
Abstract
Ankyrin G (ANK3) is a member of the Ankyrin family, which functions to provide cellular stability by anchoring the cytoskeleton to the plasma membrane. Deregulation of ANK3 expression has been observed in multiple human cancers but its mechanism remains unknown. ANK3 expression in relation to disease progression and patients’ outcome was investigated in two cohorts of prostate cancer (PCA). Mechanistic studies were carried out in vitro and in vivo using several PCA cell lines and the avian embryo model. Silencing ANK3 resulted in significant reduction of cell proliferation through an AR-independent mechanism. Decreased ANK3 expression delayed S phase to G2/M cell cycle transition and reduced the expression of cyclins A and B. However, cells with knocked-down ANK3 exhibited significant increase in cell invasion through an AR-dependent mechanism. Furthermore, we found that ANK3 is a regulator of AR protein stability. ANK3 knockdown also promoted cancer cell invasion and extravasations in vivo using the avian embryo model (p < 0.01). In human samples, ANK3 expression was dramatically upregulated in high grade intraepithelial neoplasia (HGPIN) and localized PCA (p < 0.0001). However, it was downregulated castration resistant stage (p < 0.0001) and showed inverse relation to Gleason score (p < 0.0001). In addition, increased expression of ANK3 in cancer tissues was correlated with better cancer-specific survival of PCA patients (p = 0.012).
Key message
-
Silencing ANK3 results in significant reduction of cell proliferation through an AR-independent mechanism.
-
ANK3 knockdown results in significant increase in cell invasion through an AR-dependent mechanism.
-
ANK3 is a regulator of AR protein stability.
-
ANK3 knockdown also promotes cancer cell invasion and extravasation in vivo using the avian embryo model.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prostate cancer (PCA) is the most common cancer and the second leading cause of cancer deaths in men in North American [1]. Although many patients are diagnosed early in disease stage, significant number of those patients still experience disease progression, metastasis, and development of castrate resistant disease (CRPC). Even though substantial advances have been made in prostate cancer research in recent years, tumor metastasis and recurrence are considered the major reasons for poor clinical outcome and cancer deaths [2]. Therefore, studying the mechanisms of tumor invasion and metastasis will provide further insights into the development and progression of prostate cancer.
Ankyrins are family of adapter proteins that function to anchor the cytoskeleton to plasma membrane by binding cytoskeletal spectrin/fordin and integral membrane proteins, such as glycophorin and anion exchange proteins [3, 4]. They function in regulating a variety of biological activities, such as cell motility, activation, proliferation, and contact, in addition to the maintenance of specialized membrane domains [5, 6]. The newest ankyrin family member, Ankyrin G, encoded by ANK3, is broadly expressed in epithelial tissues, kidney, skeletal and cardiac muscles, and brain [7]. Similar to other ankyrins, ANK3 has three functional domains including an N-terminal membrane domain [containing 24 ankyrin repeats, responsible for binding integral membrane proteins (adhesive or ion channel)], a central spectrin/fodrin-binding domain that links ankyrin to actin-based cytoskeleton via spectrin isoform, and a C-terminal regulatory domain [8]. Based on cell type, ANK3 is expressed as multiple isoforms of 215, 200, 170, 120, and 105 kDa as a result of alternate splicing and two translational start sites [9]. The larger ANK3 isoforms are localized to the plasma membrane; however, the 120- and 105-kDa ANK3 isoforms have been shown to localize to the late endosomes and lysosomes in mouse macrophage cells [9, 10]. Since ANK3 was originally identified in the axon initial segment and the nodes of Ranvier [8, 11, 12], it was broadly studied in and identified as a risk gene for multiple neuropsychiatric disorders, such as bipolar disorder, schizophrenia, and autism spectrum disorder [13–15]. Limited data are available about the characterization of ANK3 role in human cancer. To our knowledge, we are aware of only one previous study which reported that downregulation of the ANK3 correlates with poor prognosis in diverse human tumors such as breast, prostate, lung, and ovarian cancers [16]. Consistent with this finding, our recent paper pinpointed that ANK3 expression is significantly associated with prostate cancer disease progression [17]. These data suggested a potential role of ANK3 in the pathogenesis of human cancers. However, how ANK3 contributes to the development of cancer remains to be fully elucidated.
In the present study, we demonstrated that ANK3 has different roles in cell proliferation and invasion in vitro, likely reflective of the molecular background of such cells. Knockdown of ANK3 in prostate cancer cell lines resulted in growth repression. However, at the same time, these cells exhibited increased invasion potential. Moreover, ANK3 expression is significantly associated with prostate cancer progression and has patients’ prognostic implications.
Materials and methods
Cell lines
The human prostate cancer cell lines, LNCaP, VCaP, and PC-3 cells, were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were characterized by ATCC using short tandem repeat (STR) DNA analysis and were used for experiments within 6 months. Cells were also tested with the DDC Medical through STR profiling. LNCaP and PC-3 cells were grown in the RPMI 1640 medium supplemented with 10 % FBS (Invitrogen, Burlington, ONT, Canada), while VCaP cells were maintained in DMEM medium (Invitrogen) with 10 % FBS.
Western blot analysis
Total cellular proteins were prepared by lysing cells in RIPA buffer (Sigma-Aldrich, Oakville, ON, Canada) supplemented with protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN), followed by quantification using BCA Protein Assay Reagent Kit (Thermo Scientific, Pittsburgh, PA). Samples were resolved by SDS-PAGE, transferred to PVDF membranes (Bio-Rad Laboratories, Mississauga, Ontario), and immunoblotted with primary antibodies: anti-ANK3 antibody, AR antibody, ERG antibody, Cyclin A antibody, Cyclin B1 antibody, Cyclin E, Cyclin D1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), Caspase3, and PARP (Cell Signaling Technology Inc., Danvers, MA). Densitometry analysis was performed by Quantity One Software (Bio-Rad).
Transfections and RNA silencing
For siRNA knockdown of ANK3, cells were transiently transfected with On-Target Plus SMARTpool ANK3 siRNA (Dharmacon, Pittsburgh, PA, USA) [18]. Transfections were performed with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Cells were harvested 48 h later for western blot and real-time qPCR.
Proliferation assay
For proliferation assay, cells transfected with individual and pooled ANK3 siRNAs or control siRNA were seeded in 96-well plate. The reagent of 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2Htetrazolium (MTS, Promega, Madison, WI) was added at the indicated days. Cell proliferation rates were measured according to the manufacturer’s protocol.
Co-immunoprecipitation (Co-IP) assay
ANK3 antibody (sc-28561, Santa Cruz) was used to pull down and probe for ANK3 in LNCaP cells. The co-immunoprecipitate was then eluted and analyzed by the western blot assay along with the 5 % input controls. AR antibody (sc-7305,Santa Cruz), Ubiquitin antibody (20458, DAKO), and HSP-90 antibody ( SPA-830, Enzo) were analyzed by western blot. Rabbit IgG (sc-2027, Santa Cruz) was used as a negative control for IP.
Fluorescence-activated cell sorting (FACS)
After 48 h of siRNA transfections, cell cycle was analyzed by FACS after 40 μg/ml propidium iodide staining according to the standard protocol, http://www.meduniwien.ac.at/user/johannes.schmid/PIstaining3.htm. Relative DNA contents from at least 10,000 cells were analyzed by FACSCantoII flow cytometer and BD FACSDiva software version 5.0.3 (Becton Dickinson, San Diego, CA).
Invasion assay
Cell invasion assay was performed with CytoSelect Cell Invasion Kit (Cell Biolabs, Inc., San Diego, CA) as described previously [19]. Briefly, 1.5 × 105 cells were suspended in 300 μl serum-free medium and plated into the top chamber. The chambers were then placed into 24-well plates with 500 μl of (20 %) serum-containing medium in each well. After 48-h incubation, cells that invaded through the basement membrane and reached to the lower chamber were detached with Cell Detachment Solution and were incubated for 30 min at 37 °C. Then, 75 μl of a 4X Lysis Buffer/CyQuant GR dye solution was added and 200 μl of the mixture was transferred into a 96-well plate and was read for fluorescence at 480/520 nm. Experiments were performed in triplicate and repeated three times.
The avian embryo model of cancer cell extravasations
The avian embryo extravasations assay was performed as described previously [20]. Briefly, fertilized avian embryos were cultured ex ovo for 14 days and intravenously injected with control or ANK3 shRNA (Santa Cruz Biotechnology) expressing LNCaP and PC-3 cells that were stably labeled with GFP or RFP fluorescent proteins (0.1 × 106 cells/embryo). Four to ten hours post tumor cell injection Lectin-Alexa 647 (50 μl/embryo, Vector Laboratories) was intravenously injected to label the blood vessel luminal surface. After 10 min of lectin microinjection, embryos were transferred into temperature/humidity controlled imaging chamber and embryonic chorioallantoic membrane (CAM) vasculature was imaged using Nikon A1r MP confocal microscope to monitor the extravasations of cancer cells. Cancer cell extravasations were quantified using Nikon Elements imaging software as previously described [20]. At least 10 animals were used for each experiment.
Quantitative real-time PCR
Total RNA was extracted using Trizaol (Invitrogen) according to the manufacturer’s instructions. One microgram of total RNA was subjected to a random-primed reverse transcription using qScript™ cDNA SuperMix (Quanta Bioscience, USA). Real-time PCR was conducted in triplicates using Applied Biosystem 7500 Fast Real-time PCR system with qScript™ One-Step SYBR® Green qRT-PCR Kit (Quanta Bioscience, USA). Relative mRNA levels were normalized to GUSB.
Study population and tissue microarray construction
ANK3 expression was assessed in tow cohorts in relation to disease progression and lethal outcome. The first cohort (n = 218) men treated by retro-pubic radical prostatectomy for localized prostate cancer. Samples from selected tissue blocks reflective of the predominant two Gleason patterns were embedded onto three tissue microarray (TMA) blocks using a manual tissue arrayer (Beecher Instruments, Silver Spring, MD). Between one and nine cores (average 3.3), 0.6 mm in diameter were sampled including benign, high grade intraepithelial neoplasia (HGPIN) and prostate cancer (PCA). The second cohort (n = 312) men diagnosed with PCA by transurethral resection of prostate (TURP) and managed expectantly by observation, radiotherapy, or hormonal manipulation from 2005 to 2009. This cohort included men who met the criteria of CRPC (n = 160), who were initially treated with LH-RH agonist as monotherapy, but subsequently progressed to require channel TURP to relieve obstructive symptoms. Clinical follow-up of this cohort was collected from the Alberta Tumor Registry in regard to overall survival, cancer-specific mortality and dates of hormonal treatment implementation. Prostate cancer-specific mortality (PCSM) was defined as patients with evidence of metastatic disease that progressed while on hormonal therapy and died of PCA based on medical records. This cohort was assembled onto two tissue microarrays (TMAs) with an average of two cancer cores (2–6) per patient and adjacent benign prostate tissue as control for a total of 714 cores using the manual tissue arrayer as above. In both cohorts, all pathological and clinical data were obtained with approval of the institutional review board at University of Calgary, Faculty of Medicine, Calgary, Alberta, Canada.
Pathological analysis
After construction, 4 μm hematoxylin and eosin stained sections were examined by two study pathologists to verify the histological diagnosis of TMA cores. All Gleason scores were assessed according to the 2005 ISUP criteria [21]. For each patient, the predominant two patterns of PCA were sampled. Protein expression of ANK 3 was evaluated semi quantitatively based on 4 tired system (0; negative, 1; weak, 2; moderate, and 3; high). For each core intensity, expression was based on more than 50 % of cells expressing such intensity. For each patient, the final intensity was based on the average per search diagnosis for all available cores. Cases with equivocal intensity were mutually resolved by the study pathologists.
Tissue immunohistochemistry (IHC)
IHC was performed on Leica Bond Max platform (Leica Microsystems, Wetzlar, Germany). Four-μm-thick formalin-fixed paraffin-embedded sections were subjected to heat-induced antigen retrieval for 30 min using Leica Epitope Retrieval Solution 2. Slides were then incubated with Ankyrin G rabbit polyclonal antibody (Santa Cruz, catalog # sc-28561) for 15 min at a 1:100 dilution. Bond Polymer Refine Detection kit (Leica Microsystems, Wetzlar, Germany) was used for HRP detection and the following hematoxylin counter stain.
Statistical analysis
Patient characteristics were presented as frequencies and percentages for categorical variables and as means and ranges for continuous variables. Chi-square test was used to test associations between ANK3 protein expression and other pathological parameters. The Kaplan-Meier approach along with the log-rank test was used for survival analyses to test associations between ANK3 expression and PCSM, and time till development of castrate resistant disease. In all statistical tests, a p value <0.05 was considered significant. For in vitro experiments, data were presented as means ± SD that were calculated from three or more independent experiments. Statistical significances were calculated by using one-way ANOVA and student’s t-test with the level of significance set at *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Knockdown of ANK3 represses prostate cancer cell proliferation independently of AR
To determine the importance of ANK3 expression for cancer cell growth, we carried out cell proliferation assays in a panel of prostate cancer cell lines following ANK3 knockdown. In the LNCaP cells, we found that ANK3 knockdown dramatically repressed cell growth (Fig. 1a). To further confirm this, we conducted ANK3 knockdown in VCaP cells. For the RNA interference (RNAi) analyses, we utilized four synthetic siRNAs targeting different regions of the ANK3 transcript. All four siRNAs efficiently inhibited ANK3 expression (Supplementary Fig. 1). Since potential off-target effects of individual siRNAs can be reduced by siRNA pooling [Ref], we used a pool consisting of all four -targeting siRNAs. This siRNA pool also efficiently blocked ANK3 expression (Fig. 1) and was used for further functional experiments. Consistently, cell proliferation assays manifested significantly reduced VCaP cell growth following ANK3 knockdown (Fig. 1b). As both LNCaP and VCaP are AR expressing cells, we sought to investigate whether this role of ANK3 is dependent on the AR function. To test this, we analyzed an AR negative cell line, PC-3. The results demonstrated that PC-3 cells were still showing decreased cells growth with ANK3 knockdown (Fig. 1c), indicating that the proliferative role of ANK3 is independent of AR.

Knockdown of ANK3 inhibits cell proliferation independent of the AR. MTS assay of AR-positive LNCaP (a) and VCaP (b) control siRNA (siCtrl) and siRNA-treated (siANK3) cells. ANK3 protein knockdown was confirmed by Western blot analysis (upper). c AR-negative PC-3 was transfected with siCtrl and siANK3. Total protein was blotted (upper) and cell growth assayed. d and e LNCaP cells were transfected with either siCtrl, siANK3, or siAR or both. AR and ANK3 proteins were determined by immunoblotting (d) and cell proliferation by MTS (e). f and g PC-3 cells were transfected with AR vector with or without siANK3. Protein expression was confirmed by Western blot (f) and cell growth was measured (g). Results were repeated from triplicate experiments and expressed as mean ± SEM. *p < 0.05, **p < 0.01
To verify this, LNCaP cells were co-transfected with siANK3 and siAR. As shown in Fig. 1d and e, knockdown of ANK3 significantly decreased cell proliferation regardless of the AR status, i.e., both in the presence and absence of siAR (p < 0.001). Interestingly, we observed a synergistic effect in decreasing cell proliferation with ANK3 loss and AR signal blockage. At same time, we overexpressed AR in PC-3 cell combined with the siANK3. AR expression cannot completely overcome the effect of ANK3 knockdown. Therefore, the proliferation role of ANK3 is independent of the AR pathway.
ANK3 regulates prostate cancer cell cycle
Because knockdown of ANK3 led to reduction of cell numbers in vitro, we thought to determine whether apoptosis played a role in this process. Western blot for PARP and Caspase-3 did not show cleaved PARP or activated Caspase-3 (Fig. 2a). These results indicated that no apoptosis was induced by ANK3 depletion.

ANK3 regulates cell cycle and cyclins expression. a Caspase3 and PARP protein levels were analyzed by Western blot after cells were transfected with siCtrl or siANK3 for 48 h. b and c LNCaP cells were transfected with siCtrl or siANK3 for 48 h. b FACS assay measured cell populations in G0/G1, S, and G2/M phase. c Cyclin A, B1, E, and D1 protein levels were measured and representative Western blots are shown. d and e VCaP cells were transfected with siCtrl or siANK3 for 72 h. d FACS assay measured cell populations in G0/G1, S, and G2/M phase. e Cyclin A, B1, E, and D1 protein levels were measured and representative Western blots are shown. Results were expressed as mean ± SEM. **p = 0.01, n = 3 independent experiments per group
To further confirm the proliferative action of ANK3, we performed FACS assays to measure the cell populations at each cell cycle phase, and data were plotted over the time course. At 48 h after siRNA transfections in LNCaP cells, there was no alteration in the G0/G1 cell population, but there was an increase in the percentage of cells in S-phase, with concomitant decrease in G2/M cell population, compared with samples transfected with control siRNA (Fig. 2b). Similar observations were also noted from VCaP cells after 72 h of transfections (Fig. 2d). Consistent with cell cycle changes, protein levels of cyclin A and B1, but not cyclin D1 and E, were dramatically decreased in cells transfected with siANK3 (Fig. 2c and e). Together, these results indicated that ANK3 targeted the S phase and suppressed S to G2/M transition to inhibit prostate cancer growth.
ANK3 inhibits prostate cancer cell invasion through AR
Next, we investigated the potential function of ANK3 in cell invasion. To do this, we performed a basement membrane cell invasion assay. We first examined the effect of ANK3 knockdown on the low invasive LNCaP cells. Unexpectedly, the results showed a dramatic increase in the number of invaded cells upon ANK3 knockdown (Fig. 3a). This inhibitory effect of ANK3 on cell invasion was also observed in another cell line VCaP (Fig. 3b). Interestingly, knockdown of ANK3 in PC-3 cells showed no effect on cell invasion (Fig. 3c). Taken together, these results strongly suggested that ANK3 regulation of cell invasion is mediated by and dependent on AR pathway.

Knockdown of ANK3 promotes prostate cancer cells invasion dependent of AR. Cell invasion was analyzed in LNCaP (a), VCaP (b), and PC-3 (c) cell lines after transfection with siCtrl or siANK3. d Invasion assay of PC-3 cells transfected with AR-vector, siANK3, or AR + siANK3. e Invasion assay of LNCaP cells transfected with siAR, siANK3, or siAR + siANK3. Results were repeated from triplicate experiments and expressed as mean ± SEM. *p < 0.05, **p < 0.01
We next examined whether the expression of AR might sensitize PC-3 cells to ANK3 regulation. To do this, we overexpressed AR in PC-3. Remarkably, we found that ANK3 knockdown indeed significantly increased the invasion in AR-expressing PC-3 cells but not in the control cells (Fig. 3d). We then investigated whether blockade of AR signaling in LNCaP might attenuate the ability of ANK3 to regulate LNCaP cell invasion. We used siRNA AR to block AR signaling in LNCaP cells, which led to a reduced cell invasion as expected. Importantly, without AR expression, ANK3 knockdown no longer had a significant effect on LNCaP cell invasion (Fig. 3e). These data, therefore, suggest that ANK3 is an inhibitory factor for cell migration and invasion.
ANK3 knockdown increases AR protein stability
We measured both AR mRNA and protein levels in LNCaP and VCaP cells transfected with siANK3 in comparison to their controls. We did not observe any changes in AR mRNA levels in ANK3 knockdown cells compared with cells transfected with the control siRNA (Fig. 4a and b). However, ANK3 knockdown resulted in an increase of AR protein in LNCaP and VCaP cells (Fig. 4c and d); suggesting that ANK3 regulates AR protein stability rather than its transcription. Next, we investigated ANK3 regulation of AR protein stability by using cycloheximide (CHX) to block protein synthesis in both LNCaP and VCaP cells. We found that within 48 h of treatment, the AR protein level was reduced by only about 40 % in LNCaP cells with ANK3 knockdown; however, cells transfected with control siRNA were more sensitive to CHX treatment, where the AR protein level was reduced by up to 90 % (Fig. 4e and f). Similar results were obtained in VCaP cells (Fig. 4g and h). Real-time qPCR analysis confirmed induced expression of known AR target genes such as PSA and TMPRSS2 after depleting ANK3, supporting the activation of AR signaling pathway (Fig. 4i). Taken together, our results suggested that ANK3 is required to regulate AR protein stability in prostate cancer cells.

ANK3 regulates AR protein stability. AR mRNA levels (a and b) and protein levels (c and d) were measured by real-time qPCR and Western blot in LNCaP and VCaP cells (respectively) transfected with siCtrl or siANK3 for 48 h. E-H, LNCaP, and VCaP cells were treated with 50 μg/ml CHX for 0, 8, 24, and 48 h. Representative Western blot of AR and actin in LNCaP (e) and VCaP (g) were shown. Densitometry of AR protein levels from triplicate experiments in LNCaP (f) and VCaP (h). i Real-time qPCR analysis of AR targets in LNCaP cells transfected with siCtrl or siANK3. Results were repeated from triplicate experiments and expressed as mean ± SEM. **p < 0.01
We then ask whether endogenous ANK3 binds to AR, to this end, we performed co-IP experiment in LNCaP cells. Results shown in Fig. 5 demonstrated that ANK3 is unlikely associate with AR protein. We further investigated if ANK3-AR complex is mediated through an intermediary protein such as Ubiquitin or heat shock protein 90 (HSP-90). However, interactions between ANK3 and either Ubiquitin or HSP-90 were not detected.

The biochemical interaction among ANK3, AR, HSP-90, and Ubiquitin in LNCaP cells was examined by immunoprecipitation (IP) with anti-ANK3 antibody, whereas rabbit IgG served as a negative control. The levels of ANK3, AR, HSP-90, and Ubiquitin proteins (input) were evaluated by western immunoblotting with cognate antibodies. n = 3 independent experiments
ANK3 knockdown effect on tumor cell extravasations in vivo
Recent findings show that invasive cell behavior plays key role during cancer cell extravasations. It was shown that extravasating cancer cells utilize invadopodia to break through the vascular wall and exit into the surrounding tissue. To investigate if ANK3 expression has an effect on cancer cell invasion in vivo, that is similar to what we observed in vitro, we utilized recently developed avian embryo model of cancer cell extravasations and metastasis [20]. Fluorescently labeled PC3 or LNCaP cells that either express control, scrambled shRNA construct or shRNA, that targets ANK3 expression, were injected intravenously into the avian embryos at 14 days. Post fertilization and percentage of extravasated cancer cells were quantified at 10 h post tumor cell injection. Interestingly, we found that while ANK3 knockdown has no effect on PC3 cells extravasations it produces significant increase (p < 0.01) in extravasations of LNCaP cells supporting our in vitro findings (Fig. 6a, b). Taken together, our results suggested that ANK3 may promote cancer cell extravasations and invasion in vivo.

a Representative images showing control and shANK3 LNCaP cells (green, GFP) extravasating out of avian embryo CAM vasculature (white, Alexa 647). Upper panels show single optical slices (10× objective). Lower panels show corresponding side views that were digitally cut along the thick orange lines. Insets show 3D reconstruction of areas highlighted by the dashed squares. Red arrows point to the same cell within shCtrl or shANK3 panels. b Quantification of ANK3 expression knockdown effect on PC3 and LNCaP cancer cell extravasations. Scale bar = 100 μm. Results were repeated from triplicate experiments and expressed as mean ± SEM. **p < 0.01
ANK3 is a prognostic factor for the survival of the prostate cancer patients
To explore whether the above findings are clinically relevant for prostate cancer patients, we analyzed the ANK3 expression level and the pathologic parameters in prostate cancer patients. Interestingly, we found that ANK3 is significantly upregulated in high grade intraepithelial neoplasia (HGPIN) (p < 0.0001) and localized prostate cancer (p < 0.0001) when compared with benign prostate samples (Fig. 7a, b). However, ANK3 is significantly downregulated in castration resistant cancer (CRCP) (p < 0.0001) relative to localized prostate cancer (Fig. 7a, b). This disease stage-dependent expression pattern of ANK3 precisely reflects the dual role of ANK3 in positively regulating cell proliferation, a cellular process that dominates primary tumors, and in negatively regulating cell invasiveness, a process essential to tumor metastasis and progression. Furthermore, significant inverse correlation between ANK3 expression and Gleason Score (GS) was noted. Moderate/high ANK3 protein expression was identified in 188/194 (97 %) patients of GS < 7 compared with 108/120 (90 %) in GS7 and 265/324 (81.7 %) patients of GS > 7 (Fig. 6c) (p < 0.0001). Intensity levels of ANK3 in various tissue types and in relation to Gleason score are presented in Table 1. Also, we found that ANK3 levels are positively correlated with the overall survival of prostate cancer patients. Patients with moderate and high higher ANK3 intensity had better survival of prostate cancer deaths compared to patients with lower ANK3 intensity (p = 0.012) (Fig. 7d). However, in multivariate analysis and accounting for tumor volume, Gleason score, and ANK3 expression, ANK3 was not statistically associated with lethal disease (Table 2).

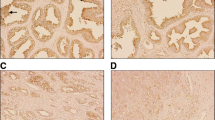
a Error bars of mean intensity value for ANK3 protein expression across various stages of prostate cancer progression (BN, HGPIN, PCA, AdvPCA, and CRPC). There is significant increase in ANK3 mean expression intensity in neoplastic epithelium compared to benign prostate tissue). b Representative images of ANK3 immunohistochemistry in benign (left panel), Gleason score 6, localized PCA (mid panel) and CRPC (right panel). c Inverse association of ANK3 intensity expression to Gleason score. d KM survival curve of ANK3 correlation to prostate cancer specific mortality. Cases with higher ANK3 expression were less likely associated with PCA specific mortality in comparison to those with lower expression
Discussion
To the best of our knowledge, this is the first study investigating the function of ANK3 in prostate cancer progression. Here, we provide evidence that knockdown of ANK3 in prostate cancer cells decreases cell proliferation by arresting the S to G2/M phase transition, while significantly promoting subsequent progression stages of metastasis dependent on AR activation. ANK3 may therefore play different roles at different stages of tumor progression, whereby tumor cells, as a response to micro-environmental signals, may switch off proliferation and start invasion.
Depending on clinical stage of the disease, ANK3 expression shows some differences in overall expression compared to benign tissue. Indeed, using the tumor progression tissue microarray, we revealed that, during prostate cancer progression from benign tissue, high grade intraepithelial neoplasia to localized prostate cancer, then to castration resistant cancer, ANK3 expression is initially upregulated (in high grade intraepithelial neoplasia and localized prostate cancer), and then it is downregulated (in advanced cancer and castration resistant cancer) compared to localized PCa. ANK3 increased expression relative to benign tissue may be associated with tumor proliferation. However, the decrease in more advanced and CRPC stages maybe associated with tumor metastasis. Furthermore, increased expression of the ANK3 predicts better overall survival of prostate cancer patients with metastatic disease. Therefore, ANK3 is demonstrated to be a novel prostate cancer prognostic maker, as recently suggested by our group [16, 17].
Our in vitro and in vivo findings of ANK3 in promoting cell proliferation and inhibiting cell invasion are also consistent with the role of ANK3 in prostate cancer patients’ progression. Tumor progression is a complex process. Although proliferation, migration, and invasion are mutually correlated, each of these is governed by a large number of tumor suppressors and oncogenes. Several genes have been reported to pose a contrasting relationship between proliferation and invasion behaviors, including cyclin D1, YB-1, Arg, mineral dust-induced gene (mdig), and FoxA1 [22–25]. In fact, actively invasive cancer cells can downregulate proliferation-associated genes while upregulating genes that encode for motility and invasion [26–28]. The changes in the pattern of ANK3 expression with tumor progression and its differential role in regulating cell proliferation and invasion are in concordance with that fact. Our findings represent ANK3 as a novel molecular target for cancer metastasis.
Androgen receptor is a well-recognized key factor for prostate cancer development/progression, including proliferation and invasion [29]. Our data relating to increased stability of AR associated with increased invasion are in line with previous studies published in prostate and osteosarcoma [30]. In addition, we provided strong evidence (both in vitro and in vivo) that ANK3 regulates cell invasion in an AR-dependent manner. However, its effect on proliferation is independent of AR despite the finding that ANK3 controls AR half-life. In previous studies, AR was found to promote prostate cancer cell proliferation by regulating G1-S phase progression and promoting G1 cyclin-dependent kinase (CDK) activity [31, 32]. These AR regulated cell cycle components are different from the ones found to be regulated by ANK3 in our study; namely (S-G2/M phase transition) and the expression of cyclins A and B, hence emphasizing that ANK3 role in PCa cell proliferation is independent of AR. In normal mouse NIH 3T3 cells, overexpression of ANK3 smaller isoforms was shown to suppress proliferation [10], which is opposite to proliferation-promoting effect of ANK3 on prostate cancer cells, suggesting that ANK3 exerts its proliferative function through multiple mechanisms depending upon cell contexts. The loss of ANK3 expression was reported to occur during epithelial-to-mesenchymal transition (EMT), which is a key event for invasion [18]. However, we could not find any changes in EMT markers (such as E-Cadherin, snail, ZEB1/2, and ZO-1) when we knocked down ANK3 (data not shown). Further studies are warranted to investigate whether the observed role of ANK3 on invasion is a result of its effects on cytoskeleton structures, cell adhesion signaling, and/or enzymes that degrade extracellular matrix. In addition, we showed that ANK3 depletion can stabilize AR protein. Since the ubiquitin-proteasomal pathway has been shown to mediate AR degradation [33], we tested if ANK3 might bind to AR through ubiquitin protein. We tested this hypothesis using immunoprecipitation but we were not able to detect such binding. Furthermore, it was shown that HSP-90 association with AR is essential for the biological function and stability of androgen receptor in prostate cancer cells [34]. However, we did not detect ANK3-HSP-90 complex in LNCaP cells. In fact, a previous study reported that 120- and 105-kDa ANK3 isoforms localize in the late endosomes and lysosomes and regulate PDGF protein degradation through lysosomal pathways (10). Future studies are needed to examine the mechanisms by which ANK3 controls protein stability.
Conclusion
In summary, our results provide new insights on ANK3, which may have a role in determining this switch between proliferation and invasion. It highlights the significance of ANK3 expression in prostate cancer progression Androgen receptor stability and cancer lethality. ANK3 may play a significant role in the development of castrate resistant disease and should be further investigated.
References
Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA Cancer J Clin 63:11–30
Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, Gasser TC, Mihatsch MJ (2000) Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol 31:578–583
Lambert S, Bennett V (1993) From anemia to cerebellar dysfunction. A review of the ankyrin gene family. Eur J Biochem 211:1–6
Peters LL, John KM, Lu FM, Eicher EM, Higgins A, Yialamas M, Turtzo LC, Otsuka AJ, Lux SE (1995) Ank3 (epithelial ankyrin), a widely distributed new member of the ankyrin gene family and the major ankyrin in kidney, is expressed in alternatively spliced forms, including forms that lack the repeat domain. J Cell Biol 130:313–330
Bennett V (1992) Ankyrins. Adaptors between diverse plasma membrane proteins and the cytoplasm. J Biol Chem 267:8703–8706
De Matteis MA, Morrow JS (1998) The role of ankyrin and spectrin in membrane transport and domain formation. Curr Opin Cell Biol 10:542–549
Bennett V, Chen L (2001) Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr Opin Cell Biol 13:61–67
Kordeli E, Lambert S, Bennett V (1995) AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270:2352–2359
Hoock TC, Peters LL, Lux SE (1997) Isoforms of ankyrin-3 that lack the NH2-terminal repeats associate with mouse macrophage lysosomes. J Cell Biol 136:1059–1070
Ignatiuk A, Quickfall JP, Hawrysh AD, Chamberlain MD, Anderson DH (2006) The smaller isoforms of ankyrin 3 bind to the p85 subunit of phosphatidylinositol 3’-kinase and enhance platelet-derived growth factor receptor down-regulation. J Biol Chem 281:5956–5964
Grubb MS, Burrone J (2010) Building and maintaining the axon initial segment. Curr Opin Neurobiol 20:481–488
Lambert S, Davis JQ, Bennett V (1997) Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J Neurosci 17:7025–7036
Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, et al. (2008) Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 40:1056–1058
Smith EN, Bloss CS, Badner JA, Barrett T, Belmonte PL, Berrettini W, Byerley W, Coryell W, Craig D, Edenberg HJ, et al. (2009) Genome-wide association study of bipolar disorder in European American and African American individuals. Mol Psychiatry 14:755–763
Pandey A, Davis NA, White BC, Pajewski NM, Savitz J, Drevets WC, McKinney BA (2012) Epistasis network centrality analysis yields pathway replication across two GWAS cohorts for bipolar disorder. Transl Psychiatry 2:e154
Glinsky GV, Berezovska O, Glinskii AB (2005) Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest 115:1503–1521
Bismar TA, Alshalalfa M, Petersen LF, Teng LH, Gerke T, Bakkar A, Al-Mami A, Liu S, Dolph M, Mucci LA, et al. (2014) Interrogation of ERG gene rearrangements in prostate cancer identifies a prognostic 10-gene signature with relevant implication to patients’ clinical outcome. BJU Int 113:309–319
Kumar S, Park SH, Cieply B, Schupp J, Killiam E, Zhang F, Rimm DL, Frisch SM (2011) A pathway for the control of anoikis sensitivity by E-cadherin and epithelial-to-mesenchymal transition. Mol Cell Biol 31:4036–4051
Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, MC W, Shimamura T, Perera SA, et al. (2007) LKB1 modulates lung cancer differentiation and metastasis. Nature 448:807–810
Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, Hague MN, Ablack A, Carmine-Simmen K, McPherson VA, et al. (2014) Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep 8:1558–1570
Epstein JI, Allsbrook WC Jr, Amin MB, Egevad LL, Committee IG (2005) The 2005 International Society of Urological Pathology (ISUP) consensus conference on Gleason grading of prostatic carcinoma. Am J Surg Pathol 29:1228–1242
Yu M, Sun J, Thakur C, Chen B, Lu Y, Zhao H, Chen F (2014) Paradoxical roles of mineral dust induced gene on cell proliferation and migration/invasion. PLoS One 9:e87998
Gil-Henn H, Patsialou A, Wang Y, Warren MS, Condeelis JS, Koleske AJ (2013) Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene 32:2622–2630
Evdokimova V, Tognon C, Ng T, Sorensen PH (2009) Reduced proliferation and enhanced migration: two sides of the same coin? Molecular mechanisms of metastatic progression by YB-1. Cell Cycle 8:2901–2906
Lehn S, Tobin NP, Berglund P, Nilsson K, Sims AH, Jirstrom K, Harkonen P, Lamb R, Landberg G (2010) Down-regulation of the oncogene cyclin D1 increases migratory capacity in breast cancer and is linked to unfavorable prognostic features. Am J Pathol 177:2886–2897
Wang W, Goswami S, Sahai E, Wyckoff JB, Segall JE, Condeelis JS (2005) Tumor cells caught in the act of invading: their strategy for enhanced cell motility. Trends Cell Biol 15:138–145
Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, Singer RH, Segall JE, Condeelis JS (2004) Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res 64:8585–8594
Wang W, Wyckoff JB, Goswami S, Wang Y, Sidani M, Segall JE, Condeelis JS (2007) Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res 67:3505–3511
Shiota M, Yokomizo A, Naito S (2011) Increased androgen receptor transcription: a cause of castration-resistant prostate cancer and a possible therapeutic target. J Mol Endocrinol 47:R25–R41
Wagle S, Park SH, Kim KM, Moon YJ, Bae JS, Kwon KS, Park HS, Lee H, Moon WS, Kim JR, et al. (2015) DBC1/CCAR2 is involved in the stabilization of androgen receptor and the progression of osteosarcoma. Sci Rep 5:13144
Ramos-Montoya A, Lamb AD, Russell R, Carroll T, Jurmeister S, Galeano-Dalmau N, Massie CE, Boren J, Bon H, Theodorou V, et al. (2014) HES6 drives a critical AR transcriptional programme to induce castration-resistant prostate cancer through activation of an E2F1-mediated cell cycle network. EMBO Mol Med 6:651–661
Balk SP, Knudsen KE (2008) AR, the cell cycle, and prostate cancer. Nucl Recept Signal 6:e001
Vanaja DK, Mitchell SH, Toft DO, Young CY (2002) Effect of geldanamycin on androgen receptor function and stability. Cell Stress Chaperones 7:55–64
Linn DE, Yang X, Xie Y, Alfano A, Deshmukh D, Wang X, Shimelis H, Chen H, Li W, Xu K, et al. (2012) Differential regulation of androgen receptor by PIM-1 kinases via phosphorylation-dependent recruitment of distinct ubiquitin E3 ligases. J Biol Chem 287:22959–22968
Acknowledgment
This work was supported in part by the Prostate Cancer Foundation Young Investigator Award (T.A. B). This work was also supported by Prostate cancer Canada and is proudly funded by the Movember Foundation-Grant no. B2013-01.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare in this study.
Additional information
Tingting Wang and Hatem Abou-Ouf these authors contributed equally
Electronic supplementary material
ESM 1
(PDF 306 kb)
Rights and permissions
About this article

Cite this article
Wang, T., Abou-Ouf, H., Hegazy, S.A. et al. Ankyrin G expression is associated with androgen receptor stability, invasiveness, and lethal outcome in prostate cancer patients. J Mol Med 94, 1411–1422 (2016). https://doi.org/10.1007/s00109-016-1458-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-016-1458-4