolar cells which, in turn, are in contact with the retinal ganglion cells (RGCs) located in the inner retina. RGC somata occupy ganglion cell layers; their dendrites project into the inner plexiform layer and their axons extend through the optic disk to exit the eye to reach the optic nerve, which transfers light signals to the brain’s visual centres (the lateral geniculate nucleus and the superior colliculus) [109,110,111,112].
RGCs are the first cells to develop from the multipotent progenitor cells in the dorsocentral retina and migrate to the diencephalon. In mice, these cells develop between E11 and P0, peaking in E14.5, while in humans, they develop between the 5th and 18th weeks of gestation [109, 113]. These cells’ development in mice depend on fibroblast growth factors Fgf3 and Ffg8 expression in the neural retina as well as Notch signalling inhibition by secreted frizzled-related proteins and the Delta signalling pathway [109, 114,115,116]. Transcription factors such as Atoh7, Neurog2, Ascl1 and downstream genes such as Brn3b and Isl-1 are expressed to enhance RGCs survival and differentiation [109, 117,118,119]. RGC growth through the optic nerve is achieved by a cAMP-dependent mechanism and the Eph/Ephrin signalling pathway.
Five per cent of the RGCs’ axons remain ipsilateral while 95% decussate to the contralateral optic tract once they approach the optic chiasm in mice, around E10 and E11 [109, 113]. Conversely, 50% of nasal retina RGC axons in humans decussate to the contralateral optic tract, while the remaining 50% of temporal retina RGC axons continue in the ipsilateral tract. It has been stated that activating Shh signalling and transcription factor Isl2 expression are needed for proper decussation. Transcription factor Zic2 promotes Ephb1 expression for RGC ipsilateral projection.
Foxd1 and Foxg1 play a key role during binocular vision development as they participate in regulating RGC ipsilateral and contralateral projection. Foxd1 is expressed in the temporal retina and Foxg1 in nasal retina, as well as in the ventral diencephalon (Fig. 3) [45, 51, 70, 113, 120]. Zic2, a zinc-finger transcription factor, regulates RGC ipsilateral projection from the ventrotemporal (VT) retina to the optic chiasm, while EphB1/ephrin-B2 guides RGC axons ipsilateral projection through the optic tract [121, 122]. Foxd1 is expressed predominantly in the VT retina, near to the midline of the optic chiasm, where it regulates Zic2 and EphB1 expression (Figs. 1 and 3) [51].

WT and mutant forms of mouse Foxd1 affecting retina development. a Foxd1 WT. Foxd1 is expressed in the temporal retina and Foxg1 in nasal retina. Isl2 expression is needed for proper decussation. Transcription factor Zic2 promotes Ephb1 expression for RGC ipsilateral projection. Zic2 regulates RGC ipsilateral projection from the ventrotemporal (VT) retina to the optic chiasm, while EphB1/ephrin-B2 guides RGC axons through the optic tract. b Foxd1 KO model. Foxd1-deficient mice showed RGC axons growing arrested toward the midline of the optic chiasma. These animals had alterations regarding the crossed/uncrossed ratio of RGC axons projecting ipsi- or contralaterally and abnormalities in chiasma shape. Mice also showed ectopic expression of Foxg1 and downregulation of Zic2 and Ephb1 expression. Other genes such as Islet1, Slit2. Boc and EphA/ephrinA also had altered expression. Dashed lines: genes with altered expression due to Foxd1 KO
Herrera et al. used a Foxd1lacZ/lacZ murine model to study the retinofugal pathway, and this transcription factor’s role during chiasma development in embryos from E12.5 to P0 (Table 1; Fig. 3). This model showed that RGC axons grew in the same way as those from wild-type mice when they exited the retina to enter the optic stalk during E12.5 and E13.5. However, following this embryonic age, Foxd1-deficient mice showed RGC axons growing toward the midline of the optic chiasma where their development became subsequently arrested. These animals had alterations regarding the crossed/uncrossed ratio of RGC axons projecting ipsi- or contralaterally and abnormalities in chiasma shape. These results demonstrated that Foxd1 is a key regulator of RGC axon migration through the optic chiasm [51]. Moreover, there was an increase of ipsilateral projections as well as the ectopic expression of Foxg1 in areas previously occupied by Foxd1, thereby affecting cell type organisation and the expression of guide factors such as Slit2 (Fig. 3). It was shown that Zic2 and Ephb1 expression was downregulated in the VT retina, indicating that these molecules might be Foxd1 targets in the retina (Fig. 1) [51, 123].
Other genes such as Islet1, Slit2. Boc and EphA/ephrinA also had altered expression, suggesting that Foxd1 is a key regulator of signalling pathways involved in VT RGC axons’ ipsilateral retinal projection [51, 70, 109, 124, 125].
Takahashi et al. studied genetic FoxD1 mechanisms for establishing temporal specificity in chick retina using in ovo electroporation and in situ hybridisation (Table 1). They found that FoxD1 played a crucial role in determining temporal projection from the retina. Moreover, these authors showed that Fgf8 and Wnt signalling repressed FoxD1 expression in optic vesicles (Fig. 1). Altered FoxD1 expression resulted in abnormal projection of RGC axons throughout the nasotemporal retina by dysregulation of downstream target proteins such as GH6, SOHo1 and EphA3 located in the temporal retina [70, 77, 80]. Foxd1 promotes temporal characteristics in the retina during optic vesicle protrusion in response to Fgf and Shh activity in the zebrafish model (Table 1; Fig. 1) [126]. Inhibiting Shh activity produced Foxd1 downregulation and subsequent abnormal routing of retinal axon projections in the mesencephalon.
Interestingly, FOXD1 (xbf-2) can also act as a transcriptional repressor participating in the development of neural plate via Bmp-4 downregulation in the Xenopus model [127]. More recently, Yaklichkin et al., (2007) have predicted the transcriptional repressor nature of Fox TFs in metazoan organisms. The authors showed that the eh1 repressor motif interacts with the Groucho transcriptional co-repressors at the C-terminal region of different Fox family members, thus potentially repressing their transcriptional activity [128]. It is highly probable that FOXD1 also functions in mammalian species as a transcriptional repressor of genes related to various processes involved in embryo development and organogenesis.
All the aforementioned results reported in different species have supported the fact that Foxd1 might be a key player during retinal development and is crucial for the suitable routing of ipsilateral RGC axon projections in mammals. Mutations within FOXD1 in humans might thus alter its functionality, causing alterations in binocular vision due to abnormal ipsilateral projections through the brain’s visual centres.
FOXD1 and the biology of cancer
The biology of cancer is genetically based on the accumulation of mutations and epigenetic modifications leading to cell metabolism dysfunction and uncontrolled proliferation. Genetic abnormalities (e.g. point mutations, gene amplifications, chromosomal translocations) and disturbances in FOX gene expression have been linked to a variety of cancer types [18, 33, 129,130,131]. FOXD1 expression disturbances, rather than mutations in its open reading frame, have been related to the biology of different types of cancer, such as prostate, lung, gastric, hepatocellular carcinoma, glioma, medulloblastoma, ovary, breast and Hodgkin lymphoma [47, 48, 52, 55, 60, 61, 63, 66, 69, 72, 75, 132].
FOXD1 expression (and that of 11 further FOX genes) in prostate cancer has been assessed by qPCR in neoplastic (malignant) prostate tissue, lymph node metastases, benign prostatic hyperplasia, xenografts and prostate cell lines [52]. That study showed that FOXD1 was very poorly expressed in normal prostate tissue but overexpressed in cancer cells and lymph node metastases, thereby seeming to contribute tumour biology (Fig. 1; Table 2). However, differential FOXD1 levels could not be related to the disease’s progression and their quantification was not defined as a prognostic marker of clinical usefulness. Comparative analysis (i.e. microarray analysis) and knockdown assays concerning lung cancer transcriptome have shown that FOXD1 expression was related to abnormal cell proliferation [61].
Microarray analysis has revealed that FOXD1 expression in non-small-cell lung cancer was related to the PTEN signature. Patients having high FOXD1 expression levels had lower survival rates than those having normal FOXD1 transcription. Indeed, high FOXD1 expression levels were linked to different clinical features, such as being male, having a history of smoking, the presence of squamous cell carcinoma and a lack of EGFR mutations (Fig. 1; Table 2) [61].
FOXD1 has been found to be differentially expressed in gastric cancer between metastatic and non-metastatic tumours [47]. These findings led to proposing it as a factor having an important role in complex regulatory networks, especially those involved in cell differentiation. A more recent study using previous gene expression patterns in gastric cancer and robust computational analysis has shown that FOXD1 is a key transcription factor, regulating numerous downstream target genes related to the disease’s progression [63]. A similar study using 8 hepatocellular carcinoma expression profiles proposed that at least ten TFs (including FOXD1) were responsible for most downstream differentially expressed genes [132] (Fig. 1; Table 2).
FOXD1 downregulation in the ovary was related to chemoresistant tumours [55]. Three studies have reported an association between FOXD1 expression and cancer biology in CNS tumours (glioma and medulloblastoma) [48, 66, 72]. Cheng et al. reported a key role for FOXD1-aldehyde dehydrogenase 1A3 (ALDH1A3) signalling in the development of the mesenchymal glioma stem-like cell (MES) population (a malignant glioblastoma subtype) (Fig. 1; Table 2). In vitro and in vivo assays have shown that FOXD1 knockdown led to MES clonogenic properties. Clinically, high FOXD1 expression levels in high-grade glioma were related to patients’ poorest prognosis [72]. FOXD1 was found to be overexpressed in glioma tissue compared to normal brain samples using primary tumours and gene knockdown experiments involving cell lines [48]. Furthermore, siRNA transfection against FOXD1 has led to a decrease in cell proliferation/migration rates and apoptotic features. A very recent study involving mice described Foxd1 as a relevant regulator of the Shh/Gli2-Nkx2-2 molecular pathway regarding medulloblastoma biology (Fig. 1; Table 2) [66]. FOXD1 has also been studied in haematopoietic malignancies, being overexpressed in six Hodgkin lymphoma cell lines and in primary cancer tissues [60]. Bioinformatics functional clustering of transcriptomic data has indicated a potential activator output for the JAK-STAT pathway in FOXD1 transcription. Specific molecules (i.e. TGFβ, WNT5B) belonging to signal transduction pathways involved in Hodgkin lymphoma biology have had a transcriptional effect on FOXD1 expression [60]. FOXD1 has been associated with tumourigenesis and the disease’s progression in malignant breast neoplasms [75]. It has been found to be upregulated in human breast cancer samples [75]. Functional in vitro studies shown that reduced FOXD1 expression has led to decreased proliferation and chemoresistance in breast cancer cell lines. Interestingly, this study also described an important role for FOXD1 in cell cycle regulation as it induces G1 to S transition via p27 direct regulation (Fig. 1; Table 2) [75].
All the above findings might suggest that FOXD1’s could have a major role in cancer biology and provide the basis for proposing it as a coherent target for future therapeutic approaches.
FOXD1 in mammalian implantation and recurrent pregnancy loss
Mammalian reproductive success can be considered the result of several finely regulated physiological and cellular processes in terms of gene expression. Numerous molecular pathways participate in the steps involved, from gamete generation/fecundation to birth. Embryo implantation (a critical stage for fertility and reproduction) involves hundreds of genes participating in complex regulatory networks [133,134,135]. Different mouse models have been used to describe its molecular basis due to inherent ethical limitations in studying implantation in human species. The interspecific recombinant congenic strains (IRCS) model, enabling quantitative trait loci (QTL) mapping, has been particularly successful, as it has led to identifying short genomic regions and genes (e.g. Foxd1 and Alpp) as being relevant molecular actors involved in implantation [36, 136,137,138,139,140,141].
Briefly, the IRCS model was created from an initial cross between two evolutionarily distant parental species of mice (Mus musculus and Mus spretus), followed by two backcrosses involving Mus musculus which reduced the percentage donor (M. spretus) genome introgressed into the receptor’s genetic background (Mus musculus) [137, 139, 140]. Consanguineous (brother-sister) crosses were then performed during > 30 generations to establish 53 IRCS strains, each having an average of 98% M. musculus and 2% M. spretus genomes (fixed at homozygous state). Complex traits can be studied in this model and QTL mapped (using genome markers) by comparing specific phenotypes between strains and parental Mus. musculus animals.
Laissue et al. studied embryonic death/resorption in 39 IRCS strains and 4 sub-strains via in vivo ultrasonography of pregnant females and identified 3 QTL potentially related to the phenotype [138]. One of these (~ 5 Mb in length), located on murine chromosome 13 (MMU13), encompassed 31 genes, three of which were transcription factors (Btf3, PolK and Foxd1). A more recent study determined that the drastic Foxd1-p.Thr152Ala mutation, located in the Foxd1-DBD, became fixed during M. spretus genome evolution (Table 1) [36]. This finding was related to transcriptomic disturbances of endometrial/placental tissues in IRCS-MMU13 females and evoked a potentially major role for Foxd1 in embryonic resorption and litter size modulation.
Interestingly, the Foxd1-p.Thr152Ala mutation has led to transactivation disturbances of direct target genes such as Pgf and C3, two key molecules involved in embryo implantation physiology (Fig. 1). These findings led to proposing FOXD1 as a coherent RPL-related candidate gene. FOXD1 open reading frame direct sequencing involving a panel of 556 RPL-affected women revealed 10 non-synonymous heterozygous mutations potentially related to the phenotype [36]. RPL-related FOXD1-p.Ala356Gly, FOXD1-p.Ile364Met and FOXD1-p.Ala428_Ala429InsAlaAla mutations have shown transactivation disturbances in PGF and/or C3 promoters, similarly to the Foxd1-p.Thr152Ala change. Interestingly, it has been stated that RPL individuals carrying FOXD1 mutations have 10.3 times higher relative risk than women having wild-type alleles, while the FOXD1-p.Ala88Gly variant seems to be related to a protective effect. Taken together, these results have demonstrated that FOXD1 might be a key molecule participating in human implantation and pregnancy maintenance and could be used as a molecular marker in clinical environments.
Concluding remarks and future directions
FOXD1 has demonstrated multi-level roles during normal development, adult physiology and several diseases’ pathogenesis. It is crucial for kidney and retina organogenesis, facial growth centers development in normal conditions, as well as for female fertility. Interestingly, coding mutations in human species have been uniquely associated with RPL, while different kinds of murine models have shown retina, kidney and fertility dysfunction. These differences might have been due to inherent interspecific genomic/transcriptomic/proteomic differences and to the nature of FOXD1 mutant forms (e.g. KO, point mutations, in or out the DBD).
It should be noted that the embryonic resorption phenotype was not studied in Foxd1-KO animals, and kidney and optic dysfunctions were not assessed in animals carrying the Foxd1-p.Thr152Ala mutation [36, 50, 51, 58, 74, 76, 90]. Thus, it cannot be ruled out that these animals could have dysfunctions affecting multiple organs which have not yet been described. FOXD1 variants found in human RPL patients were located exclusively outside the DBD, arguing in favour of drastic mutations possibly being negatively selected or related to another phenotype (e.g. renal or visual). It might also be possible that severe mutations could cause a syndromic disorder affecting various tissues. Consequently, FOXD1 encoding regions should be directly sequenced in patients having different combinations of renal, visual and/or implantation dysfunction.
FOXD1 expression disturbances have been associated with the disease’s pathogenesis in cancer. Specific transcriptome studies and promoter sequence analysis concerned with enlarged types of tumours would thus be interesting. Genome-editing approaches using somatic cells might be considered as a future therapeutic alternative. For instance, CRISPR technology might be applied in vitro and in vivo to modifying FOXD1-relevant genomic regions’ underlying functional characteristics. FOXD1 upstream and downstream regulation might also be explored using additional experimental techniques, such as yeast one-hybrid (Y1H) screening for DNA-protein interactions and chromatin immunoprecipitation sequencing (ChIP-Seq). Finally, it is worth stressing that FOXD1 structure elucidation and novel protein partner discoveries are needed at proteomic level for a better understanding of its function.
References
Lee TI, Young RA (2013) Transcriptional regulation and its misregulation in disease. Cell 152:1237–1251
Fuda NJ, Ardehali MB, Lis JT (2009) Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461:186–192
Zhou Q, Li T, Price DH (2012) RNA polymerase II elongation control. Annu Rev Biochem 81:119–143
Siggers T, Duyzend MH, Reddy J, Khan S, Bulyk ML (2011) Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol Syst Biol 7:555
Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Zhou T, Rohs R, Honig B, Bussemaker HJ, Mann RS (2011) Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell 147:1270–1282
Spitz F, Furlong EEM (2012) Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13:613–626
Smith NC, Matthews JM (2016) Mechanisms of DNA-binding specificity and functional gene regulation by transcription factors. Curr Opin Struct Biol 38:68–74
Schleif RF (2013) Modulation of DNA binding by gene-specific transcription factors. Biochemistry 52:6755–6765
Adachi K, Schöler HR (2012) Directing reprogramming to pluripotency by transcription factors. Curr Opin Genet Dev 22:416–422
Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT (2018) The human transcription factors. Cell 172:650–665
Osório J (2016) Gene regulation: landscape and mechanisms of transcription factor cooperativity. Nat Rev Genet 17:5
Yusuf D, Butland SL, Swanson MI, Bolotin E, Ticoll A, Cheung WA, Cindy Zhang X, Dickman CTD, Fulton DL, Lim JS, Schnabl JM, Ramos OHP, Vasseur-Cognet M, de Leeuw CN, Simpson EM, Ryffel GU, Lam EWF, Kist R, Wilson MSC, Marco-Ferreres R, Brosens JJ, Beccari LL, Bovolenta P, Benayoun BA, Monteiro LJ, Schwenen HDC, Grontved L, Wederell E, Mandrup S, Veitia RA, Chakravarthy H, Hoodless PA, Mancarelli MM, Torbett BE, Banham AH, Reddy SP, Cullum RL, Liedtke M, Tschan MP, Vaz M, Rizzino A, Zannini M, Frietze S, Farnham PJ, Eijkelenboom A, Brown PJ, Laperrière D, Leprince D, de Cristofaro T, Prince KL, Putker M, del Peso L, Camenisch G, Wenger RH, Mikula M, Rozendaal M, Mader S, Ostrowski J, Rhodes SJ, van Rechem C, Boulay G, Olechnowicz SWZ, Breslin MB, Lan MS, Nanan KK, Wegner M, Hou J, Mullen RD, Colvin SC, Noy P, Webb CF, Witek ME, Ferrell S, Daniel JM, Park J, Waldman SA, Peet DJ, Taggart M, Jayaraman PS, Karrich JJ, Blom B, Vesuna F, O'Geen H, Sun Y, Gronostajski RM, Woodcroft MW, Hough MR, Chen E, Europe-Finner GN, Karolczak-Bayatti M, Bailey J, Hankinson O, Raman V, LeBrun DP, Biswal S, Harvey CJ, DeBruyne JP, Hogenesch JB, Hevner RF, Héligon C, Luo XM, Blank M, Millen K, Sharlin DS, Forrest D, Dahlman-Wright K, Zhao C, Mishima Y, Sinha S, Chakrabarti R, Portales-Casamar E, Sladek FM, Bradley PH, Wasserman WW (2012) The transcription factor encyclopedia. Genome Biol 13:R24
Deplancke B, Alpern D, Gardeux V (2016) The genetics of transcription factor DNA binding variation. Cell 166:538–554
Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM (2009) A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10:252–263
Fulton DL, Sundararajan S, Badis G, Hughes TR, Wasserman WW, Roach JC, Sladek R (2009) TFCat: the curated catalog of mouse and human transcription factors. Genome Biol 10:R29
Wingender E, Schoeps T, Haubrock M, Dönitz J (2015) TFClass: a classification of human transcription factors and their rodent orthologs. Nucleic Acids Res 43:D97–D102
Ehsani R, Bahrami S, Drabløs F (2016) Feature-based classification of human transcription factors into hypothetical sub-classes related to regulatory function. BMC Bioinformatics 17:459
Hannenhalli S, Kaestner KH (2009) The evolution of Fox genes and their role in development and disease. Nat Rev Genet 10:233–240
Golson ML, Kaestner KH (2016) Fox transcription factors: from development to disease. Development 143:4558–4570
Laissue P, Vinci G, Veitia RA, Fellous M (2008) Recent advances in the study of genes involved in non-syndromic premature ovarian failure. Mol Cell Endocrinol 282:101–111
Martins R, Lithgow GJ, Link W (2016) Long live FOXO: unraveling the role of FOXO proteins in aging and longevity. Aging Cell 15:196–207
Benayoun BA, Caburet S, Veitia RA (2011) Forkhead transcription factors: key players in health and disease. Trends Genet 27:224–232
Le Fevre AK, Taylor S, Malek NH et al (2013) FOXP1 mutations cause intellectual disability and a recognizable phenotype. Am J Med Genet A 161:3166–3175
Webb AE, Brunet A (2014) FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci 39:159–169
Coomans de Brachène A, Demoulin J-B (2016) FOXO transcription factors in cancer development and therapy. Cell Mol Life Sci 73:1159–1172
Maiese K (2016) Forkhead transcription factors: new considerations for Alzheimer’s disease and dementia. J Transl Sci 2:241–247
Siper PM, De Rubeis S, Trelles MDP et al (2017) Prospective investigation of FOXP1 syndrome. Mol Autism 8:57
Elzaiat M, Todeschini A-L, Caburet S, Veitia RA (2017) The genetic make-up of ovarian development and function: the focus on the transcription factor FOXL2. Clin Genet 91:173–182
Link W, Fernandez-Marcos PJ (2017) FOXO transcription factors at the interface of metabolism and cancer. Int J Cancer 141:2379–2391
Weigel D, Jürgens G, Küttner F, Seifert E, Jäckle H (1989) The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell 57:645–658
Lai E, Prezioso VR, Smith E, Litvin O, Costa RH, Darnell JE (1990) HNF-3A, a hepatocyte-enriched transcription factor of novel structure is regulated transcriptionally. Genes Dev 4:1427–1436
Clark KL, Halay ED, Lai E, Burley SK (1993) Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 364:412–420
Katoh M, Katoh M (2004) Human FOX gene family (review). Int J Oncol 25:1495–1500
Shimeld SM, Degnan B, Luke GN (2010) Evolutionary genomics of the Fox genes: origin of gene families and the ancestry of gene clusters. Genomics 95:256–260
Georges AB, Benayoun BA, Caburet S, Veitia RA (2010) Generic binding sites, generic DNA-binding domains: where does specific promoter recognition come from? FASEB J 24:346–356
Laissue P, Lakhal B, Vatin M, Batista F, Burgio G, Mercier E, Santos ED, Buffat C, Sierra-Diaz DC, Renault G, Montagutelli X, Salmon J, Monget P, Veitia RA, Méhats C, Fellous M, Gris JC, Cocquet J, Vaiman D (2016) Association of FOXD1 variants with adverse pregnancy outcomes in mice and humans. Open Biol 6:160109
Everson JL, Fink DM, Yoon JW, Leslie EJ, Kietzman HW, Ansen-Wilson LJ, Chung HM, Walterhouse DO, Marazita ML, Lipinski RJ (2017) Sonic hedgehog regulation of Foxf2 promotes cranial neural crest mesenchyme proliferation and is disrupted in cleft lip morphogenesis. Development 144:2082–2091
Leslie EJ, Liu H, Carlson JC, Shaffer JR, Feingold E, Wehby G, Laurie CA, Jain D, Laurie CC, Doheny KF, McHenry T, Resick J, Sanchez C, Jacobs J, Emanuele B, Vieira AR, Neiswanger K, Standley J, Czeizel AE, Deleyiannis F, Christensen K, Munger RG, Lie RT, Wilcox A, Romitti PA, Field LL, Padilla CD, Cutiongco-de la Paz EMC, Lidral AC, Valencia-Ramirez LC, Lopez-Palacio AM, Valencia DR, Arcos-Burgos M, Castilla EE, Mereb JC, Poletta FA, Orioli IM, Carvalho FM, Hecht JT, Blanton SH, Buxó CJ, Butali A, Mossey PA, Adeyemo WL, James O, Braimah RO, Aregbesola BS, Eshete MA, Deribew M, Koruyucu M, Seymen F, Ma L, de Salamanca JE, Weinberg SM, Moreno L, Cornell RA, Murray JC, Marazita ML (2016) A genome-wide association study of nonsyndromic cleft palate identifies an etiologic missense variant in GRHL3. Am J Hum Genet 98:744–754
Moreno LM, Mansilla MA, Bullard SA, Cooper ME, Busch TD, Machida J, Johnson MK, Brauer D, Krahn K, Daack-Hirsch S, L’Heureux J, Valencia-Ramirez C, Rivera D, López AM, Moreno MA, Hing A, Lammer EJ, Jones M, Christensen K, Lie RT, Jugessur A, Wilcox AJ, Chines P, Pugh E, Doheny K, Arcos-Burgos M, Marazita ML, Murray JC, Lidral AC (2009) FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Hum Mol Genet 18:4879–4896
Lammer EJ, Mohammed N, Iovannisci DM, Ma C, Lidral AC, Shaw GM (2016) Genetic variation of FOXE1 and risk for orofacial clefts in a California population. Am J Med Genet A 170:2770–2776
Mohamad Shah NS, Salahshourifar I, Sulong S, Wan Sulaiman WA, Halim AS (2016) Discovery of candidate genes for nonsyndromic cleft lip palate through genome-wide linkage analysis of large extended families in the Malay population. BMC Genet 17:39
Lennon CJ, Birkeland AC, Nuñez JAP, Su GH, Lanzano P, Guzman E, Celis K, Eisig SB, Hoffman D, Rendon MTG, Ostos H, Chung WK, Haddad J Jr (2012) Association of candidate genes with nonsyndromic clefts in Honduran and Colombian populations. Laryngoscope 122:2082–2087
Bahuau M, Houdayer C, Tredano M, Soupre V, Couderc R, Vazquez MP (2002) FOXC2 truncating mutation in distichiasis, lymphedema, and cleft palate. Clin Genet 62:470–473
Ozturk F, Li Y, Zhu X, Guda C, Nawshad A (2013) Systematic analysis of palatal transcriptome to identify cleft palate genes within TGFβ3-knockout mice alleles: RNA-Seq analysis of TGFβ3 mice. BMC Genomics 14:113
Hatini V, Tao W, Lai E (1994) Expression of winged helix genes, BF-1 and BF-2, define adjacent domains within the developing forebrain and retina. J Neurobiol 25:1293–1309
Ernstsson S, Pierrou S, Hulander M, Cederberg A, Hellqvist M, Carlsson P, Enerbäck S (1996) Characterization of the human forkhead gene FREAC-4. Evidence for regulation by Wilms’ tumor suppressor gene (WT-1) and p53. J Biol Chem 271:21094–21099
Feng D, YE X, Zhu Z et al (2015) Comparative transcriptome analysis between metastatic and non-metastatic gastric cancer reveals potential biomarkers. Mol Med Rep 11:386–392
Gao Y-F, Zhu T, Mao X-Y, Mao CX, Li L, Yin JY, Zhou HH, Liu ZQ (2017) Silencing of Forkhead box D1 inhibits proliferation and migration in glioma cells. Oncol Rep 37:1196–1202
Gumbel JH, Patterson EM, Owusu SA, Kabat BE, Jung DO, Simmons J, Hopkins T, Ellsworth BS (2012) The forkhead transcription factor, Foxd1, is necessary for pituitary luteinizing hormone expression in mice. PLoS One 7:1–10
Hatini V, Huh SO, Herzlinger D, Soares VC, Lai E (1996) Essential role of stromal mesenchyme in kidney morphogenesis revealed by targeted disruption of winged helix transcription factor BF-2. Genes Dev 10:1467–1478
Herrera E, Marcus R, Li S, Williams SE, Erskine L, Lai E, Mason C (2004) Foxd1 is required for proper formation of the optic chiasm. Development 131:5727–5739
van der Heul-Nieuwenhuijsen L, Dits NF, Jenster G (2009) Gene expression of forkhead transcription factors in the normal and diseased human prostate. BJU Int 103:1574–1580
Hung C, Linn G, Chow YH, Kobayashi A, Mittelsteadt K, Altemeier WA, Gharib SA, Schnapp LM, Duffield JS (2013) Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med 188:820–830
Jeong J (2004) Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev 18:937–951
Ju W, Yoo BC, Kim I-J, Kim JW, Kim SC, Lee HP (2009) Identification of genes with differential expression in chemoresistant epithelial ovarian cancer using high-density oligonucleotide microarrays. Oncol Res 18:47–56
van Mens TE, Liang H-PH, Basu S, Hernandez I, Zogg M, May J, Zhan M, Yang Q, Foeckler J, Kalloway S, Sood R, Karlson CS, Weiler H (2017) Variable phenotypic penetrance of thrombosis in adult mice after tissue-selective and temporally controlled Thbd gene inactivation. Blood Adv 1:1148–1158
Kobayashi A, Mugford JW, Krautzberger AM, Naiman N, Liao J, McMahon AP (2014) Identification of a multipotent self-renewing stromal progenitor population during mammalian kidney organogenesis. Stem Cell Reports 3:650–662
Levinson RS (2005) Foxd1-dependent signals control cellularity in the renal capsule, a structure required for normal renal development. Development 132:529–539
Millington G, Elliott KH, Y-TT C et al (2017) Cilia-dependent GLI processing in neural crest cells is required for tongue development. Dev Biol 424:124–137
Nagel S, Meyer C, Kaufmann M, Drexler HG, MacLeod RAF (2014) Deregulated FOX genes in Hodgkin lymphoma. Genes Chromosom Cancer 53:917–933
Nakayama S, Soejima K, Yasuda H, Yoda S, Satomi R, Ikemura S, Terai H, Sato T, Yamaguchi N, Hamamoto J, Arai D, Ishioka K, Ohgino K, Naoki K, Betsuyaku T (2015) FOXD1 expression is associated with poor prognosis in non-small cell lung cancer. Anticancer Res 35:261–268
Newman EA, Kim DW, Wan J, Wang J, Qian J, Blackshaw S (2018) Foxd1 is required for terminal differentiation of anterior hypothalamic neuronal subtypes. Dev Biol 439:102–111
Xu G, Li K, Zhang N, Zhu B, Feng G (2016) Screening driving transcription factors in the processing of gastric cancer. Gastroenterol Res Pract 2016:1–9
Yeo HC, Ting S, Brena RM, Koh G, Chen A, Toh SQ, Lim YM, Oh SKW, Lee DY (2016) Genome-wide transcriptome and binding sites analyses identify early FOX expressions for enhancing cardiomyogenesis efficiency of hESC cultures. Sci Rep 6:31068
Zhang H, Palmer R, Gao X, Kreidberg J, Gerald W, Hsiao L, Jensen RV, Gullans SR, Haber DA (2003) Transcriptional activation of placental growth factor by the forkhead/winged helix transcription factor FoxD1. Curr Biol 13:1625–1629
Zhang Y, Wang T, Wang S, Xiong Y, Zhang R, Zhang X, Zhao J, Yang AG, Wang L, Jia L (2018) Nkx2-2as suppression contributes to the pathogenesis of sonic hedgehog medulloblastoma. Cancer Res 78:962–973
Baek J-I, Choi SY, Chacon-Heszele MF, Zuo X, Lipschutz JH (2014) Expression of Drosophila forkhead transcription factors during kidney development. Biochem Biophys Res Commun 446:15–17
Zhao M, Zhou Y, Zhu B, Wan M, Jiang T, Tan Q, Liu Y, Jiang J, Luo S, Tan Y, Wu H, Renauer P, del Mar Ayala Gutiérrez M, Castillo Palma MJ, Ortega Castro R, Fernández-Roldán C, Raya E, Faria R, Carvalho C, Alarcón-Riquelme ME, Xiang Z, Chen J, Li F, Ling G, Zhao H, Liao X, Lin Y, Sawalha AH, Lu Q (2016) IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann Rheum Dis 75:1998–2006
Zhou H, Lv Q, Guo Z (2018) Transcriptomic signature predicts the distant relapse in patients with ER+ breast cancer treated with tamoxifen for five years. Mol Med Rep 17:3152–3157
Carreres MI, Escalante A, Murillo B, Chauvin G, Gaspar P, Vegar C, Herrera E (2011) Transcription factor Foxd1 is required for the specification of the temporal retina in mammals. J Neurosci 31:5673–5681
Cederberg A, Hulander M, Carlsson P, Enerbäck S (1999) The kidney-expressed winged helix transcription factor FREAC-4 is regulated by Ets-1: a possible role in kidney development. J Biol Chem 274:165–169
Cheng P, Wang J, Waghmare I, Sartini S, Coviello V, Zhang Z, Kim SH, Mohyeldin A, Pavlyukov MS, Minata M, Valentim CLL, Chhipa RR, Bhat KPL, Dasgupta B, la Motta C, Kango-Singh M, Nakano I (2016) FOXD1-ALDH1A3 signaling is a determinant for the self-renewal and tumorigenicity of mesenchymal glioma stem cells. Cancer Res 76:7219–7230
Koga M, Matsuda M, Kawamura T, Sogo T, Shigeno A, Nishida E, Ebisuya M (2014) Foxd1 is a mediator and indicator of the cell reprogramming process. Nat Commun 5:1–9
Fetting JL, Guay JA, Karolak MJ, Iozzo RV, Adams DC, Maridas DE, Brown AC, Oxburgh L (2014) FOXD1 promotes nephron progenitor differentiation by repressing decorin in the embryonic kidney. Development 141:17–27
Zhao Y-F, Zhao J-Y, Yue H, Hu KS, Shen H, Guo ZG, Su XJ (2015) FOXD1 promotes breast cancer proliferation and chemotherapeutic drug resistance by targeting p27. Biochem Biophys Res Commun 456:232–237
Song R, Lopez MLSS, Yosypiv IV (2017) Foxd1 is an upstream regulator of the renin-angiotensin system during metanephric kidney development. Pediatr Res 82:855–862
Yuasa J, Hirano S, Yamagata M, Noda M (1996) Visual projection map specified by topographic expression of transcription factors in the retina. Nature 382:632–635
Dahle MK, Grønning LM, Cederberg A, Blomhoff HK, Miura N, Enerbäck S, Taskén KA, Taskén K (2002) Mechanisms of FOXC2- and FOXD1-mediated regulation of the RI alpha subunit of cAMP-dependent protein kinase include release of transcriptional repression and activation by protein kinase B alpha and cAMP. J Biol Chem 277:22902–22908
Berg DT, Myers LJ, Richardson MA, Sandusky G, Grinnell BW (2005) Smad6s regulates plasminogen activator inhibitor-1 through a protein kinase C-β-dependent up-regulation of transforming growth factor-β. J Biol Chem 280:14943–14947
Takahashi H, Sakuta H, Shintani T, Noda M (2009) Functional mode of FoxD1/CBF2 for the establishment of temporal retinal specificity in the developing chick retina. Dev Biol 331:300–310
Fink DM, Sun MR, Heyne GW, Everson JL, Chung HM, Park S, Sheets MD, Lipinski RJ (2018) Coordinated d-cyclin/Foxd1 activation drives mitogenic activity of the sonic hedgehog signaling pathway. Cell Signal 44:1–9
Piscione TD, Waters AM (2008) Structural and functional development of the kidney. In: Geary DF, Schaefer F (eds) Comprehensive pediatric nephrology, Mosby, Philadelphia, pp 91–129. https://doi.org/10.1016/B978-0-323-04883-5.50012-X
Hendry C, Rumballe B, Moritz K, Little MH (2011) Defining and redefining the nephron progenitor population. Pediatr Nephrol 26:1395–1406
Mugford JW, Sipilä P, McMahon JA, McMahon AP (2008) Osr1 expression demarcates a multi-potent population of intermediate mesoderm that undergoes progressive restriction to an Osr1-dependent nephron progenitor compartment within the mammalian kidney. Dev Biol 324:88–98
Li W, Hartwig S, Rosenblum ND (2014) Developmental origins and functions of stromal cells in the normal and diseased mammalian kidney. Dev Dyn 243:853–863
Davies J (2017) Pax2: a “keep to the path” sign on Waddington’s epigenetic landscape. Dev Cell 41:331–332
Nagy II, Xu Q, Naillat F et al (2016) Impairment of Wnt11 function leads to kidney tubular abnormalities and secondary glomerular cystogenesis. BMC Dev Biol 16:30
Paroly SS, Wang F, Spraggon L, Merregaert J, Batourina E, Tycko B, Schmidt-Ott KM, Grimmond S, Little M, Mendelsohn C (2013) Stromal protein Ecm1 regulates ureteric bud patterning and branching. PLoS One 8:e84155
Yallowitz AR, Hrycaj SM, Short KM, Smyth IM, Wellik DM (2011) Hox10 genes function in kidney development in the differentiation and integration of the cortical stroma. PLoS One 6:e23410
Hum S, Rymer C, Schaefer C, Bushnell D, Sims-Lucas S (2014) Ablation of the renal stroma defines its critical role in nephron progenitor and vasculature patterning. PLoS One 9:e88400
Mukherjee E, Maringer KV, Papke E et al (2017) Endothelial markers expressing stromal cells are critical for kidney formation. Am J Physiol Ren Physiol. https://doi.org/10.1152/ajprenal.00136.2017
Sequeira-Lopez MLS, Lin EE, Li M, Hu Y, Sigmund CD, Gomez RA (2015) The earliest metanephric arteriolar progenitors and their role in kidney vascular development. Am J Phys Regul Integr Comp Phys 308:R138–R149
Humphreys BD, Lin S-L, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176:85–97
Chang YT, Yang CC, Pan SY, Chou YH, Chang FC, Lai CF, Tsai MH, Hsu HL, Lin CH, Chiang WC, Wu MS, Chu TS, Chen YM, Lin SL (2016) DNA methyltransferase inhibition restores erythropoietin production in fibrotic murine kidneys. J Clin Invest 126:721–731
Ohmori T, Tanigawa S, Kaku Y, Fujimura S, Nishinakamura R (2015) Sall1 in renal stromal progenitors non-cell autonomously restricts the excessive expansion of nephron progenitors. Sci Rep 5:1–11
Gomez IG, Duffield JS (2014) The FOXD1 lineage of kidney perivascular cells and myofibroblasts: functions and responses to injury. Kidney Int Suppl 4:26–33
Kobayashi H, Liu Q, Binns TC, Urrutia AA, Davidoff O, Kapitsinou PP, Pfaff AS, Olauson H, Wernerson A, Fogo AB, Fong GH, Gross KW, Haase VH (2016) Distinct subpopulations of FOXD1 stroma-derived cells regulate renal erythropoietin. J Clin Invest 126:1926–1938
Fanni D, Gerosa C, Vinci L, Ambu R, Dessì A, Eyken PV, Fanos V, Faa G (2016) Interstitial stromal progenitors during kidney development: here, there and everywhere. J Matern Fetal Neonatal Med 29:1–6
Junttila S, Saarela U, Halt K, Manninen A, Parssinen H, Lecca MR, Brandli AW, Sims-Lucas S, Skovorodkin I, Vainio SJ (2015) Functional genetic targeting of embryonic kidney progenitor cells ex vivo. J Am Soc Nephrol 26:1126–1137
Sims-Lucas S, Schaefer C, Bushnell D, Ho J, Logar A, Prochownik E, Gittes G, Bates CM (2013) Endothelial progenitors exist within the kidney and lung mesenchyme. PLoS One 8:1–8
Lemos DR, Marsh G, Huang A, Campanholle G, Aburatani T, Dang L, Gomez I, Fisher K, Ligresti G, Peti-Peterdi J, Duffield JS (2016) Maintenance of vascular integrity by pericytes is essential for normal kidney function. Am J Physiol Ren Physiol 311:F1230–F1242
Gerl K, Steppan D, Fuchs M, Wagner C, Willam C, Kurtz A, Kurt B (2017) Activation of hypoxia signaling in stromal progenitors impairs kidney development. Am J Pathol 187:1496–1511
Lin EE, Sequeira-Lopez MLS, Gomez RA (2014) RBP-J in FOXD1+ renal stromal progenitors is crucial for the proper development and assembly of the kidney vasculature and glomerular mesangial cells. Am J Physiol Renal Physiol 306:F249–F258
Boyle SC, Liu Z, Kopan R (2014) Notch signaling is required for the formation of mesangial cells from a stromal mesenchyme precursor during kidney development. Development 141:346–354
Duffield JS, Humphreys BD (2011) Origin of new cells in the adult kidney: results from genetic labeling techniques. Kidney Int 79:494–501
Schrimpf C, Duffield JS (2011) Mechanisms of fibrosis: the role of the pericyte. Curr Opin Nephrol Hypertens 20:297–305
Nakagawa N, Duffield JS (2013) Myofibroblasts in fibrotic kidneys. Curr Pathobiol Rep 1:189–198
Duffield JS (2014) Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest 124:2299–2306
Erskine L, Herrera E (2014) Connecting the retina to the brain. ASN Neuro 6:175909141456210
Zhou ZJ, McCall MA (2008) Retinal ganglion cells in model organisms: development, function and disease. J Physiol 586:4343–4345
Sernagor E, Eglen SJ, Wong RO (2001) Development of retinal ganglion cell structure and function. Prog Retin Eye Res 20:139–174
Sanes JR, Masland RH (2015) The types of retinal ganglion cells: current status and implications for neuronal classification. Annu Rev Neurosci 38:221–246
Petros TJ, Rebsam A, Mason CA (2008) Retinal axon growth at the optic chiasm: to cross or not to cross. Annu Rev Neurosci 31:295–315
Austin CP, Feldman DE, Ida JA, Cepko CL (1995) Vertebrate retinal ganglion cells are selected from competent progenitors by the action of Notch. Development 121:3637–3650
Henrique D, Hirsinger E, Adam J, Roux IL, Pourquié O, Ish-Horowicz D, Lewis J (1997) Maintenance of neuroepithelial progenitor cells by Delta-Notch signalling in the embryonic chick retina. Curr Biol 7:661–670
Esteve P, Sandonìs A, Cardozo M, Malapeira J, Ibañez C, Crespo I, Marcos S, Gonzalez-Garcia S, Toribio ML, Arribas J, Shimono A, Guerrero I, Bovolenta P (2011) SFRPs act as negative modulators of ADAM10 to regulate retinal neurogenesis. Nat Neurosci 14:562–569
Maurer KA, Riesenberg AN, Brown NL (2014) Notch signaling differentially regulates Atoh7 and Neurog2 in the distal mouse retina. Development 141:3243–3254
Pacal M, Bremner R (2014) Induction of the ganglion cell differentiation program in human retinal progenitors before cell cycle exit. Dev Dyn 243:712–729
Prasov L, Glaser T (2012) Dynamic expression of ganglion cell markers in retinal progenitors during the terminal cell cycle. Mol Cell Neurosci 50:160–168
Pratt T (2004) The winged helix transcription factor Foxg1 facilitates retinal ganglion cell axon crossing of the ventral midline in the mouse. Development 131:3773–3784
Herrera E, Brown L, Aruga J et al (2003) Zic2 patterns binocular vision by specifying the uncrossed retinal projection. Cell 114:545–557
Williams SE, Mann F, Erskine L, Sakurai T, Wei S, Rossi DJ, Gale NW, Holt CE, Mason CA, Henkemeyer M (2003) Ephrin-B2 and EphB1 mediate retinal axon divergence at the optic chiasm. Neuron 39:919–935
Tian NM, Pratt T, Price DJ (2008) Foxg1 regulates retinal axon pathfinding by repressing an ipsilateral program in nasal retina and by causing optic chiasm cells to exert a net axonal growth-promoting activity. Development 135:4081–4089
Sanchez-Arrones L, Nieto-Lopez F, Sanchez-Camacho C, Carreres MI, Herrera E, Okada A, Bovolenta P (2013) Shh/Boc signaling is required for sustained generation of ipsilateral projecting ganglion cells in the mouse retina. J Neurosci 33:8596–8607
Wang Q, Marcucci F, Cerullo I, Mason C (2016) Ipsilateral and contralateral retinal ganglion cells express distinct genes during decussation at the optic chiasm. eNeuro 3. https://doi.org/10.1523/ENEURO.0169-16.2016
Hernández-Bejarano M, Gestri G, Spawls L, Nieto-López F, Picker A, Tada M, Brand M, Bovolenta P, Wilson SW, Cavodeassi F (2015) Opposing Shh and Fgf signals initiate nasotemporal patterning of the zebrafish retina. Development 142:3933–3942
Mariani FV, Harland RM (1998) XBF-2 is a transcriptional repressor that converts ectoderm into neural tissue. Development 125:5019–5031
Yaklichkin S, Vekker A, Stayrook S, Lewis M, Kessler DS (2007) Prevalence of the EH1 Groucho interaction motif in the metazoan Fox family of transcriptional regulators. BMC Genomics 8:201
Myatt SS, Lam EW-F (2007) The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer 7:847–859
Lakhal B, Philibert P, Laissue P, Benayoun B, Dipietromaria A, Braham R, Elghezal H, Saad A, Feellous M, Veitia RA, Sultan C (2009) Molecular genetics of secondary amenorrhea: functional analysis of an heterozygous variant of FOX-L2 gene (G187D) supports its involvement in non-syndromic premature ovarian failure. Horm Res 72:54–54
Katoh M, Igarashi M, Fukuda H, Nakagama H, Katoh M (2013) Cancer genetics and genomics of human FOX family genes. Cancer Lett 328:198–206
Chen J, Qian Z, Li F, Li J, Lu Y (2017) Integrative analysis of microarray data to reveal regulation patterns in the pathogenesis of hepatocellular carcinoma. Gut Liver 11:112–120
Cha J, Sun X, Dey SK (2012) Mechanisms of implantation: strategies for successful pregnancy. Nat Med 18:1754–1767
Koot YEM, Teklenburg G, Salker MS, Brosens JJ, Macklon NS (2012) Molecular aspects of implantation failure. Biochim Biophys Acta 1822:1943–1950
White MD, Plachta N (2015) How adhesion forms the early mammalian embryo. Curr Top Dev Biol 112:1–17
L’Hôte D, Serres C, Laissue P et al (2007) Centimorgan-range one-step mapping of fertility traits using interspecific recombinant congenic mice. Genetics 176:1907–1921
Burgio G, Szatanik M, J-LL G et al (2007) Interspecific recombinant congenic strains between C57BL/6 and mice of the Mus spretus species: a powerful tool to dissect genetic control of complex traits. Genetics 177:2321–2333
Laissue P, Burgio G, L’Hôte D et al (2009) Identification of quantitative trait loci responsible for embryonic lethality in mice assessed by ultrasonography. Int J Dev Biol 53:623–629
Laissue P, L’Hôte D, Serres C, Vaiman D (2009) Mouse models for identifying genes modulating fertility parameters. Animal 3:55–71
L’Hôte D, Laissue P, Serres C et al (2010) Interspecific resources: a major tool for quantitative trait locus cloning and speciation research. BioEssays 32:132–142
Vatin M, Burgio G, Renault G, Laissue P, Firlej V, Mondon F, Montagutelli X, Vaiman D, Serres C, Ziyyat A (2012) Refined mapping of a quantitative trait locus on chromosome 1 responsible for mouse embryonic death. PLoS One 7:e43356. https://doi.org/10.1371/journal.pone.0043356
Funding
The present study was supported by the Universidad del Rosario (Grant CS/Genetics/ABN062-2018). Laissue’s lab is supported by the Universidad del Rosario.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Fig. S1.
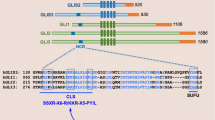
FOXD1 interspecific alignment (Homo sapiens and Mus musculus). An alignment with the nucleotide and amino acid sequences from human and mouse are shown. FOXD1 protein consists of 465 and 456 amino acids in mouse and human, respectively. The bold letters within the purple box indicate the conserved DBD sequence between both species. The yellow and blue boxes show the poly-Ala and poly-Pro stretches, respectively, located within the COOH-terminal domain. (PDF 263 kb)
Rights and permissions
About this article

Cite this article
Quintero-Ronderos, P., Laissue, P. The multisystemic functions of FOXD1 in development and disease. J Mol Med 96, 725–739 (2018). https://doi.org/10.1007/s00109-018-1665-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-018-1665-2