
Abstract
Tumor immune escape and the initiation of metastasis are critical steps in malignant progression of tumors and have been implicated in the failure of some clinical cancer immunotherapy. Tumors develop numerous strategies to escape immune surveillance or metastasize: Tumors not only modulate the recruitment and expansion of immunosuppressive cell populations to develop the tumor microenvironment or pre-metastatic niche but also switch the phenotype and function of normal immune cells from a potentially tumor-reactive state to a tumor-promoting state. Immunosuppressive cells facilitate tumor immune escape by inhibiting antitumor immune responses and furthermore promote tumor metastasis by inducing immunosuppression, promoting tumor cell invasion and intravasation, establishing a pre-metastatic niche, facilitating epithelial-mesenchymal transition, and inducing angiogenesis at primary tumor or metastatic sites. Numerous translational studies indicate that it is possible to inhibit tumor immune escape and prevent tumor metastasis by blocking immunosuppressive cells and eliminating immunosuppressive mechanisms that are induced by either immunosuppressive cells or tumor cells. Furthermore, many clinical trials targeting immunosuppressive cells have also achieved good outcome. In this review, we focus on the underlying mechanisms of immunosuppressive cells in promoting tumor immune escape and metastasis, discuss our current understanding of the interactions between immunosuppressive cells and tumor cells in the tumor microenvironment, and suggest future research directions as well as potential clinical strategies in cancer immunotherapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Understanding the relationship between the immune system and cancer progression has been a hot topic in the field of tumor immunology. Tumors can drive the generation of immunosuppressive or regulatory immune cell subtypes as well as recruit massive number of tumor-promoting myeloid cells to establish the tumor microenvironment, thus promoting tumor progression. Compelling evidence indicates that myeloid-derived suppressor cell (MDSC), regulatory T cell (Treg), tumor-associated macrophage (TAM), regulatory dendritic cell (DCreg), neutrophil, T helper 17 cell (Th17), and regulatory B cell (Breg) are key immunosuppressive cells that promote tumor progression [1–5]. However, open questions still remain regarding the functions and the underlying mechanisms of the immunosuppressive cells in tumor immune escape and metastasis. Critical steps in the malignant progression of tumors are the evasion of immune destruction and the initiation of tumor cell metastasis, and both have been described previously as two hallmarks of cancer [6]. These steps can be achieved by inhibiting the host’s immune system, especially by induction, expansion, and recruitment of the immunosuppressive cells.
The immune system plays a dual role in cancer: It can not only suppress tumor development and progression by destroying cancer cells or inhibiting their outgrowth, but it can also promote tumor progression by establishing favorable conditions within the tumor microenvironment that facilitate tumor outgrowth and metastasis [2, 7]. During tumor progression, innate and adaptive immune molecules and cells can recognize and eliminate the transformed cancer cells, previously known as cancer immunosurveillance. However, tumors can change the host immune system to escape immune control, grow progressively, and emerge as clinically apparent disease [8–10]. Moreover, growing tumors and their metastatic derivatives also develop strategies to overcome the immune response in future metastatic sites via multiple mechanisms, including recruitment of immunosuppressive cells and formation of a pre-metastatic niche, thereby promoting metastasis and the establishment of metastatic disease [2].
A variety of the mechanisms for tumor immune escape and metastasis are thought to be involved in the failure of some clinical cancer immunotherapy. Many considerable clinical evidence also suggest that the immunosuppressive cell subsets play a key role in tumor progression and thus have strong predictive value for clinical outcome in cancer patients. For example, tumor-infiltrating immune cells with immunosuppressive signatures exploit the inhibitory molecules, such as programmed death 1 (PD-1), to limit T cell-mediated immune responses [11]. Therefore, suppressing the suppressor may be an attractive approach to block tumor immune escape or inhibit tumor metastasis by reversing immunosuppression and enhancing antitumor immune response at the various stages of tumor progression. Moreover, identifying cellular and molecular suppressors for antitumor immune response, elucidating their functional mechanisms in promoting tumor immune escape and metastasis, and consequently suppressing the activities of immunosuppressive cells or even eliminating the immunosuppressive cells in the tumor microenvironment will provide new strategies for cancer immunotherapy.
In this review, we will focus on the underlying mechanisms by which immunosuppressive cells promote tumor immune escape and metastasis. We will also summarize our current understanding of the interactions between immunosuppressive cells and cancer cells in the tumor microenvironment and discuss future research directions and potential clinical strategies in cancer immunotherapy.
Tumor immune escape
Tumor immune escape always leads to malignant progression and failure of cancer immunotherapy. Cancer cells have developed several different mechanisms by which to escape the immune system. For example, cancer cells undergo continuous remodeling at the genetic, epigenetic, and metabolic levels to resist apoptosis, reduce immune recognition, and select tumor variants resistant to immune effectors. More importantly, tumors effectively induce and recruit distinct suppressive immune cells within the tumor tissue to escape from antitumor immune responses.
In the past years, our understanding as to how immunosuppressive cells contribute to tumor immune escape has been extensively improved. During tumor progression, the immunosuppressive cells are mobilized in response to tumor-induced cytokine axes, such as transforming growth factor-β (TGF-β) and CXCL5-CXCR2. These tumor-infiltrating immunosuppressive cells evade immune surveillance via mechanisms such as (a) disruption of antigen presentation by DC; (b) inhibition of T and B cell proliferation and activation by expression of immunoregulatory molecules such as arginase, iNOS, and indoleamine-2,3-dioxygenase (IDO), or secretion of immunosuppressive cytokines such as IL-10 and TGF-β; or (c) inhibition of cytotoxicity of cytotoxic T lymphocytes (CTL), natural killer (NK) cells [12].
Tregs play important roles in promoting tumor immune escape. They are a physiologically suppressive subset that limits autoimmune responses and maintains the homeostasis of immune response. Tregs can inhibit the function of T lymphocytes by (a) producing the immunosuppressive cytokines IL-10 and TGF-β; (b) expressing the negative costimulatory molecules such as cytotoxic T lymphocyte antigen 4 (CTLA-4), PD-1, or the ligand PD-L1; or (c) consuming the cytokine IL-2 [13, 14]. Tregs are often hijacked by tumors to promote immune escape so as to favor tumor outgrowth. For example, Tregs can suppress tumor-associated antigen presentation and interfere with cytotoxic T cell function by inhibiting cytolytic granule release during tumor progression [15]. Tumors under hypoxia conditions promote the recruitment of CD4+CD25+Foxp3+ Tregs by inducing the expression of the chemokine CCL28, which dampens effector T cell function to maintain tumor immune escape and promote tumor angiogenesis by secreting VEGFA [16]. Meanwhile, human hepatocellular carcinoma-infiltrating CD4+CD69+Foxp3− Tregs are reported to suppress antitumor T cell immune response via membrane-bound TGF-β1 (mTGF-β1) thus promoting tumor immune escape [17].
MDSCs can facilitate tumor immune escape through multiple mechanisms. For example, tumor-infiltrating MDSCs impair the function of T cells, NK cells, DCs, thus facilitating tumor escape from immune attack; this tumor-promoting role of MDSCs is mediated by cellular stress sensor C/EBP homologous protein (Chop) and by interleukin-6 (IL-6) derived from MDSCs. The immunosuppressive function of MDSCs can be reversed in 3LL lung carcinoma-bearing mice if Chop is deleted in these cells [18]. In a murine rhabdomyosarcoma tumor model, production of CXCR2 ligands by cancer cells induced robust expansion of CXCR2+CD11b+Ly6Ghi MDSCs, which could mediate local immunosuppression and inhibit T cell proliferation via L-arginine depletion [19]. This is one of the key mechanisms utilized by tumors to escape immune surveillance. At the same time, the suppressive activity of these MDSCs also limits the efficacy of checkpoint blockade [19]. Besides, MDSCs that differentiated from bone marrow progenitor cells and that are activated by HMGB1 can mediate tumor immune escape by producing high levels of IL-10, suppressing antigen-driven activation of CD4+ and CD8+ T cells, and downregulating the expression of L-selectin on circulating naive T cells [20].
TAMs can inhibit CD8+ T cell-mediated antitumor immune responses to facilitate tumor immune escape [21, 22]. TAMs within mammary tumor tissue can produce high levels of the immunosuppressive cytokine IL-10 to suppress IL-12 expression in intratumoral DCs and consequently block CD8+ T cell-dependent antitumor immune responses [23, 24]. These tumor-promoting TAMs are derived from CCR2+ inflammatory monocytes, and their terminal differentiation depends on the Notch signaling pathway via transcription factor RBPJ [25, 26]. Mechanistically, TAMs can also suppress the cytotoxic activity of CD8+ T cells either directly through their expression of inhibitor ligands such as PD-L1 and B7-H4 (also known as VTCN1) or indirectly via CCL22-mediated recruitment of Tregs [27]. Moreover, clinical data show that greater accumulation of TAMs in tumor tissue can predict the poor prognosis of cancer patients. For instance, an increased number of CD68+ macrophages in classic Hodgkin’s lymphoma biopsies correlated strongly with shortened survival and could even be used as a clinical predictor for relapse after therapy, which validated the finding that patients who fail to respond to primary treatment always overexpress a macrophage gene signature [28].
Other immunosuppressive cells are also involved in promoting tumor immune escape. For example, protumoral “N2” neutrophils can suppress CD8+ T cell function by secreting stored arginase 1 (ARG1) to degrade extracellular arginine, a factor needed for the proper function of T cells [5]. DCreg is a regulatory DC subset that can be induced by tumors and then suppresses antitumor immune response [29, 30]. Tolerogenic DC (Tol-DC) is the term usually used to designate the immature, maturation-resistant DC or alternatively activated DC subset that express low levels of surface MHC and costimulatory molecules in tolerance induction. Tol-DC has tolerogenic characteristic and is closely related to the induction of immune tolerance and can prevent the pathogenesis of autoimmune diseases in normal physiological condition [31]. In hepatocellular carcinoma, a new subset of human CD14+ CTLA-4+ DCreg has been identified to significantly suppress T cell response through CTLA-4-dependent IL-10 and IDO production [29], which indicates the important role for DC-derived CTLA-4 in tumor immune escape or immunosuppression.
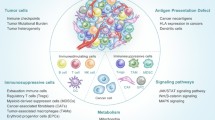
Taken together, the immunosuppressive cells have functional roles in promoting tumor immune escape by producing immunosuppressive cytokines and generating an immunosuppressive network (Fig. 1). Therefore, identifying the origin and functional characteristics of immunosuppressive cells will add the insight into tumor immune escape and help design new approaches of cancer immunotherapy by blocking tumor immune escape.

Immunosuppressive cells in the promotion of tumor immune escape. Cancer cells secrete chemokines and cytokines to recruit immunosuppressive cells including myeloid-derived suppressor cell (MDSC), regulatory T cell (Treg), tumor-associated macrophage (TAM), T helper 17 cell (Th17), regulatory dendritic cell (DCreg), tumor-associated neutrophil (TAN), and regulatory B cell (Breg) to generate an immunosuppressive tumor microenvironment. Tumors also actively switch the phenotype and function of normal immune cells from potentially tumor-reactive states to potentially tumor-promoting or immunosuppressive states. These immunosuppressive cells directly suppress the cytotoxic functions of natural killer (NK) cells and CD8+ cytotoxic T lymphocytes through the expression and production of various factors, such as PD-L1, B7-H4, and β-galactoside-binding protein (βGBP), which inhibit antitumor immune responses, thereby promoting tumor immune escape
Development of immunosuppressive tumor microenvironment
Interactions between cancer cells and the non-tumor components in tumor microenvironment may emerge as important determinants of tumor behavior. Molecular profiling of cellular components from various human tumor specimens has yielded valuable information on patient prognosis, which further highlights the critical role of the tumor microenvironment in directing tumor development and progression [3]. Importantly, local immunosuppression in tumor microenvironment seems to be the primary cause of tumor immune escape and also has a promoting effect on tumor metastasis.
The immunosuppressive tumor microenvironment is established, at least partially, by recruiting immunosuppressive cells to function as effectors to inhibit immune response and by tumor cell-derived immunosuppressive cytokines such as vascular endothelial growth factor (VEGF), TGF-β, galectin, or IDO [5]. It is well known that tumors, especially at the late stage of their development, can actively drive the generation of immunosuppressive or regulatory immune cell subtypes and recruit the cells to the tumor microenvironment. MDSCs, TAMs, and Tregs are the major immunosuppressive cells that populate in this immunosuppressive microenvironment. MDSCs are a heterogeneous group of myeloid progenitor cells and immature myeloid cells, which can inhibit antitumor immune response in tumor microenvironment by (a) inducing Tregs; (b) producing immunosuppressive cytokines such as TGF-β; (c) depleting or sequestering the amino acids arginine, tryptophan, or cysteine required for T cell function; (d) or nitrating the T cell receptor (TCR) or chemokine receptor on tumor-specific T cells [5, 32]. Chemokines expressed by lymphoma cells can recruit immunosuppressive cells including TAMs, MDSCs, Tregs, and Th2-polarized cells to maintain a tolerogenic microenvironment and allow the lymphoma cells to escape detection [33]. In addition, the production and elaboration of GM-CSF, IL-1β, VEGF, and prostaglandin E2 (PGE2) by tumors lead to the expansion and accumulation of MDSCs in the tumor microenvironment [32]. In human cancer patients, immunosuppression of lymphocytes within the tumor microenvironment has also been widely observed for a variety of cancer types [4].
Immunosuppressive cells in the tumor microenvironment can secrete or express a number of immunosuppressive molecules to promote tumor immune escape. For example, TAMs, one of the most abundant cell types in the tumor microenvironment, are capable of secreting an array of cytokines, chemokines, and enzymes that suppress CD4+ and CD8+ T cell effector function directly or indirectly by recruiting natural Tregs to the tumor microenvironment as well as by inducing CD4+ iTregs and sustaining their survival. TAMs can also suppress T cell activity by depleting L-arginine in the tumor microenvironment [21, 22]. Besides, tumor cell-mediated chemokine production is linked with immunosuppressive cell recruitment into the tumor microenvironment. For instance, melanomas-derived CCL21 recruits lymphoid tissue-inducer (LTi) cells to develop an immunosuppressive tertiary lymphoid structure within the tumor mass, which could polarize monocytes to M2 macrophages and recruit MDSCs and Tregs into the microenvironment to promote tumor immune escape [34]. In addition to these major immunosuppressive cells, DCs are also recruited to the tumor microenvironment and become important players in the immunosuppressive network. Hypoxia and apoptotic cells in the tumor microenvironment can induce the generation of Tol-DCs via IL-10 and TGF-β, and these Tol-DCs also promote the expansion of CD4+ Foxp3+ Tregs via PD-L1 on their surface [31]. Moreover, a high level of Fas expressed in CD11bhiIalow DCreg can be induced by endothelial stromal cell-derived TGF-β via ERK/β-catenin pathway activation and thus empowering DCregs to inhibit CD4+ T cell proliferation significantly so as to exert their immunosuppressive function in the immune microenvironment [30].
The development of an immunosuppressive tumor microenvironment can also contribute to the failure of antitumor immunity. Many subtypes of immunosuppressive cells trafficked or recruited to the tumor microenvironment can communicate with cancer cells via the secretion of cytokines, growth factors, and proteases that remodel the tumor microenvironment. Thus, deciphering the molecular and cellular components of the tumor microenvironment and their functional mechanisms that facilitate tumor immune escape and promote tumor metastasis will contribute to a better understanding of tumor-host interactions and help us overcome challenges in developing new powerful strategies for cancer immunotherapy.
The phenotypic and functional switch of immune cells
Tumors can manipulate the immune system by actively switching the phenotype and function of immune cells to help cancer cells evade the host immune response and achieve further metastasis. Tumors educate or reprogram the immune cells in different ways, including (a) the regulation of myeloid cell differentiation, (b) subversion of the antitumor activity of immune cells, and (c) re-education of immune cells with immunosuppressive/pro-tumorigenic functions [35].
Accumulating data suggest that tumors polarize the recruited immune cells from a potentially tumor-reactive state to a tumor-promoting state. Macrophages, which comprise a very plastic cell population, exhibit distinct functions in response to environmental signals. They can be conventionally divided into two subgroups, M1 and M2. M1-type macrophages are classically activated and play important roles in the innate response to invading pathogens. By contrast, M2-type macrophages can be alternatively activated by IL-4, IL-10, IL-13, and glucocorticoid, all of which play important roles in tissue repair and facilitate tumor progression by expressing high levels of IL-10 and low levels of IL-12. The conversion of M1 to M2 macrophages, and vice versa, is determined by the tumor microenvironment. For example, tumor-driven granulo-monocytopoiesis mediates TAM differentiation and M2 polarization based on the expression of retinoic acid-related orphan receptor (RORC1/RORγ) in tumor-bearing mice and cancer patients. Accordingly, the ablation of RORC1 in the hematopoietic compartment prevents tumor-driven myelopoiesis, resulting in inhibition of tumor growth and metastasis [36]. In many kinds of tumors, M2-type macrophages comprise most TAMs and promote tumor progression and metastasis by expressing high levels of soluble factors and cytokines such as MMP9, VEGF, EGF, and TGF-β. Furthermore, tumor-derived TGF-β and PGE2 can promote the differentiation of macrophages that express low levels of markers associated with M1-type macrophages [37]. Cytokines including IL-1, IL-4, IL-6, IL-10, CSF, and TNF-α in the tumor microenvironment can promote the polarization of TAMs by activating the STAT signaling pathway. A local hypoxic tumor microenvironment polarizes the M1 to M2 macrophages and promotes their expression of the angiopoietin receptor TIE2, which downregulates antitumor function of macrophages and facilitates tumor metastasis [38].
Many tumor-derived factors, such as TGF-β, VEGF, IL-4, IL-6, and IL-13, regulate multiple pathways likely related to myeloid cell differentiation and polarization of myeloid cell function. For example, neutrophils have been shown to shift from an antitumoral N1 phenotype to a protumoral N2 phenotype in the tumor microenvironment. In lung adenocarcinoma and mesothelioma models, TGF-β induced tumor-infiltrating neutrophils to develop an N2 phenotype, which is characterized by ARG1 expression and low levels of TNF, CCL3, and intercellular adhesion molecule 1 (ICAM1) [39]. Furthermore, TGF-β production by tumor cells can also convert effector T cells into Tregs that, in turn, suppress other effector T cells infiltrating the tumor mass [40].
Similarly to M1/M2 macrophages and N1/N2 neutrophils, DCs may also be induced by tumor cells to switch to a regulatory state. Tumor cell-derived factors, including IL-10, TGF-β, VEGF, and PGE2, can induce DCs with low expression of MHC and costimulatory molecules (e.g., CD40, CD80, CD86) and low production of TNF-α, interferon-α (IFN-α), IL-12, and CCL5, thus ultimately leading to T cell anergy and Treg induction [40, 41]. Abnormal DC differentiation and defective DC function are systemic phenomena that affect the myeloid cell lineage during cancer progression. For example, human lung carcinoma cells can convert mature DCs into TGF-β-producing cells [41], whereas mouse lung cancer can drive DCs to express high levels of IL-10, nitric oxide, VEGF, and ARG1. MHC-II+CD11b+CD11c+ tumor-infiltrating mouse DCs have been shown to suppress CD8+ T cells and antitumor immune responses through ARG1 production [42], an immunosuppressive mechanism previously attributed to TAMs and MDSCs.
Tumor-derived factors can also redirect myeloid differentiation toward the accumulation and expansion of potent immunosuppressive cells. It has been demonstrated that tumor-secreted factors, such as TGF-β and IL-6, upregulate inhibitor of differentiation 1 (Id1) to redirect bone marrow-derived DC differentiation toward MDSCs that express high levels of Id1, with a reciprocal decrease in DC numbers, and Id1 overexpression results in a systemic immunosuppressive phenotype that inhibits CD8+ T cell proliferation and increases primary tumor growth and metastatic progression. Furthermore, advanced melanoma patients have increased plasma TGF-β levels and express higher levels of Id1 in myeloid peripheral blood cells [43]. Moreover, the immunosuppressive tumor microenvironment can switch a Th1 tumor-suppressive phenotype such as CD4+ “helper” T cells (which aid cytotoxic CD8+ T cells in tumor rejection) to a Th2 tumor-promoting “regulatory” phenotype (which blocks CD8+ T cell activity) [38, 44]. Other regulatory lymphocyte populations re-educated by cancer cells can be found in subsets of NKT cells and B cells, which can inhibit immune effector responses against cancer.
It is clear that tumors alter immune cells and that these alterations involve all terminally differentiated myeloid lineages and their pathologically activated immature progenitors. The phenotype and function of immune cells depend on the dynamic change of the tumor microenvironment to some degree. Indeed, these “tumor-educated” immune cells contribute to tumor immune escape by inhibiting an antitumor immune response and contribute to every stage of metastasis by promoting tumor cell invasion into the surrounding tissue, aiding intravasation and survival in the circulation, and facilitating tumor cell extravasation and persistent growth at metastatic sites. Interestingly, some of the switches in these cells are governed by common tumor-derived factors and transcriptional programs, but there are also diverse phenotypical and functional changes in different myeloid cell subpopulations. Unveiling the underlying mechanisms of immune cell phenotypic and functional switch mediated or driven by tumors will provide an opportunity for therapeutic interventions that may concomitantly normalize the abnormality of immune cells.
Immunosuppressive cells promote tumor metastasis
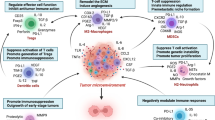
Tumor progression always ends in metastatic disease, which is responsible for >90 % of cancer mortality. Successful metastatic spread requires several steps including epithelial-to-mesenchymal transition (EMT), local invasion, intravasation into the vasculature, transit through the circulatory system, extravasation and seeding at the pre-metastatic niche, and finally, cancer cell survival and growth at the metastatic site. During each step of metastasis, immunogenic tumor cells are being exposed to the immune system and cleared by certain immune cells such as cytotoxic T cells and NK cells. Nevertheless, growing tumors and their metastatic derivatives have developed strategies to overcome these immune mechanisms, at least partially, by recruiting immunosuppressive cells and re-programming the differentiation of tumor-infiltrating immune cells into immunosuppressive and tumor-promoting cells during metastasis [45]. It is now well accepted that the interactions between tumor cells and immunosuppressive cells are complicated and dynamic, as many subtypes of immunosuppressive cells trafficking to the tumor microenvironment can promote tumor metastasis at multiple stages via different mechanisms as follows (Fig. 2).

Immunosuppressive cells in promotion of tumor metastasis. Immunosuppressive cells influence every step of tumor metastasis via multiple mechanisms. a Immunosuppressive cells such as MDSCs, TAMs, and Tregs directly suppress the cytotoxic functions of CD8+ T cells and NK cells to favor cancer cell survival during metastasis. b Immunosuppressive cells facilitate cancer cell invasion, egress from the primary site, and intravasation through secretion of cytokines such as EGF, TNF-α, CXCL12, IL-1, and IL-6. c Immunosuppressive cells play important roles in creating and populating favorable pre-metastatic niches in distant sites. d, e Immunosuppressive cells can promote tumor metastasis by facilitating the epithelial-mesenchymal transition (d) and by inducing angiogenesis through the production of pro-angiogenic factors, cytokines, and growth factors such as ANG2, VEGF-A (e)
Inhibition of antitumor immune response
In a primary tumor, cancer cells secrete chemokines and cytokines to recruit MDSCs, TAMs, Tregs, tumor-associated neutrophils (TANs), and other immunosuppressive cells to primary and secondary tumor sites. These cells directly suppress the cytotoxic functions of CD8+ T cells and NK cells, thus facilitating cancer cell egress from the primary site and survival in the circulation [46]. For example, IL-1β elicits IL-17 expression from gamma delta (γδ) T cells, resulting in systemic, granulocyte colony-stimulating factor (G-CSF)-dependent expansion, and polarization of neutrophils in mice bearing mammary tumors. These TANs acquire the ability to suppress CTL, thereby facilitating the establishment of tumor metastases [47]. Furthermore, Th17 cell recruitment to tumors seems to indirectly suppress antitumor immunity in the primary sites and to enhance metastasis. In a subcutaneous EL4 lymphoma model, IL-17 from Th17 cells promoted cancer-associated fibroblasts to secrete G-CSF so as to recruit MDSCs to the tumor site, and tumor metastasis was promoted via the immunosuppressive function of these immature myeloid cells [48]. Besides, Tregs are also required and recruited for lung metastases in experimental breast tumors [49] as well as in lymph node metastases [50, 51]. In a spontaneous metastasis model of hepatocellular carcinoma, the suppression of venous metastases was accompanied by the reduced numbers of CD4+ CD25+ Tregs in primary tumors, which suggested that Treg recruitment to the primary tumor is necessary for tumor metastasis. Treg recruitment has been shown to rely on tumor-derived CCL22 and also depends on the type of tumor model [52]. In addition to recruiting immunosuppressive cells, tumors can also regulate the function of immunosuppressive cells to strengthen metastasis. For instance, tumor-derived factors such as galectin-1 can promote systemic immunosuppression by regulating the clonal expansion and function of Tregs, probably by increasing the expression of the Treg molecule LAT (linker for activation of T cells), thus enhancing breast cancer metastasis [53]. Other immunosuppressive mechanisms of Tregs in promoting tumor metastasis include but are not limited to (a) the induction of NK cell apoptosis by secreting β-galactoside-binding protein (βGBP) or by direct cell-to-cell contact and (b) TGF-β secretion [52, 54]. Furthermore, tumor-infiltrating plasmacytoid DCs (pDCs) can increase the numbers of MDSCs and Tregs in a transplanted breast cancer tissue and, in turn, to promote tumor bone metastasis by reducing CD8+ T cell cytotoxicity [55]. Breg, which is a specific B cell population expressing CD25 and B220, may also promote tumor metastasis through suppression of immune response. Intravenous injection of 4 T1 breast cancer cells increases the number of circulating CD25+ B cells, and their depletion using a B20-specific antibody could reduce lung metastasis [49]. Tumor-evoked Breg in lung metastases can induce the conversion of CD4+ T cells into Foxp3+ Tregs via TGF-β secretion [49].
Promotion of tumor cell invasion and intravasation
Immunosuppressive cells can facilitate tumor metastasis by promoting tumor cell invasion and intravasation. They promote cancer cell invasion through the secretion of cytokines such as IL-1, TNF-α, and IL-6 that stimulate matrix metalloproteinase (MMP) expression in cancer cells via NF-κB and STAT3 signaling. For example, macrophages can stimulate cancer cell migration by secreting factors such as migration-stimulating factor. Furthermore, TAM-derived EGF activates the EGF receptor in cancer cells, which directly enhances their invasion capability, motility, and intravasation by increasing invadopodium formation and matrix degradation [56]. This TAM-mediated tumor invasion mechanism is reportedly triggered by HRGβ1 (a ligand of ErbB3 expressed by MTLn3 breast cancer cells) and CXCL12 dependent on the EGF/CSF-1 paracrine loop between cancer cells and TAMs [57]. Indeed, the “tumor-educated” macrophages promote tumor metastasis through several steps: These TAMs in primary tumors can promote cancer cell egress from the primary sites, invasion of the surrounding tissue, and intravasation and survival in the circulation; whereas metastasis-associated macrophages (MAMs) within secondary sites support cancer cell extravasation and persistent growth at metastatic sites [58].
Formation of pre-metastatic niche
Recent evidence suggests that primary tumors can prepare the distant secondary sites by promoting the formation of supportive metastatic environments, termed the pre-metastatic niche, prior to the arrival of metastatic cancer cells. Immunosuppressive cells play important roles in creating and populating favorable pre-metastatic niches as well as in facilitating the dissemination, proliferation, and colonization of metastatic cancer cells to promote further metastasis. For instance, MDSCs have frequently been shown to accumulate in the pre-metastatic niche. MDSCs can secrete IL-6, IL-23, and TGF-β to recruit Th17 cells, and these Th17 cells secrete IL-17 to promote the recruitment of MDSCs and secretion of G-CSF by cancer-associated fibroblasts, which in turn promote the immunosuppressive function of MDSCs in the pre-metastatic niche [59]. The immunosuppressive environment is one of the underlying mechanisms by which the pre-metastatic niche promotes tumor metastasis. Furthermore, Th17 and Th2 lymphocytes can program TAMs to foster a pre-metastatic niche via secretion of several cytokines including IL-6 and IL-17 [60].
Facilitating epithelial-mesenchymal transition
The morphogenetic process of epithelial-mesenchymal transition (EMT) continues to be recognized as an important mechanism that promotes tumor metastasis. Tumor-infiltrating immunosuppressive cells such as TAMs, MDSCs, and Tregs can produce large quantities of cytokines which are important inducers of EMT. TAMs are found to promote tumor metastasis by secreting TGF-β, and the JAK/STAT3 pathway is required for TGF-β-induced EMT and cancer cell migration and invasion. This cancer cell migration and invasion occur when the expression of p-Smad3 and Snail is upregulated, such that JAK/STAT3 and TGF-β/Smad signaling synergistically enhances EMT in lung carcinomas and facilitates further metastasis [61].
Induction of angiogenesis
Immunosuppressive cells also promote tumor metastasis through the induction of angiogenesis, which is dependent on recruitment of the immunosuppressive cells to tumor lesions and on the production of pro-angiogenic factors, cytokines, and growth factors such as angiopoietin 2 (ANG2) and VEGF-A. Immunosuppressive cells such as TAMs, MDSCs, TANs, and even mast cells have all been shown to contribute to angiogenesis. For example, a study in metastatic MMTV-PyMT mammary carcinomas and RIP1-Tag2 pancreatic insulinomas showed that the MRC1(+) TIE2-expressing TAMs could enhance tumor angiogenesis and metastatic dissemination of cancer cells via cell-to-cell interactions with endothelial cells; the interactions were mediated by an endothelial cell-derived ANG2-TIE2 axis [62]. These findings are consistent with the clinical association found between high TAM infiltration and poor outcome in cancer patients. Moreover, metastasis has also been shown to require close association and collaboration among cancer cells, immunosuppressive cells, and other stromal and inflammatory components of the tumor microenvironment. These critical components highlight key areas for future investigation of immunosuppressive cells’ function in the metastasis process.
Cancer immunotherapy via suppressing the suppressor
The recruitment of immunosuppressive cells to the primary tumor microenvironment as well as the pre-metastatic niche promotes tumor immune escape and is critical for the establishment of tumor metastasis. Tumor infiltration of certain immunosuppressive cell types always correlates with poor prognosis for cancer patients. For instance, one of the most prevalent mechanisms of immune escape in patients is through the suppressive activity of MDSCs arising as a consequence of the aberrant myelopoiesis that occurs in cancer [63]. This is supported by the observation that the elevated numbers of peripheral MDSCs positively correlate poor prognosis of cancer patients with advanced disease and lack of therapeutic efficacy [64]. In some cancer types including breast cancer and hepatocellular carcinoma, the increased numbers of Tregs correlate with reduced overall survival of cancer patients [65]. It was shown in clinic that DCs could induce a pre-metastatic niche during lymph node metastasis (LNM) through COX-2/EP3-dependent induction of SDF-1, suggesting that inhibition of this signaling axis may be an effective strategy to suppress pre-metastatic niche formation and LNM [51]. Therefore, suppressing the suppressor may be a promising immunotherapy strategy to block tumor immune escape and metastasis by reversing the immunosuppression and triggering an effective antitumor immune response at the different stages of cancer progression. Various cancer therapies that attempt to block mechanisms of tumor immune escape and metastasis by targeting the immunosuppressive cells have shown promise. Some clinically available drugs that act against cancers not only have the ability to eliminate tumor cells but can also block the tumor-promoting activity of immunosuppressive cells in tumor microenvironments and consequently favor antitumor immune responses [66]. These findings explain their rapid applications in cancer immunotherapy. The therapeutic approaches that are based on suppressing the suppressor strategy can be summed up as: (a) reversing the inhibitory pathways or blocking immune checkpoint to enhance effective antitumor T cell responses; (b) suppressing the function of immunosuppressive cells by targeting these cells directly or reducing the number of immunosuppressive cells recruited via tumors; (c) targeting the specific molecular and cellular components involved in immunosuppressive tumor microenvironment, instead of targeting the tumor cells.
Immune enhancement by immune checkpoint blockade
Many immunosuppressive cells and cancer cells express inhibitory molecules PD-1, PD-L1, and CTLA-4, and some activated immune cells also express other inhibitory molecules such as the lymphocyte-activated gene-3 (LAG-3, CD223) [67, 68], T cell immunoglobulin and mucin-containing protein 3 (TIM-3) [69], the V-domain Ig suppressor of T cell activation (VISTA) [70], and B and T lymphocyte attenuator (BTLA) [71]. These inhibitory signals (known as immune checkpoints) downregulate T cell activities and prevent cancer cells from being eliminated by effector T cells. Therefore, targeting these checkpoints may boost antitumor immune responses [72]. Immune enhancement strategies that target tumor immune escape mechanisms are currently in clinical trials, including antibody blockade or neutralization of these immune checkpoints [73].
Indeed, antibodies against CTLA-4 or PD-1 have been clinically tested and applied [11, 72, 74–76] and have resulted in prolonged overall survival of cancer patients. For example, application of PD-1-blocking monoclonal antibody nivolumab could inhibit tumor immune evasion and have substantial therapeutic effect in patients with Hodgkin’s lymphoma [11]. Two humanized antibodies against CTLA-4, ipilimumab, and tremelimumab have undergone Phase III clinical trials in patients with advanced melanoma and have shown overall survival benefits [75]. Furthermore, the recent success of the clinical combination of ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) [11, 77] may have revolutionized the treatment strategy for melanoma patients. The use of this combination of antibodies to block immune checkpoints has shown responses in almost half of metastatic melanoma patients for whom conventional therapies have failed [76]. In 2011, ipilimumab was approved by the US Food and Drug Administration (FDA) because of its benefit to long-term survival of patients with metastatic melanoma. In addition, nivolumab was also approved by FDA in March 2015 for patients with advanced or metastatic non-small cell lung cancer, showing an improvement in overall survival in patients as compared to docetaxel chemotherapy [74]. Many other antibodies against PD-1 or PD-L1 have also been developed and clinically tested in clinical trials for advanced solid tumors, including renal cell carcinoma, prostate cancer, and colon cancer [72].
Now, multiple additional negative checkpoint regulators that restrict the ability of T cells to effectively attack tumors have been discovered. These new negative checkpoints include TIM-3, BTLA, LAG-3, VISTA, and the T cell immunoreceptor with Ig and ITIM domains (TIGIT) [78], which are possibly new and promising targets for cancer therapeutic manipulation. TIM-3 was initially identified as a specific marker of fully differentiated IFN-γ producing CD4 T helper 1 (Th1) and cytotoxic CD8 T cells, and TIM3 is also highly expressed on Tregs, monocytes, macrophages, and DCs. TIM-3, which regulates these immune cells’ function in several ways, emerged as a key regulator of dysfunctional or exhausted CD8+ T cells arising in cancer [69]. In acute myelogenous leukemia, blocking TIM-3 ex vivo or in vivo increases the functionality of exhausted T cells, synergizes with PD-1 blockade and inhibits tumor growth more significantly [79]. Besides, intratumoral Foxp3+ Tregs that co-express PD-1 and TIM-3 are highly suppressive and comprise a specialized subset of tissue Tregs. Depleting Treg by TIM-3 blockade also has a synergistic effect on tumor growth inhibition [80]. In addition to TIM-3 blockade, treatments are also being developed to enable blockade of other inhibitory pathways. For example, BTLA and PD-1 are co-expressed on tumor-specific CD8+ T cells in melanoma patients and promote tumor immune escape. BTLA can be downregulated by vaccination with peptide and CpG oligodeoxynucleotides, which reduces BTLA’s immunosuppressive function mediated by herpes virus entry mediator, and partially improve the efficiency of immunotherapy [81]. Another immune-inhibitory molecule LAG-3 can be expressed, together with PD-1, on tumor-infiltrating lymphocytes to promote tumor immune escape. Blockade of LAG-3 and PD-1 can synergistically inhibit tumor growth and enhance antitumor immunity in the established Sa1N fibrosarcoma and MC38 colorectal adenocarcinoma-bearing mice, thus representing a promising combinatorial immunotherapy strategy for cancer [82].
A better understanding of how to unleash T cell responses by targeting immune checkpoints has led to clinical successes in the field of cancer immunotherapy. These new findings will inspire us to rethink the interaction between host immune system and tumor and identify the predictive biomarkers for effective targeting of the immunosuppressive molecules, more importantly, empower the approaches of cancer immunotherapy based on immune checkpoints.
Targeting immunosuppressive cells
As mentioned above, immunosuppressive cells play important roles in promoting tumor immune escape and metastasis. Many targeted therapeutic agents have been demonstrated to function as immune modulators, and some are being explored as inhibitors of immunosuppressive cells such as all trans retinoic acid (ATRA) and sunitinib (both inhibit MDSCs) [66]. Identification of the immunosuppressive cells in specific tumor types or within the tumor microenvironment and inhibition of their immunosuppressive function by targeting these cells directly or indirectly will provide new opportunities to the design of cancer immunotherapy.
Multiple researches targeting immunosuppressive cells are ongoing and may guide the future cancer immunotherapy strategies. More recently, a new therapeutic peptide called peptibody (a kind of peptide-Fc fusion protein) has been generated through adapting a competitive peptide phage display platform to identify candidate peptides that specifically bind MDSCs. These peptibodies successfully depleted blood, splenic MDSCs, and intratumoral MDSCs in multiple syngeneic tumor models, without affecting myeloid-lineage cell types. Importantly, peptibodies deplete both granulocytic and monocytic MDSC subsets and can retard tumor growth in vivo with limited off-target effects [83]. Besides, a type of tyrosine kinase inhibitor, sunitinib, is being used in renal cell carcinoma to induce apoptosis of MDSC or to suppress Treg function [66]. Inhibition of Tregs and MDSCs by inactivation of the p110δ isoform of phosphoinositide 3-kinase (PI3Kδ) can induce immune-mediated rejection of various types of tumors, in addition to their remarkable therapeutic efficacy in some human leukemia. This p110δ inactivation in Tregs can unleash CD8+ cytotoxic T cells and break tumor-induced immune tolerance, thus inducing tumor regression [84, 85]. Consistent with these findings, immunosuppressive cells can be depleted in other ways. For instance, CD4+ Tregs are susceptible to the ATP-binding cassette-transporter B1 (ABCB1)-substrate cyclophosphamide (CPA). The ABCB1-substrate CPA was cytotoxic for Treg cells at a 100-fold lower dose than for non-regulatory counterparts [86]. In this way, new immunotherapeutic strategies can use CPA to boost antitumor immunity by selectively depleting Treg cells. Moreover, the CC chemokine receptor 4 (CCR4) that was highly expressed on Tregs can be targeted by an anti-CCR4 antibody mogamulizumab, resulting in the reduction of the numbers of CCR4+ malignant T cells and CCR4+ Treg cells in cutaneous T cell lymphoma [87]. Such exciting pre-clinical data are indicative of the great potential of cancer immunotherapy.
The successes of these basic and translational studies imply that it is possible to inhibit tumor immune escape and prevent metastasis by enhancing antitumor immunity through blockade of immunosuppressive cells or eliminating immunosuppressive mechanisms that are induced via immunosuppressive cells or tumor cells. Many pre-clinical and clinical cancer immunotherapy approaches, especially immune checkpoint blockade, have been extensively reviewed [72, 73, 88], and a number of promising approaches are soon to enter the clinic. However, there are still some problems and challenges worthy of our future attention. For example, we do not exactly know which cell types induce CTLA-4- or PD-1-mediated immunosuppression in different kinds of tumors because multiple immunosuppressive cell populations within the tumor microenvironment, such as TAMs, express both CTLA-4 ligand and PD-L1. In addition, blocking inhibitory signals such as CTLA-4 on all cells that express this receptor may result in increased autoimmune disease. Therefore, the particular roles of immunosuppressive cells in tumor immune escape and metastasis call for further research on identification of cellular and molecular suppressors for immune response against cancer and the development of new cancer treatment strategies.
Conclusions and perspectives
Immunosuppressive cells play important roles in promoting tumor immune escape and metastasis via different mechanisms. A large number of basic and clinical advancements bring us a few steps closer to our goal of inhibiting tumor progression and metastasis. Nonetheless, many unanswered questions and new challenges still remain. To date, cancer immunotherapy is not well accepted as a standard therapy for cancers because there is a wide chasm between impressive pre-clinical results and limited clinical results. In fact, the mechanisms for tumor immune escape and metastasis are always implicated in the ineffectiveness of cancer immunotherapy and even in chemotherapy or radiation therapy. Several important questions in this field need to be answered: (1) What is the relationship among the different immunosuppressive cells, host stromal cells, and cancer cells during tumor progression? (2) How might the tumor microenvironment be educated and sculpted by cancer cells and how might this result in myeloid cell diversity even within the same tissue? (3) Unlike other therapies that target cancer cells, treatments aiming to inhibit immunosuppression target the immune system itself. How can we most effectively boost the antitumor effects by inhibiting the function of immunosuppressive cells at the tumor site without disrupting the remaining immune system or concomitantly inducing life-threatening autoimmunity? (4) Are we able to develop the personalized or precision cancer immunotherapy designed specifically for an individual cancer patient and the individual tumor type?
Multiple forms of immunotherapies are being designed and explored to answer these questions, including adoptive cellular immunotherapy that re-activates antitumor immune response through adoptive transfer of T cells or NK cells. Moreover, suppressing the suppressors may be another promising strategy for active cancer immunotherapy. We present potential research directions and clinical strategies in cancer immunotherapy in the remaining sections.
Suppressing the immunosuppressive cells
The identification of immunosuppressive cells in certain cancers and their roles in tumor progression will pave the way for development of therapies by inactivation of these cells. Targeting key immunosuppressive cells such as MDSCs, TAMs, and Tregs at different stages are promising options that can be approached via four specific strategies: (1) preventing the differentiation of myeloid cells into mature immunosuppressive cells; (2) blocking immunosuppressive cell recruitment, expansion, and activation; (3) inhibiting the suppressive function of immunosuppressive cells; and (4) depleting intratumoral immunosuppressive cells.
For example, immunosuppressive cells can be therapeutically targeted with drugs that prevent their proliferation in the bone marrow [2] and avoid their mobilization and recruitment to the tumor microenvironment or pre-metastatic organs. Designing strategies that aim to re-educate the immunosuppressive activity of MDSCs is another attractive approach because MDSCs are composed of mixed subpopulations of cells with varying maturity and plasticity and can also differentiate into multiple immunosuppressive cell types. Other therapeutic approaches that induce immunosuppressive cell apoptosis, maintain their immature state, and inhibit their immunosuppression functions may also take effect. As such, targeting Tregs via a CD25-blocking monoclonal antibody has been beneficial in improving immunotherapy responses in patients with metastatic breast cancer [15, 89]. The remarkable suppressive effects observed in the above-mentioned studies strongly suggest that an immunosuppressive cell-targeting therapy may be an effective strategy. Thus, a more detailed understanding of the molecular mechanisms governing immunosuppressive cell recruitment, activation, and function in pre-clinical models of specific cancer types may guide more rational designs for future trials.
Re-programming tumor microenvironment
Another possible therapeutic strategy is withdrawal of the tumor microenvironment that supports tumor cells’ immune escape and metastasis. Tumor microenvironment alterations and immunosuppressive cell accumulation can subvert the therapeutic efficacy of traditional anticancer therapy and ultimately abrogate patient outcome [90]. Accumulating evidence has also shown overwhelming heterogeneity at every level in cancer cells [91], such that new cancer therapy strategies targeting the tumor microenvironment could be an even more promising option compared to targeting cancer cells directly. Consequently, targeting different components of the tumor microenvironment or eliminating its immunosuppressive phenotype may effectively prevent cancer cell immune escape and metastasis. Moreover, given the paradoxical capacity of the tumor microenvironment to both promote and impair tumor progress, therapeutic strategies that manipulate and re-educate the tumor microenvironment from pro-tumor to antitumor behavior is well worth exploring. Some immunotherapies based on this re-programming approach have generated much excitement in the clinic recently [92, 93] and inspires us to investigate the global tumor microenvironment more comprehensively.
Reversing immunosuppression
The success of current clinical trials testing CTLA-4 and PD-1 blockade stimulate our interest in new approaches that block other potential effectors of immunosuppression, including the soluble factors (such as IDO and TGF-β) and cellular mediators (such as Bregs and Th17 cells) of the tumor process. Little is known about the recruitment of some other types of immune cells to the developing tumor microenvironment or their contribution to tumor immune escape and metastasis. This is an emerging research field, and future studies are required to investigate the role of immune cells as well as their interactions with cancer cells. Better understanding of the polarization of myeloid cells to a tumor-promoting phenotype and their potential in suppressing cellular mediators of the antitumor immune responses will be critical in inhibiting tumor immune escape and metastatic progression.
Personalized and precision immunotherapy of cancer
Tumors from different individuals display unique characteristics depending on the individual’s genetic makeup, the cellular components of the host immune system, and the tumor stage at the time of diagnosis. Certain types of tumors may predominantly recruit specific types of immunosuppressive cells, via distinct chemoattractants, to promote tumor progression. For example, a high number of intratumoral TAMs always correlates with poor prognosis in breast cancer patients, which is recruited through the high expression of the monocyte chemoattractant CCL2, and these macrophages can accelerate breast cancer metastasis by promoting angiogenesis [94, 95]; besides, rhabdomyosarcoma-derived CXCR2 ligands induce robust accumulation of MDSCs in the tumor bed to mediate local immunosuppression and escape immune defenses [19], while hepatocellular carcinoma predominantly recruits Tregs by secreting CCL22 [52]. Therefore, adaptation of personalized and precision therapies targeting unique tumor signatures should be one of the goals. On one hand, personalized treatment selection will require analysis of the immunosuppressive cell type, the entire tumor microenvironment, and cancer types to determine specific therapies. On the other hand, precise therapeutic approaches to cancer could assess the cancer cell and immunosuppressive cell profile in individual cancers, so that the targeting drug can be precisely tailored to maximize the response.
Combination therapy of cancer
Because tumors use multiple mechanisms to escape antitumor immune responses and the metastasis mechanisms may be interactive and mutually compensatory, a combination strategy rather than a single approach will be more efficient to overcome tumor immune escape and metastasis. Optimal results will require combination of different immune checkpoint blockade with important signal pathway inhibitors and other established therapies. For instance, only a population of melanoma patients responds to the treatment which blocks immune-inhibitory receptors because of the absence of a spontaneous antitumor T cell response in tumor sites. A more recent study revealed a correlation between activation of the WNT/β-catenin signaling pathway and absence of a T cell gene expression signature by molecular analysis of human metastatic melanoma samples. Tumor-intrinsic active β-catenin signaling results in T cell exclusion and resistance to anti-PD-L1/anti-CTLA-4 monoclonal antibody therapy and also suppresses the recruitment of CD103+ DCs by impairing production of the chemokine CCL4 [96]. These findings suggest that targeting of β-catenin could be combined with immune checkpoint blockade to enforce antitumor immune response in melanoma patients. Another recent clinical trial reported that treatment with an anti-CTLA4 antibody plus radiation in a subset of patients with metastatic melanoma results in major tumor regressions, and this effect was also reproduced in mouse models, but the efficacy can be inhibited by PD-L1 on tumor cells. Later combination of radiation with both anti-CTLA-4 and anti-PD-L1 antibodies simultaneously could increase the treatment responses in clinical trials [88]. Furthermore, when trafficking of MDSCs to tumor was inhibited by CXCR2 deficiency or when the patients had undergone anti-CXCR2 monoclonal antibody therapy, delayed anti-PD1 treatment could induce significant antitumor effects [19].
Taken together, these studies suggest that (1) combining therapies that target mechanisms of tumor immune escape (for example, inhibiting immunosuppressive cell types via antibody) with activation of normal immune T cell function may provide more benefits for patients; (2) optimal results will require a combination of various immune checkpoint inhibitors as well as a combination of these inhibitors with other traditional therapies; (3) blockade of an inhibitory pathway in combination with a simultaneous agonistic signal through a stimulatory pathway, such as ICOS, OX40, and CD137(4-1BB) [72, 97]; (4) concomitant targeting of cancer cells and their local tumor microenvironment as well as targeting the pre-metastatic niche may have robust consequences for suppressing tumor progression and metastasis. Therefore, the most successful cancer therapy strategies may be to simultaneously target multiple immune escape pathways and metastasis mechanisms or to combine them with more conventional treatments such as chemotherapy and radiation.
A better understanding of the mechanisms by which immunosuppressive cells promote tumor immune escape and metastasis is promising enough as to suggest a breakthrough in the area of cancer immunotherapy and, more importantly, will provide a scientific rationale for clinical trials to improve clinical outcomes for patients.
References
Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14:1014–1022
Kitamura T, Qian BZ, Pollard JW (2015) Immune cell promotion of metastasis. Nat Rev Immunol 15:73–86
McAllister SS, Weinberg RA (2014) The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 16:717–727
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19:1423–1437
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ (2011) Natural innate and adaptive immunity to cancer. Annu Rev Immunol 29:235–271
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Gomez Perdiguero E, Geissmann F (2014) Cancer immunology. Identifying the infiltrators. Science 344:801–802
Kim R, Emi M, Tanabe K (2007) Cancer immunoediting from immune surveillance to immune escape. Immunology 121:1–14
Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331:1565–1570
Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HM et al. (2015) Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol DOI 10.1016/j.semcancer.2015.03.004.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ et al (2015) PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 372:311–319
Mittal D, Gubin MM, Schreiber RD, Smyth MJ (2014) New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol 27:16–25
Colonna M (2013) Immunology: an innate regulatory cell. Nature 498:42–43
Wolf D, Wolf AM (2015) CCR 20th Anniversary Commentary: from regulatory T cells to checkpoint monoclonal antibodies-immuno-oncology advances clinical cancer research. Clin Cancer Res 21:2657–2659
von Boehmer H, Daniel C (2013) Therapeutic opportunities for manipulating T(Reg) cells in autoimmunity and cancer. Nat Rev Drug Discov 12:51–63
Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L et al (2011) Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475:226–230
Han Y, Yang Y, Chen Z, Jiang Z, Gu Y, Liu Y, Xu S, Lin C, Pan Z, Zhou W et al (2014) Human hepatocellular carcinoma-infiltrating CD4(+)CD69(+)Foxp3(−) regulatory T cell suppresses T cell response via membrane-bound TGF-beta1. J Mol Med (Berl) 92:539–550
Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, Ochoa AC, Cui Y, Del Valle L, Rodriguez PC (2014) The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity 41:389–401
Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, Kaplan RN, Mackall CL (2014) Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med 6:237ra267
Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, Tracey KJ, Ostrand-Rosenberg S (2014) HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res 74:5723–5733
Qian BZ, Pollard JW (2010) Macrophage diversity enhances tumor progression and metastasis. Cell 141:39–51
Noy R, Pollard JW (2014) Tumor-associated macrophages: from mechanisms to therapy. Immunity 41:49–61
Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM (2014) Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26:623–637
Liu Y, Cao X (2015) Intratumoral dendritic cells in the anti-tumor immune response. Cell Mol Immunol 12:387–390
Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO (2014) The cellular and molecular origin of tumor-associated macrophages. Science 344:921–925
Liu Y, Cao X (2015) The origin and function of tumor-associated macrophages. Cell Mol Immunol 12:1–4
Alderton GK (2014) Tumour immunology: turning macrophages on, off and on again. Nat Rev Immunol 14:136–137
Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, Delaney A, Jones SJ, Iqbal J, Weisenburger DD et al (2010) Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med 362:875–885
Han Y, Chen Z, Yang Y, Jiang Z, Gu Y, Liu Y, Lin C, Pan Z, Yu Y, Jiang M et al (2014) Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 59:567–579
Qian C, Qian L, Yu Y, An H, Guo Z, Han Y, Chen Y, Bai Y, Wang Q, Cao X (2013) Fas signal promotes the immunosuppressive function of regulatory dendritic cells via the ERK/beta-catenin pathway. J Biol Chem 288:27825–27835
Wu C, Zhang Y, Jiang Y, Wang Q, Long Y, Wang C, Cao X, Chen G (2013) Apoptotic cell administration enhances pancreatic islet engraftment by induction of regulatory T cells and tolerogenic dendritic cells. Cell Mol Immunol 10:393–402
Marvel D, Gabrilovich DI (2015) Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest DOI 10.1172/JCI80005: 1–9.
Upadhyay R, Hammerich L, Peng P, Brown B, Merad M, Brody JD (2015) Lymphoma: immune evasion strategies. Cancers (Basel) 7:736–762
Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA (2010) Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 328:749–752
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268
Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG, Porta C, Anselmo A, Tartari S, Doni A et al (2015) RORC1 regulates tumor-promoting “emergency” granulo-monocytopoiesis. Cancer Cell 28:253–269
Torroella-Kouri M, Silvera R, Rodriguez D, Caso R, Shatry A, Opiela S, Ilkovitch D, Schwendener RA, Iragavarapu-Charyulu V, Cardentey Y et al (2009) Identification of a subpopulation of macrophages in mammary tumor-bearing mice that are neither M1 nor M2 and are less differentiated. Cancer Res 69:4800–4809
Galli SJ, Borregaard N, Wynn TA (2011) Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol 12:1035–1044
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16:183–194
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P (2010) The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol 10:554–567
Dumitriu IE, Dunbar DR, Howie SE, Sethi T, Gregory CD (2009) Human dendritic cells produce TGF-beta 1 under the influence of lung carcinoma cells and prime the differentiation of CD4 + CD25 + Foxp3+ regulatory T cells. J Immunol 182:2795–2807
Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, Allen PM (2009) Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res 69:3086–3094
Papaspyridonos M, Matei I, Huang Y, do Rosario Andre M, Brazier-Mitouart H, Waite JC, Chan AS, Kalter J, Ramos I, Wu Q et al (2015) Id1 suppresses anti-tumour immune responses and promotes tumour progression by impairing myeloid cell maturation. Nat Commun 6:6840
Sato K (2014) Helper T cell diversity and plasticity. Circ J 78:2843–2844
Yaguchi T, Sumimoto H, Kudo-Saito C, Tsukamoto N, Ueda R, Iwata-Kajihara T, Nishio H, Kawamura N, Kawakami Y (2011) The mechanisms of cancer immunoescape and development of overcoming strategies. Int J Hematol 93:294–300
Smith HA, Kang Y (2013) The metastasis-promoting roles of tumor-associated immune cells. J Mol Med (Berl) 91:411–429
Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJ, Ciampricotti M, Hawinkels LJ, Jonkers J et al (2015) IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522:345–348
Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV et al (2013) An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med 19:1114–1123
Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, Malchinkhuu E, Wersto RP, Biragyn A (2011) Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res 71:3505–3515
Lee HE, Park DJ, Kim WH, Kim HH, Lee HS (2011) High FOXP3+ regulatory T-cell density in the sentinel lymph node is associated with downstream non-sentinel lymph-node metastasis in gastric cancer. Br J Cancer 105:413–419
Ogawa F, Amano H, Eshima K, Ito Y, Matsui Y, Hosono K, Kitasato H, Iyoda A, Iwabuchi K, Kumagai Y et al (2014) Prostanoid induces premetastatic niche in regional lymph nodes. J Clin Invest 124:4882–4894
Yang P, Li QJ, Feng Y, Zhang Y, Markowitz GJ, Ning S, Deng Y, Zhao J, Jiang S, Yuan Y et al (2012) TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 22:291–303
Dalotto-Moreno T, Croci DO, Cerliani JP, Martinez-Allo VC, Dergan-Dylon S, Mendez-Huergo SP, Stupirski JC, Mazal D, Osinaga E, Toscano MA et al (2013) Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res 73:1107–1117
Zou W (2006) Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 6:295–307
Sawant A, Hensel JA, Chanda D, Harris BA, Siegal GP, Maheshwari A, Ponnazhagan S (2012) Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. J Immunol 189:4258–4265
Zhou ZN, Sharma VP, Beaty BT, Roh-Johnson M, Peterson EA, Van Rooijen N, Kenny PA, Wiley HS, Condeelis JS, Segall JE (2014) Autocrine HBEGF expression promotes breast cancer intravasation, metastasis and macrophage-independent invasion in vivo. Oncogene 33:3784–3793
Hernandez L, Smirnova T, Kedrin D, Wyckoff J, Zhu L, Stanley ER, Cox D, Muller WJ, Pollard JW, Van Rooijen N et al (2009) The EGF/CSF-1 paracrine invasion loop can be triggered by heregulin beta1 and CXCL12. Cancer Res 69:3221–3227
Valastyan S, Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147:275–292
Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, Katsanis E, Larmonier N (2014) Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res 74:104–118
DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM (2009) CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16:91–102
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu Z, Zhao J, Zhang HT (2014) JAK/STAT3 signaling is required for TGF-beta-induced epithelial-mesenchymal transition in lung cancer cells. Int J Oncol 44:1643–1651
Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L et al (2011) Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19:512–526
Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI (2001) Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol 166:678–689
Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ (2009) Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother 58:49–59
Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH (2006) Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 24:5373–5380
Kim SJ, Ha GH, Kim SH, Kang CD (2014) Combination of cancer immunotherapy with clinically available drugs that can block immunosuppressive cells. Immunol Invest 43:517–534
Castelli C, Triebel F, Rivoltini L, Camisaschi C (2014) Lymphocyte activation gene-3 (LAG-3, CD223) in plasmacytoid dendritic cells (pDCs): a molecular target for the restoration of active antitumor immunity. Oncoimmunology 3, e967146
Goldberg MV, Drake CG (2011) LAG-3 in cancer immunotherapy. Curr Top Microbiol Immunol 344:269–278
Japp AS, Kursunel MA, Meier S, Malzer JN, Li X, Rahman NA, Jekabsons W, Krause H, Magheli A, Klopf C et al. (2015) Dysfunction of PSA-specific CD8 T cells in prostate cancer patients correlates with CD38 and Tim-3 expression. Cancer Immunol Immunother DOI 10.1007/s00262-015-1752-y
Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, Noelle RJ, Wang L (2014) VISTA regulates the development of protective antitumor immunity. Cancer Res 74:1933–1944
Gertner-Dardenne J, Fauriat C, Orlanducci F, Thibult ML, Pastor S, Fitzgibbon J, Bouabdallah R, Xerri L, Olive D (2013) The co-receptor BTLA negatively regulates human Vgamma9Vdelta2 T-cell proliferation: a potential way of immune escape for lymphoma cells. Blood 122:922–931
Sharma P, Allison JP (2015) The future of immune checkpoint therapy. Science 348:56–61
Korman AJ, Peggs KS, Allison JP (2006) Checkpoint blockade in cancer immunotherapy. Adv Immunol 90:297–339
Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E et al (2015) Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 373:123–135
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K et al (2013) Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369:122–133
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454
Le Mercier I, Lines JL, Noelle RJ (2015) Beyond CTLA-4 and PD-1, the generation Z of negative checkpoint regulators. Front Immunol 6:418
Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, Murphy WJ, Azuma M, Anderson AC, Kuchroo VK et al (2011) Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 117:4501–4510
Sakuishi K, Ngiow SF, Sullivan JM, Teng MW, Kuchroo VK, Smyth MJ, Anderson AC (2013) TIM3FOXP3 regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology 2, e23849
Derre L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, Michielin O, Olive D, Speiser DE (2010) BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest 120:157–167
Woo SRTM, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S et al (2012) Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72:917–927
Qin H, Lerman B, Sakamaki I, Wei G, Cha SC, Rao SS, Qian J, Hailemichael Y, Nurieva R, Dwyer KC et al (2014) Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med 20:676–681
Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H et al (2014) Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510:407–411
Killock D (2014) Immunotherapy: PI3Kdelta inhibition lifts the breaks on antitumour immunity. Nat Rev Clin Oncol 11:442
Dimeloe S, Frick C, Fischer M, Gubser PM, Razik L, Bantug GR, Ravon M, Langenkamp A, Hess C (2014) Human regulatory T cells lack the cyclophosphamide-extruding transporter ABCB1 and are more susceptible to cyclophosphamide-induced apoptosis. Eur J Immunol 44:3614–3620
Ni X, Langridge T, Duvic M (2015) Depletion of regulatory T cells by targeting CC chemokine receptor type 4 with mogamulizumab. Oncoimmunology 4, e1011524
Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM et al (2015) Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520:373–377
Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, Colligon TA, Trosko JA, Leinbach LI, Pletcher CH et al (2012) CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med 4:134ra162
De Palma M, Lewis CE (2013) Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 23:277–286
Fang H, Declerck YA (2013) Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res 73:4965–4977
Restifo NP, Dudley ME, Rosenberg SA (2012) Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12:269–281
Vanneman M, Dranoff G (2012) Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer 12:237–251
Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, Bentires-Alj M (2014) Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515:130–133
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475:222–225
Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523:231–235
Kohrt HE, Colevas AD, Houot R, Weiskopf K, Goldstein MJ, Lund P, Mueller A, Sagiv-Barfi I, Marabelle A, Lira R et al (2014) Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest 124:2668–2682
Acknowledgments
This work was supported by grants from National Key Basic Research Program of China (2014CB542100, 2011CB965202).
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Liu, Y., Cao, X. Immunosuppressive cells in tumor immune escape and metastasis. J Mol Med 94, 509–522 (2016). https://doi.org/10.1007/s00109-015-1376-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-015-1376-x