
Abstract
Inflammatory bowel disease (IBD) is a devastating disease that is associated with excessive inflammation in the intestinal tract in genetically susceptible individuals and potentially triggered by microbial dysbiosis. This illness markedly predisposes patients to thrombophilia and chronic debility as well as bowel, lymphatic, and liver cancers. Development of new therapies is needed to re-establish long-term immune tolerance in IBD patients without increasing the risk of opportunistic infections and cancer. Aberrant purinergic signaling pathways have been implicated in disordered thromboregulation and immune dysregulation, as noted in the pathogenesis of IBD and other gastrointestinal/hepatic autoimmune diseases. Expression of CD39 on endothelial or immune cells allows for homeostatic integration of hemostasis and immunity, which are disrupted in IBD. Our focus in this review is on novel aspects of the functions of CD39 and related NTPDases in IBD. Regulated CD39 activity allows for scavenging of extracellular nucleotides, the maintenance of P2-receptor integrity and coordination of adenosinergic signaling responses. CD39 together with CD73, serves as an integral component of the immunosuppressive machinery of dendritic cells, myeloid cells, T and B cells. Genetic inheritance and environental factors closely regulate the levels of expression and phosphohydrolytic activity of CD39, both on immune cells and released microparticles. Purinergic mechanisms associated with T regulatory and supressor T helper type 17 cells modulate disease activity in IBD, as can be modeled in experimental colitis. As a recent example, upregulation of CD39 is dependent upon ligation of the aryl hydrocarbon receptor (AHR), as with natural ligands such as bilirubin and 2-(1′ H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE). Decreased expression of CD39 and/or dysfunctional AHR signaling, however, abrogates the protective effects of immunosuppressive AHR ligands. These factors could also serve as biomarkers of disease activity in IBD. Heightened thrombosis, inflammation, and immune disturbances as seen in IBD appear to be associated with aberrant purinergic signaling. Ongoing development of therapeutic strategies augmenting CD39 ectonucleotidase bioactivity via cytokines or AHR ligands offers promise for management of thrombophilia, disordered inflammation, and aberrant immune reactivity in IBD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel disease (IBD) is an important illness of unclear pathogenesis associated with major defects in mucosal immunoregulation and develops in genetically susceptible individuals. These abnormalities often occur in association with microbial dysbiosis and result in unfettered inflammation of the intestine and extraintestinal tissues. Such events result in long-term morbidity and possibly even death, in otherwise healthy adults and children.
Dampening inflammation and re-establishing immune tolerance in IBD remain the major therapeutic goal. However, existing IBD therapies albeit providing recent advances, still largely rely on broad-based immunosuppression. For example, only around half of the patients treated with anti-TNF agents show substantive clinical responses. These improvements are often self-limited, while unfortunately increasing the risk of opportunistic infections.
The goal of our laboratory has been to investigate the control of mucosal immune responses, which are based on fundamental signaling pathways. Our own long-term interests in the regulation of purinergic signaling are now being leveraged to develop innovative and hopefully non-toxic therapies for IBD. This review and the accompanying articles in this special issue address new therapeutic concepts in IBD, as based on recent, linked work in hypoxia and purinergic signaling, mucosal barrier functions and microRNA biology.
In several recent, comprehensive reviews [1,2,3,4], we have already addressed the biological functions of ecto-enzymes, such as CD39, CD73, and CD38, in the regulation of purinergic signaling and control of extracellular adenosine levels. Others, and we, have noted the importance of these mechanisms in immunomodulation, as in cancer and inflammation. The ectonucleotidases of the CD39 family, in particular, have major impacts on the dynamic equilibrium of proinflammatory extracellular ATP, ADP nucleotides vs. the immunosuppressive potential of adenosine nucleosides. CD39 plays a dominant role in purinergic regulation of vascular inflammation, thrombosis, and the immune response in such settings. As such, the relevance and importance of these purinergic signaling pathways in selected neoplastic states (lymphoma and chronic leukemia) and inflammatory diseases (sepsis and autoimmunity) have been already alluded to in recent work.
In this update, we first provide a brief synopsis of the major components of purinergic signaling; chiefly for those not familiar to this field. We will focus on very recent work detailing the immunomodulation of CD39 on T cells and other immune cells by both genetic and environmental factors in the setting of IBD and experimental colitis, inclusive of the new roles for natural metabolites such as bilirubin. We will also briefly cover the role of CD39 expression on exosomes and microparticles, in control of inflammation in the gut and touch on the relevance of the microbiome. Lastly, we cover the emerging importance of other NTPDases of the CD39 family and speculate on their role in controlling gut inflammation.
Overview of purinergic signaling
Extracellular nucleotides (e.g., ATP, UTP, ADP, NAD), and the derivative nucleosides (e.g., adenosine from ATP), are released in a regulated manner by most cells to provide the initiators and primary components for purinergic responses [5]. In such settings, this process involves pannexins, which are conserved transmembrane channels that allow the passage of ions and small molecules. The pannexin-1 channels, as an example, mediate the release of ATP from activated T cells and dendritic cells; or even operate following apoptosis.
Under conditions of inflammatory stress, much higher levels of ATP and other nucleotides or nucleosides are released to the extracellular space by pannexins, gap junction hemichannels, e.g., connexin 43, following exocytosis as from dense platelet granules, and with active cell death. These extracellular nucleotide/nucleoside mediators bind specific purinergic receptors, which comprise an essential requirement for this signaling network.
Almost all immune and vascular cells express multiple type-2 purinergic/pyrimidinergic (P2) receptors for nucleotides and adenosine or type-1 purinergic (P1) receptors [6]. There are at least seven ionotropic (P2X1-7), eight metabotropic (P2Y1,2,4,6,11–14), and four adenosine receptor subtypes (A1, A2A, A2B, A3), which have been identified (more if one tallies in heteromers) [7]. P2X and P2Y11 receptors are chiefly activated by extracellular ATP; P2Y2 by ATP and UTP; P2Y1, P2Y12, and P2Y13 by ADP; P2Y4 by UTP; P2Y6 by UDP; and P2Y14 by UDP-glucose. (Nomenclature:http://www.guidetopharmacology.org/targets.jsp).
Extracellular ATP and the related nucleotide derivatives play important roles as signaling molecules. These pro inflammatory mediators participate in both autocrine and paracrine circuits to regulate cellular metabolism, migration, proliferation and apoptosis through signaling pathways triggered by P2Y and P2X receptors. Extracellular nucleotides also serve as substrates for ectonucleotidases, which generate the immunosupressive nucleoside, adenosine, after phosphohydrolysis via the ecto-enzymes on the cell membrane with catalytic domains located in the extracellular compartment.
CD39 is the prototype of the ecto-nucleoside triphosphate diphosphohydrolase (E-NTPDase) family (EC 3.6.1.5). These proteins comprise a group of ecto-enzymes that hydrolyze extracellular nucleoside tri- and diphosphates. One important ecto-nucleotidase chain or cascade, is initiated by these NTPDases, and is then terminated by ecto-5′-nucleotidase (CD73; EC 3.1.3.5) [8, 9].
As another example, AMP can be phosphorylated by ecto-adenylate kinase, or dephosphorylated by ecto-alkaline phosphatase [1, 9]. These and other ecto-enzymes hydrolyze extracellular nucleotides to generate nucleotides and nucleosides, which in turn differentially activate other P2, and then ultimately adenosine receptors. Whereas extracellular ATP generally provides pro-inflammatory signals, the extracellular adenosine produced from ATP/ADP/AMP degradation has potent immunosuppressive effects mediated by adenosinergic responses and cAMP-mediated effects. Hence, events triggered by adenosine generation may often have opposing effects to those seen with the initial P2-mediated effects [10].
Other cell surface-located nucleotide hydrolyzing and interconverting ectoenzymes have been described. These include the ecto-nucleotide pyrophosphatase phosphodiesterases (E-NPPs; EC 3.1.4.1, EC 3.6.1.9 and the autotaxin group), CD38, NAD-glycohydrolases, alkaline and acid phosphatases, diadenosine polyphosphate hydrolases, adenylate kinases, nucleoside diphosphate kinase, and potentially ecto-F1-Fo ATP synthases [9]. These other ectonucleotidases are not covered in detail here, given space constraints.
Recent work has also shown that E-NPP1 appears to catalyze transformation of NAD and ADP-ribose to generate AMP [11]. Moreover, it is also apparent that ecto-5′-nucleotidase/CD73 together with adenosine deaminase-1 and 2 (ADA1 and 2 (latter only in man, as no orthologous mouse gene exists); EC 3.5.4.4 and 3.5.4.2) generate and then convert adenosine to inosine. These pathways closely regulate local, pericellular, and extracellular concentrations of adenosine and are also required for intracellular salvage with synthesis of nucleotides derived from intermediates produced by these and other degradative pathways [9].
In the short term, P2RATP-mediated effects and the linked signaling pathways receptors trigger sudden pathophysiological processes, impacting acute inflammatory processes. Ongoing activation of P2R-signaling pathways impact later immune responses resulting in chronic inflammatory disease and exacerbating fibrosis [3, 12]. Immunosuppression in the short term is mediated by generation of adenosine; whereas, the fibrosis seen in chronic inflammation may be linked to unfettered adenosine generation. The challenge is to dissect out beneficial effects of adenosine, preclude resistance to these vs. preventing the deleterious signaling pathways of adenosine that cause chronic disease. Aberrant control of these purinergic activities could impact inflammation as well as fibrogenic reactions, as in chronic disease processes such as IBD.
Cellular immunomodulation and CD39
There is evidence that innate and adaptive immunity can be modulated by extracellular adenosine, released in response to tissue disturbing signals and extracellular nucleotides such as ATP or nicotinamide adenine nucleotide. Upon binding to multiple type 2 purinergic/pyrimidinergic (P2Y and P2X) receptors, ATP can have effects on cellular metabolism, migration, proliferation, and apoptosis. Transcriptional upregulation of CD39 and CD73 ectonucleotidases with increased immune cell infiltration at sites of injury results in conversion of a dominant P2-environment to one associated with decreased levels of nucleotides and a shift over to more predominant adenosinergic responses.
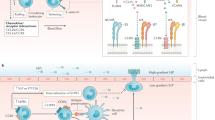
CD39 is highly expressed on vascular endothelial cells and T regulatory cells, where this ecto-enzyme contributes to suppressive functionality through the generation of adenosine [13] (see Fig. 1).CD39 is also expressed by subsets of memory cells with effector function [14] and by M2 anti-inflammatory monocytes [15].

Purinergic cytoprotection. This illustrates the role of the purinergic ecto-enzyme network in gastrointestinal inflammation. T regulatory cells (Treg) express the entire ecto-enzymatic machinery necessary to convert ATP/ADP into adenosine. Increased extracellular adenosine levels contribute to creating a favorable, homeostatic microenvironment by switching off T cell responses, producing anergy, inducing cytoprotection in an autocrine fashion, and promoting resolution of inflammation in the gut. Note also that hypoxia/HIF-1 alpha may modulate FOXP3 as well as CD39 and CD73 expression via Sp1. See text for details
Further, CD39 induction on prototypic, pathogenic Th17 cells imparts regulatory properties to these cells. These transitioned Th17 cells express CD39 and select functional features of Treg, including expression of FOXP3 at high levels and suppression of responder cell proliferation and pro-inflammatory cytokine production [16].
Despite acquiring regulatory features, these “suppressor-like” Th17 (supTh17) cells also retain certain effector Th17 cell properties, including IL-17 production and low levels of A2A adenosine receptor. Because of heightened expression of adenosine deaminase, these suppressive Th17 cells effectively hydrolyze the nucleoside adenosine into the somewhat more pro-inflammatory inosine derivative and hence appear to exhibit a dualistic phenotype. Of note is that inosine can activate A3 receptors to produce mast cell degranulation, which further regulates the chemotaxis of neutrophils and macrophages [17, 18].
Previous studies from Esplugues and colleagues have shown that pathogenic Th17 cells undergo “regulation” in the small intestine. Indeed, while still expressing IL-17A and IL-17F, these cells also become capable of producing IL-10 and of “suppressing” responders.
Our own evidence that these cells maintain classical Th17 features while acquiring typical Treg properties, inclusive of CD39 expression, indicates that this lymphocyte subset may exert dual function depending on the environment within which it operates. However, it should also be noted that extracellular nucleotides may serve as negative modulators of immunity, or as immunodepressants. Indeed, chronic, repetitive exposure to lower extracellular nucleotide levels tends to suppress immunity and inflammation [19].
Lastly, our studies have indicated that altered CD39 expression and changes in the nucleotide/nucleoside balance impact insulin-sensitivity, block mTOR activation (ATP-dependent) while boosting AMPK functions (adenosine-dependent process) [20]. Although CD39 appears to be associated with enhanced T cell survival, much as rapamycin and metformin are known to do so, additional effects of CD39 include protection from P2X7-mediated apoptosis and the provision of nucleosides that activate A2A receptors, obviating activation-induced cell death (AICD), promoting intracellular anabolic as well as purine salvage pathways.
CD39 and regulation by the aryl hydrocarbon receptor (AHR) and HIF-1 alpha.
The aryl hydrocarbon receptor (AHR) is ubiquitously expressed on a variety of cells and specific patterns of activation upregulate E-NTPDase-type ectonucleotidases on immunocytes, myeloid cells, endothelium, and parenchymal cells in vivo and in vitro [21].
The ligation of AHR by dioxins in the presence of TGF-beta induces Foxp3+ inducible Treg that can suppress responder T cell functions via CD39 [22].
Activation of AHR can promote generation of CD39+ regulatory-type T helper type 17 (Th17) cells as well as type 1 regulatory T cells or Tr1 cells, which express high levels of IL-10. Upregulation of CD39 is dependent upon ligation of the AhR on immune cells. AhR is additionally controlled by hypoxia and HIF-1alpha activity, as in the case of Tr1 cells [23]. Furthermore, hypoxic conditions per se might activate the purinergic signaling by upregulating expression of CD39, as shown in the cardiac ischemia model in which transcription of CD39 was controlled by Sp-1 [24], and through HIF-1 alpha induction of CD73, which ultimately converts AMP into adenosine [25].
Recent elegant work has shown that adenosinergic A2BR-mediated responses, which are anti inflammatory and cytoprotective involve further interactions of HIF-1 alpha and the circadian rhythm protein PER2 [26, 27].
Other groups have also shown that the alternative adenosinergic A2AR pathway, together with TNF, have the capacity to regulate immune cell intrinsic “clocks” implicating involvement of circadian rhythms in clinicopathologic changes in prototypic rheumatological disease, as with morning stiffness [28].
Furthermore, there are important seasonal and latitudinal patterns linking IBD exacerbations to light exposure and circadian rhythms. There are substantive differences in the expression of circadian-type genes between normal and diseased intestinal mucosa in IBD. Such deregulated genes, e.g., PER1 and PER3 could have pathophysiological relevance and may suggest novel therapeutic approaches distinct from the facile use of melatonin in such disease settings [29, 30].
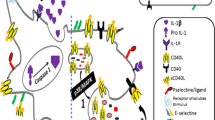
In the past few decades, bilirubin, a byproduct of heme catalysis, and a pigment also clearly altered by light exposure, has been shown to have a major salutary role as a potent antioxidant. Most recently, the molecule has been found to possess immunomodulatory properties that rival the redox capacity. These possibly explain its ability to suppress inflammation as in IBD, where development of jaundice has been shown to suppress colonic inflammation. We have recently demonstrated unconjugated bilirubin to serve as a potent immunomodulator and have shown that the molecular basis for its immunosuppressive effects is dependent upon the upregulation of CD39 by interactions with AHR [31] (Longhi et al., 2017, in press and Fig. 2).

Bilirubin metabolism and mechanism of action. a Unconjugated bilirubin (UCB) is an end product of heme catalysis and has known immunosuppressant properties. Heme-oxygenase-1 (HO-1) catalyzes heme degradation to biliverdin (BV), which is then converted to UCB by biliverdin reductase (BVR). As it is insoluble in water, UCB binds to albumin in the circulation. In the hepatocytes, UCB is conjugated with glucuronic acid by UDP glucuronosyltransferase 1 (UGT1A1) and is then excreted into the bile. Then, after being metabolized to urobilinogen and de-conjugated by the bacterial flora, it is excreted in the urine and feces. Proportions of bilirubin are, however, re-absorbed and undergo enterohepatic circulation. b UCB serves as an endogenous ligand for the aryl hydrocarbon receptor (AhR), a mediator of toxin responses and adaptive immunity. AhR engagement by UCB results in upregulation of CD39, the ectoenzyme initiating an ATP/ADP hydrolysis cascade that culminates with the generation of adenosine. Release of adenosine in the extracellular milieu leads to a decrease in cell proliferation, reduction in Th1 and Th2 development, attenuation of Th17 pathogenic potential and Treg induction
Hence, limitations in the levels of CD39 and/or dysfunction of AHR abrogate the protective effects of unconjugated bilirubin in experimental colitis and in IBD patients. Therefore, in DSS-induced colitis, the administration of unconjugated bilirubin systemically resulted in amelioration of disease activity particularly during recovery, improved histology scores, and increased IL-10 production by colonic intraepithelial CD4 cells. These salutary effects were abrogated in Entpd1 −/− and AhR d mice, in which AhR is dysfunctional [32]. Notably, unconjugated bilirubin fails to boost CD39, FOXP3, and immunosuppressive function in IBD derived Th17 cells, which additionally display defective AhR bioactivity (Longhi et al., 2017, in press). The concept that beneficial effects of AhR ligation are mediated via CD39 induction has been also supported by recent work by Goettel et al. in the context of experimental colitis [33]. Administration of ITE, another AhR endogenous ligand, prevents T cell mediated tissue damage in humanized mice. This effect is associated with an increased proportion of CD39+ CD4 lymphocytes sequestrated in the colonic wall compartment.
Overall, these findings suggest that boosting AhR signaling upon exposure to natural/endogenous ligands or otherwise enhancing CD39 ectoenzymatic properties might represent attractive strategies to correct effector Th17 dysfunction in IBD.
Purinergic/adenosinergic responses in IBD and experimental colitis
Given the immunosuppressive properties of adenosine, modulation of purinergic signaling has been evaluated in the context of IBD and experimental models of colitis, to curb inflammation. The suggestion that adenosine might be a key immune mediator controlling inflammation in IBD, was indicated first by mechanistic studies of sulfasalazine, and methotrexate. Both drugs have been shown to be operational, at least in part, through adenosine-dependent mechanisms.
Furthermore, administration of ATL313, and other direct agonists of the A2A adenosine receptor can attenuate colitis in mice with adoptive transfer of CD45RBhigh cells, and also suppress the production of pro-inflammatory cytokines (IL-2, IFNγ, and TNFα) but not the anti-inflammatory (IL-10 and TGFβ) cytokines [34].
Also, albeit controversial, direct activation of the A2B adenosine receptor can also boost IL-10 release by intestinal epithelial cells, which is linked to amelioration of DSS colitis [35].
Alterations in the generation of adenosine, such as those associated with genetic deletions of CD39 or CD73, result in more severe course of experimental colitis in mutant mice. CD39 deletion in mice results in exacerbation of DSS-induced and other experimental colitis, whereas transgenic over expression appears to ameliorate disease (unpublished observations of Maria Serena Longhi and Simon C. Robson; 2017) [36].
Expression of CD39 on endothelial or immune cells allows for homeostatic integration of immunity resulting in control of hemostatic and immunobiological reactions, which appear to be disrupted in IBD. Single nucleotide polymorphisms adjacent to the CD39 promoter region have been associated with low levels of CD39 mRNA that confer susceptibility to Crohn’s disease [37]. The associated decreases in CD39 expression levels and consequently lower adenosine generation are likely to be linked to the impairment of CD4+CD25high regulatory T cells in this disease process.
Since this publication in 2009, it has been increasingly recognized that highly heritable traits that dictate adaptive immune responsiveness include the different levels of expression of CD39 on Treg, as noted in large population analyses and twin studies [38, 39]. In contrast, CD73 expression by Treg, in humans seems to beat least in part a consequence of the environmental exposure to, e.g., pathogens, diet, or microbiome elements, shared in a household during maturation [40]. This recent work is suggestive of adaptive immune traits being more impacted by genetics, while in contrast innate immune traits are dictated more by environmental factors. Irrespectively, intrinsic or acquired defects resulting in lower levels of CD39 might lead to T cell autoreactivity because of the lack of immune-modulatory adenosine.
CD39 and exosomes in IBD
Microparticles (exosomes or extracellular vesicles; MPs) are released from cells into the blood or at sites of inflammation in the intestinal tract. These MPs can be isolated from the blood, tissue fluids, or fecal samples. Depending on the cellular origin, intestinal MP express cell surface markers and contain protein/RNA with pro- or anti-inflammatory properties. In addition, these MP constitute a mode of communication through which intestinal cells may influence the luminal microbiome. We have recently reported that extracellular vesicles, derived from the colonic luminal fluid of IBD patients, display pro-inflammatory properties as these MP contain high mRNA and protein levels of IL-6, IL-8, IL-10, and TNF-α, and promote macrophage migration [41]. We have also shown that CD39 associates with circulating plasma-derived MP and may directly or indirectly confer functional properties on cells.
Indeed, surrounding cells can absorb MPs shed from sites of inflammation. We have demonstrated the presence of E-NTPDase activity in circulating MP isolated in human plasma [42]. Most importantly, the mRNA within MPs can be taken up by these cells and further translated. We have recently shown that properties of MPs obtained from patients with IBD provide a mechanism for some of the regional variations in inflammation, as noted within the diseased intestinal tract. We have also shown modulatory roles for CD39 within MPs in the exchange of regulatory signals between leucocytes and vascular cells [43].
Particular interest exists in programming cell lines to produce MPs with phenotypic characteristics, such as IL-10 induced anti-inflammatory CD39 expressing MPs from dendritic cells. Our own work proposes that intrinsic properties of MPs suggest a role as novel biomarkers of inflammatory pathways, or even as therapeutic vehicles for local delivery of anti-inflammatory compounds and purinergic modulators in IBD, as we have previously determined in liposomal reconstitution of CD39 [44].
Microbiome elements—fecal transplants to correct dysbiosis and aberrant purinergic signaling
The commensal flora is recognized to play an important role in the control of the immune response in the context of IBD and experimental colitis [45]. Different molecules mediate effects of the microbiome on the immune response, including long-chain fatty acids and tryptophan derivatives that also trigger AHR. Extracellular ATP released by commensal bacteria has been shown to activate purinergic inflammatory signaling to promote the differentiation of intestinal Th17 cells [45, 46].
The NLRP3 inflammasome catalyzes the production of active IL-1 and IL-18 in response to diverse endogenous or exogenous danger signals. One such signal is ATP, which activates the NLRP3 inflammasome in DCs through a mechanism mediated by P2X7R [47].
Curiously, the derivative adenosine alone can also activate the A2AR/CREB/HIF-1 alpha pathway, which is also required for sustained production of IL-1 after the initial inflammasome activation [48].
The importance of the NLRP3 inflammasome in the T cell response can be highlighted by the decrease in Th1 and Th17 responses observed in NLPR3-deficient mice.CD39 also appears to impact the NLRP3-associated control of T cell immunity, as recently shown by collaborative studies of tolerogenic DCs induced with IL-27 [23].
Conversely, several pathogen bacteria express ectonucleotidases that may modulate the immune response through the effects on purinergic signaling. Several bacteria also release factors that induce CD39 expression on immune cells [49].
Taken together, these findings suggest that extracellular ATP and derivatives produced by microorganisms and by host cells in response to microbial molecules, such as TLR agonists, might play an important role in dictating the relationship between the host and the commensal flora.
This topic addressing the role of ectonucleotidases on host-pathogen interactions has been previously reviewed in Samson et al. [50].
It is generally accepted that the microbiome in IBD, in particular in Crohn’s disease, is characterized by reduced diversity, particularly of firmicutes and bacteroidetes. We recently conducted an open label study transferring the intestinal microbiota from healthy individuals into patients with IBD in order to see if this could correct dysbiosis and reverse mucosal inflammation. Those patients who had clinical responses demonstrated significant shifts in fecal microbial composition toward the respective donor’s profile and we also noted an increase in Treg in this subset [51]. Further work is ongoing to dissect out the nucleotide and purine metabolome in these patients post fecal transplant and microbiome transfer.
Recent work has shown potential relevance of the mycobiome in colitis [52]. Saccharomyces cerevisiae has been recently shown to both exacerbate experimental colitis and increase gut barrier permeability in mice. Yeast colonization was found to enhance host purine metabolism in germ free animals, leading to an increase in uric acid production. Importantly, treatment with uric acid alone worsened disease and increased gut permeability. This interesting area of research is somewhat controversial given that Saccharomyces cerevisiae can be also considered as a probiotic as it may limit adherent-invasive Escherichia coli (AIEC) in CEACAM6-expressing mice [53].
Other NTPDases expressed in the gastrointestinal tract and putative roles in IBD
The ecto-ATPase activity in the gut predominantly resides in blood vessels, immune cells, visceral smooth muscle, and the enteric nervous system [54]. While CD39 is the major E-NTPDase expressed by the endothelium and immune cells, we have noted that NTPDase2 and NTPDase3 are responsible, in large part, for the ATPase activity in the muscle layers and the nervous system. In addition, Kusu and coworkers have reported the expression of NTPDase7 by the epithelial cells of the murine small intestine [55].
NTPDase2 and NTPDase3 are two cell membranes located ecto-enzymes in the E-NTPDases family that share significant structural homology and functional similarity to CD39 [9, 56]. The enzymatic activity of NTPDase3 is similar to that of CD39; whereas, NTPDase2 has significantly weaker ADPase activity [57]. NTPDase7 is also known as LALP1 and is conventionally thought to be an endo-apyrase. Whether it is also expressed on the plasma membrane in humans is yet to be fully confirmed [58].
Both NTPDase2 and NTPDase3 are known be expressed in nerve tissues [59, 60]. In the gut, the expression of NTPDase2 has been further noted on glial cells, while NTPDase3 localizes to both glia and neurons [54, 61, 62]. In both humans and mice, NTPDase3 antibodies positively stain nerve fibers penetrating the smooth muscle layers, whereas the expression of NTPDase2 in these areas is less prominent (Feldbrügge et al., 2017, in press and see Fig. 3).

Expression of select E-NTPDases in the digestive tract. a A cross-sectional diagram of the digestive tract highlighting the three key layers. The expression of E-NTPDases in the digestive tract in relation to other cellular structures are shown in the mucosa (b), lamina propria and submucosa (c), and the muscularis (d). See text for details
We have further shown that the genetic deletion of Entpd2 results in exacerbated DSS-induced experimental colitis in these mutant mice that do not express NTPDase2. This outcome is associated with an increase in the proportion of proinflammatory macrophages in the lamina propria. Similarly, mice globally null for Entpd3, which lack all NTPDase3 expression, have more pronounced anemia compared to wild type in this same DSS-induced colitis model. We have also compared the ADPase activity in the plasma of patients with Crohn’s disease and controls, and found that Crohn’s patients have lower circulating ADPase activity. This ADPase activity is in part contributed by non-CD39 NTPDases, as suggested by sensitivity to non-CD39 NTPDase inhibitors.
The emerging roles of NTPDase2 and NTPDase3 in IBD further support the innovative concept of neuroimmune interaction [63, 64].This interaction may function at multiple levels. Hence, eATP transmits signals both among neurons in myenteric and submucosal ganglia via P2X2 and P2Y1 receptors and between nerve and smooth muscle cells via P2Y1 receptors, exerting an inhibitory effect on the muscularis [65,66,67].eATP can also activate ionotropic P2X7 receptors in macrophages, dendritic cells, and neutrophils, which in turn induces NLRP3 inflammasome assembly and the release of interleukin 1β and 18 [68]. Furthermore, extracellular ATP has been shown to mediate the communication between neurons, glia, and contribute to the maintenance of intestinal homeostasis and mucosal barrier [69, 70]. Glial cells can perpetuate the release of pro-inflammatory ATP in the setting of intestinal inflammation via the activation of P2Y1 receptors, which in turn mediates neuronal cell death via P2X7 receptors [71].A recent study by Gabanyi and coworkers also suggested that enteric neurons in the muscularis externa can mediate the polarization of tissue resident macrophages toward a tissue-protective phenotype [64]. Purinergic signaling may be a crucial mechanism modulating this interaction.
Interesting work from Kusu et al. with respect to NTPDase7, suggested that there is yet another mechanism that gut purinergic signaling can modulate host immunity [55]. The team at Osaka University observed that NTPDase7 expressed on the intestinal epithelial cells modulates the ATP content in the intestinal lumen per se. Mice null for Entpd7 and deficient in NTPDase7 cannot scavenge luminal ATP produced by commensal microbiota. As a consequence, this enhances the development of proinflammatory Th17 cells, leading to a more severe phenotype in models of experimental autoimmune encephalomyelitis. Whether such immune dysregulation is relevant to human IBD remains to be determined.
Conclusions
This manuscript has summarized the role of aberrant purinergic signaling in IBD and gastrointestinal autoimmunity and has suggested how pharmacological modulation of purinergic responses, adenosine generation, AhR and HIF-1 alpha signaling (among others) could be exploited to treat these important conditions. The purinergic signaling pathways could be targeted for IBD treatment by the use of soluble ectonucleotidases, adenosine receptor agonists, or HIF activators, inter alia, as previously addressed in two important reviews [72, 73].
In this review, we have highlighted how targeting CD39 (and related ectonucleotidases) to modulate the purinergic-adenosinergic axis could have major impacts on extent of the inflammatory infiltrate in IBD (via adenosine receptor agonists and/or boosting CD39 or related ectonucleotidases). We propose that augmentation of CD39 and related ectonucleotidase bioactivity, possibly in MPs, might also control aberrant autoimmune reactions (via pharmacological use of adenosine receptor agonists and/or regulated CD39 expression).
These purinergic mechanisms involved in both the generation of adenosine and scavenging of extracellular nucleotides have major impacts on the downstream signaling pathways critical to both thrombo regulation and most importantly to the progression of inflammation and are hence of considerable and increasing therapeutic interest.
References
Eltzschig HK, Sitkovsky MV, Robson SC (2012) Purinergic signaling during inflammation. N Engl J Med 367:2322–2333
Longhi MS, Robson SC, Bernstein SH, Serra S, Deaglio S (2013) Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states. J Mol Med (Berl) 91:165–172
Takenaka MC, Robson S, Quintana FJ (2016) Regulation of the T cell response by CD39. Trends Immunol 37:427–439
Allard B, Longhi MS, Robson SC, Stagg J (2017) The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev 276:121–144
Luthje J (1989) Origin, metabolism and function of extracellular adenine nucleotides in the blood [published erratum appears in Klin Wochenschr 1989 May 15;67(10):558]. [review]. Klin Wochenschr 67:317–327
Burnstock G, Knight G (2004) Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 240:31–304
Burnstock G (2002) Purinergic signaling and vascular cell proliferation and death. Arterioscler Thromb Vasc Biol 22:364–373
Resta R, Yamashita Y, Thompson LF (1998) Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev 161:95–109
Robson SC, Sevigny J, Zimmermann H (2006) The E-NTPDase family of ecto-nucleotidases: structure function relationshhips and pathophysiological significance. Purinergic Signal 2:409–430
Beldi G, Enjyoji K, Wu Y, Miller L, Banz Y, Sun X, Robson SC (2008) The role of purinergic signaling in the liver and in transplantation: effects of extracellular nucleotides on hepatic graft vascular injury, rejection and metabolism. Front Biosci 13:2588–2603
Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, Serra S, Malavasi F (2013) A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2:e26246
Cekic C, Linden J (2016) Purinergic regulation of the immune system. Nat Rev Immunol 16:177–192
Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M et al (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204:1257–1265
Dwyer KM, Hanidziar D, Putheti P, Hill PA, Pommey S, McRae JL, Winterhalter A, Doherty G, Deaglio S, Koulmanda M et al (2010) Expression of CD39 by human peripheral blood CD4+ CD25+ T cells denotes a regulatory memory phenotype. Am J Transplant 10:2410–2420
Cohen HB, Briggs KT, Marino JP, Ravid K, Robson SC, Mosser DM (2013) TLR stimulation initiates a CD39-based autoregulatory mechanism that limits macrophage inflammatory responses. Blood 122:1935–1945
Longhi MS, Moss A, Bai A, Wu Y, Huang H, Cheifetz A, Quintana FJ, Robson SC (2014) Characterization of human CD39+ Th17 cells with suppressor activity and modulation in inflammatory bowel disease. PLoS One 9:e87956
Jin X, Shepherd RK, Duling BR, Linden J (1997) Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest 100:2849–2857
Joos G, Jakim J, Kiss B, Szamosi R, Papp T, Felszeghy S, Saghy T, Nagy G, Szondy Z (2017) Involvement of adenosine A3 receptors in the chemotactic navigation of macrophages towards apoptotic cells. Immunol Lett 183:62–72
Di Virgilio F, Boeynaems JM, Robson SC (2009) Extracellular nucleotides as negative modulators of immunity. Curr Opin Pharmacol 9:507–513
Sun X, Han L, Seth P, Bian S, Li L, Csizmadia E, Junger WG, Schmelzle M, Usheva A, Tapper EB et al (2013) Disordered purinergic signaling and abnormal cellular metabolism are associated with development of liver cancer in Cd39/ENTPD1 null mice. Hepatology 57:205–216
Gao L, Dong L, Whitlock JP Jr (1998) A novel response to dioxin. Induction of ecto-ATPase gene expression. J Biol Chem 273:15358–15365
Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, Kozoriz D, Weiner HL, Quintana FJ (2010) Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol 11:846–853
Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, Wu Y, Mayo L, Ben-Hamo R, Efroni S et al (2013) IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol 14:1054–1063
Hart ML, Gorzolla IC, Schittenhelm J, Robson SC, Eltzschig HK (2010) SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J Immunol 184:4017–4024
Hart ML, Grenz A, Gorzolla IC, Schittenhelm J, Dalton JH, Eltzschig HK (2011) Hypoxia-inducible factor-1alpha-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J Immunol 186:4367–4374
Eltzschig HK, Bonney SK, Eckle T (2013) Attenuating myocardial ischemia by targeting A2B adenosine receptors. Trends Mol Med 19:345–354
Eckle T, Hartmann K, Bonney S, Reithel S, Mittelbronn M, Walker LA, Lowes BD, Han J, Borchers CH, Buttrick PM et al (2012) Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat Med 18:774–782
Perez-Aso M, Feig JL, Mediero A, Cronstein BN (2013) Adenosine A2A receptor and TNF-alpha regulate the circadian machinery of the human monocytic THP-1 cells. Inflammation 36:152–162
Stein AC, Gaetano JN, Jacobs J, Kunnavakkam R, Bissonnette M, Pekow J (2016) Northern latitude but not season is associated with increased rates of hospitalizations related to inflammatory bowel disease: results of a multi-year analysis of a national cohort. PLoS One 11:e0161523
Palmieri O, Mazzoccoli G, Bossa F, Maglietta R, Palumbo O, Ancona N, Corritore G, Latiano T, Martino G, Rubino R et al (2015) Systematic analysis of circadian genes using genome-wide cDNA microarrays in the inflammatory bowel disease transcriptome. Chronobiol Int 32:903–916
Jangi S, Otterbein L, Robson S (2013) The molecular basis for the immunomodulatory activities of unconjugated bilirubin. Int J Biochem Cell Biol 45:2843–2851
Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL (2008) Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453:65–71
Goettel JA, Gandhi R, Kenison JE, Yeste A, Murugaiyan G, Sambanthamoorthy S, Griffith AE, Patel B, Shouval DS, Weiner HL et al (2016) AHR activation is protective against colitis driven by T cells in humanized mice. Cell Rep 17:1318–1329
Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, Ernst PB (2006) Cutting edge: critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol 177:2765–2769
Frick JS, MacManus CF, Scully M, Glover LE, Eltzschig HK, Colgan SP (2009) Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol 182:4957–4964
Doherty GA, Bai A, Hanidziar D, Longhi MS, Lawlor GO, Putheti P, Csizmadia E, Nowak M, Cheifetz AS, Moss AC et al (2012) CD73 is a phenotypic marker of effector memory Th17 cells in inflammatory bowel disease. Eur J Immunol 42:3062–3072
Friedman DJ, Kunzli BM, Yi AR, Sevigny J, Berberat PO, Enjyoji K, Csizmadia E, Friess H, Robson SC (2009) From the cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci U S A 106:16788–16793
Orru V, Steri M, Sole G, Sidore C, Virdis F, Dei M, Lai S, Zoledziewska M, Busonero F, Mulas A et al (2013) Genetic variants regulating immune cell levels in health and disease. Cell 155:242–256
Roederer M, Quaye L, Mangino M, Beddall MH, Mahnke Y, Chattopadhyay P, Tosi I, Napolitano L, Terranova Barberio M, Menni C et al (2015) The genetic architecture of the human immune system: a bioresource for autoimmunity and disease pathogenesis. Cell 161:387–403
Mangino M, Roederer M, Beddall MH, Nestle FO, Spector TD (2017) Innate and adaptive immune traits are differentially affected by genetic and environmental factors. Nat Commun 8:13850
Mitsuhashi S, Feldbrugge L, Csizmadia E, Mitsuhashi M, Robson SC, Moss AC (2016) Luminal extracellular vesicles (EVs) in inflammatory bowel disease (IBD) exhibit proinflammatory effects on epithelial cells and macrophages. Inflamm Bowel Dis 22:1587–1595
Jiang ZG, Wu Y, Csizmadia E, Feldbrugge L, Enjyoji K, Tigges J, Toxavidis V, Stephan H, Muller CE, McKnight CJ et al (2014) Characterization of circulating microparticle-associated CD39 family ecto-nucleotidases in human plasma. Purinergic Signal. doi:10.1007/s11302-014-9423-6
Banz Y, Beldi G, Wu Y, Atkinson B, Usheva A, Robson SC (2008) CD39 is incorporated into plasma microparticles where it maintains functional properties and impacts endothelial activation. Br J Haematol 142:627–637
Haller CA, Cui W, Wen J, Robson SC, Chaikof EL (2006) Reconstitution of CD39 in liposomes amplifies nucleoside triphosphate diphosphohydrolase activity and restores thromboregulatory properties. J Vasc Surg 43:816–823
Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T et al (2015) Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell 163:367–380
Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K et al (2008) ATP drives lamina propria T(H)17 cell differentiation. Nature 455:808–812
Baron L, Gombault A, Fanny M, Villeret B, Savigny F, Guillou N, Panek C, Le Bert M, Lagente V, Rassendren F et al (2015) The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis 6:e1629
Ouyang X, Ghani A, Malik A, Wilder T, Colegio OR, Flavell RA, Cronstein BN, Mehal WZ (2013) Adenosine is required for sustained inflammasome activation via the A(2)A receptor and the HIF-1alpha pathway. Nat Commun 4:2909
Wang Y, Telesford KM, Ochoa-Reparaz J, Haque-Begum S, Christy M, Kasper EJ, Wang L, Wu Y, Robson SC, Kasper DL et al (2014) An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat Commun 5:4432
Sansom FM, Robson SC, Hartland EL (2008) Possible effects of microbial ecto-nucleoside triphosphate diphosphohydrolases on host-pathogen interactions. Microbiol Mol Biol Rev 72:765–781 table of contents
Vaughn BP, Vatanen T, Allegretti JR, Bai A, Xavier RJ, Korzenik J, Gevers D, Ting A, Robson SC, Moss AC (2016) Increased intestinal microbial diversity following fecal microbiota transplant for active Crohn’s disease. Inflamm Bowel Dis 22:2182–2190
Chiaro TR, Soto R, Zac Stephens W, Kubinak JL, Petersen C, Gogokhia L, Bell R, Delgado JC, Cox J, Voth W et al (2017) A member of the gut mycobiota modulates host purine metabolism exacerbating colitis in mice. Sci Transl Med 9
Sivignon A, de Vallee A, Barnich N, Denizot J, Darcha C, Pignede G, Vandekerckove P, Darfeuille-Michaud A (2015) Saccharomyces cerevisiae CNCM I-3856 prevents colitis induced by AIEC bacteria in the transgenic mouse model mimicking Crohn’s disease. Inflamm Bowel Dis 21:276–286
Lavoie EG, Gulbransen BD, Martin-Satue M, Aliagas E, Sharkey KA, Sevigny J (2011) Ectonucleotidases in the digestive system: focus on NTPDase3 localization. Am J Physiol Gastrointest Liver Physiol 300:G608–G620
Kusu T, Kayama H, Kinoshita M, Jeon SG, Ueda Y, Goto Y, Okumura R, Saiga H, Kurakawa T, Ikeda K et al (2013) Ecto-nucleoside triphosphate diphosphohydrolase 7 controls Th17 cell responses through regulation of luminal ATP in the small intestine. J Immunol 190:774–783
Zimmermann H, Zebisch M, Strater N (2012) Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 8:437–502
Heine P, Braun N, Heilbronn A, Zimmermann H (1999) Functional characterization of rat ecto-ATPase and ecto-ATP diphosphohydrolase after heterologous expression in CHO cells. Eur J Biochem 262:102–107
Shi JD, Kukar T, Wang CY, Li QZ, Cruz PE, Davoodi-Semiromi A, Yang P, Gu Y, Lian W, Wu DH et al (2001) Molecular cloning and characterization of a novel mammalian endo-apyrase (LALP1). J Biol Chem 276:17474–17478
Wink MR, Braganhol E, Tamajusuku AS, Lenz G, Zerbini LF, Libermann TA, Sevigny J, Battastini AM, Robson SC (2006) Nucleoside triphosphate diphosphohydrolase-2 (NTPDase2/CD39L1) is the dominant ectonucleotidase expressed by rat astrocytes. Neuroscience 138:421–432
Belcher SM, Zsarnovszky A, Crawford PA, Hemani H, Spurling L, Kirley TL (2006) Immunolocalization of ecto-nucleoside triphosphate diphosphohydrolase 3 in rat brain: implications for modulation of multiple homeostatic systems including feeding and sleep-wake behaviors. Neuroscience 137:1331–1346
Cardoso AM, Schetinger MR, Correia-de-Sa P, Sevigny J (2015) Impact of ectonucleotidases in autonomic nervous functions. Auton Neurosci 191:25–38
Braun N, Sevigny J, Robson SC, Hammer K, Hanani M, Zimmermann H (2004) Association of the ecto-ATPase NTPDase2 with glial cells of the peripheral nervous system. Glia 45:124–132
Di Giovangiulio M, Verheijden S, Bosmans G, Stakenborg N, Boeckxstaens GE, Matteoli G (2015) The neuromodulation of the intestinal immune system and its relevance in inflammatory bowel disease. Front Immunol 6:590
Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D (2016) Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell 164:378–391
Gallego D, Gil V, Martinez-Cutillas M, Mane N, Martin MT, Jimenez M (2012) Purinergic neuromuscular transmission is absent in the colon of P2Y(1) knocked out mice. J Physiol 590:1943–1956
Galligan JJ (2002) Ligand-gated ion channels in the enteric nervous system. Neurogastroenterol Motil 14:611–623
Gulbransen BD, Bashashati M, Hirota SA, Gui X, Roberts JA, MacDonald JA, Muruve DA, McKay DM, Beck PL, Mawe GM et al (2012) Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med 18:600–604
Gombault A, Baron L, Couillin I (2012) ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol 3:414
Fields RD, Stevens B (2000) ATP: an extracellular signaling molecule between neurons and glia. Trends Neurosci 23:625–633
Ruhl A (2005) Glial cells in the gut. Neurogastroenterol Motil 17:777–790
Brown IA, McClain JL, Watson RE, Patel BA, Gulbransen BD (2016) Enteric glia mediate neuron death in colitis through purinergic pathways that require connexin-43 and nitric oxide. Cell Mol Gastroenterol Hepatol 2:77–91
Eltzschig HK, Bratton DL, Colgan SP (2014) Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov 13:852–869
Ochoa-Cortes F, Linan-Rico A, Jacobson KA, Christofi FL (2014) Potential for developing purinergic drugs for gastrointestinal diseases. Inflamm Bowel Dis 20:1259–1287
Acknowledgements
The work summarized in this review article was supported by the National Institute of Health grants to SCR and MSL; R01 HL094400; R01 DK108894; P01HL107152, and P01 HL087203 as well as the generosity of the family of Jane O. Siegel; as well as a Clinical Research Award from the American Gastroenterology Association and the Alan Holfman Clinical and Translational Research Award from the American Association for the Study of Liver Disease to ZGJ. We thank Eliza Robson for the graphic design for figures.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Rights and permissions
About this article

Cite this article
Longhi, M.S., Moss, A., Jiang, Z.G. et al. Purinergic signaling during intestinal inflammation. J Mol Med 95, 915–925 (2017). https://doi.org/10.1007/s00109-017-1545-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-017-1545-1