
Abstract
The transcription factor Stat3 is an activator of systemic inflammatory genes. Two isoforms of Stat3 are generated by alternative splicing, Stat3α and Stat3β. The β isoform lacks the transactivation domain but retains other functions, including dimerization and DNA binding. Stat3β-deficient mice exhibit elevated expression of systemic inflammatory genes and are hyperresponsive to lipopolysaccharide, suggesting that Stat3β functions predominantly as a suppressor of systemic inflammation. To test whether Stat3β deficiency would provoke pathologic effects associated with chronic inflammation, we asked whether selective removal of Stat3β would exacerbate the development of atherosclerosis in apolipoprotein E-deficient mice. In apoE−/−Stat3β−/− mice atherosclerotic plaque formation was significantly enhanced relative to apoE−/−Stat3β+/+ controls. The ability of Stat3β deficiency to promote atherosclerosis was more pronounced in female mice, but could be unmasked in males by feeding a high fat diet. Infiltrating macrophages were not increased in aortas of apoE−/−Stat3β−/− mice. In contrast, the proportion of pro-inflammatory TH17 cells was significantly elevated in aortic infiltrates from apoE−/−Stat3β−/− mice, relative to paired apoE−/−Stat3β+/+ littermates. These observations indicate that Stat3β can suppress pathologic sequelae associated with chronic inflammation. Our findings further suggest that in Stat3β-deficient mice the unopposed action of Stat3α may enhance atherogenesis in part by promoting differentiation of TH17 cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atherosclerosis is the major underlying cause of cardiovascular disease (CVD), including myocardial infarction and stroke. Atherosclerotic plaque is an accumulation of lipid particles, inflammatory cells and smooth muscle cells in the arterial subendothelium. The development of atherosclerotic plaque (reviewed in [1]) begins with subendothelial retention and oxidation of low density lipoprotein (LDL). In response to oxidized LDL and other mediators, endothelial cells express adhesion molecules and chemoattractant cytokines. Subsequently, monocytes are recruited into the subendothelium, where they differentiate into macrophages and internalize modified LDL to become foam cells. Smooth muscle cells are recruited to the plaque and induced to proliferate in response to factors produced by macrophages and the foam cells derived from them.
Epidemiologic and genetic evidence has identified chronic inflammation as an etiologic factor in the development of atherosclerosis, and inflammatory markers can serve as predictors of future cardiovascular events. Such markers include IL-6 [2], C-reactive protein [3–5], sICAM1 [6], and CD40L [7]. Autoimmune disease, which is accompanied by systemic inflammation, has also been linked to CVD; patients with systemic lupus erythematosis, for example, are at increased risk for atherosclerosis [8]. Alterations in genes associated with inflammation can modulate the risk of atherosclerosis in humans. Polymorphisms in IL-6 have been associated with atherosclerotic vascular disease [9], as have polymorphisms in genes encoding inflammatory adhesion molecules such as VCAM-1, ICAM-1 and PECAM-1 [10].
Inflammation is also implicated in atherogenesis by the identification of genes that modify the atherosclerotic phenotypes of apoE-deficient or LDL receptor-deficient mice. Mice lacking apoE have elevated plasma cholesterol and develop atherosclerotic lesions while maintained on a normal diet; mice lacking the LDL receptor develop atherosclerotic lesions when fed a diet rich in fat and cholesterol [11]. Genetic lesions that impair macrophage differentiation or recruitment of monocytes and macrophages attenuate atherosclerosis in mouse models [12], as does ablation of the gene encoding MyD88, which transduces pathogenic signals in dendritic cells and macrophages [13]. Correspondingly, deletion of the gene encoding IL-1β, an inflammatory mediator produced by innate sentinel cells, is also associated with reduced severity of atherosclerosis in the apoE-deficient model [14].
The systemic inflammatory component of innate immunity, termed the acute phase response (APR), is initiated upon stimulation of monocytes, macrophages and dendritic cells through toll-like receptors, which trigger the production of IL-1β, tumor necrosis factor-α (TNFα) and IL-6. These mediators, in turn, modulate expression of APR genes in the liver and other sites [15]. IL-6 exerts its effects on APR genes primarily through the latent transcription factor Stat3. Mammals produce two alternatively spliced isoforms of Stat3, Stat3α and Stat3β. Stat3β lacks the 55 carboxy-terminal amino acid residues of Stat3α that span the transactivation domain [16–18]. Because Stat3β retains dimerization and DNA-binding functions, it can behave in a dominant-negative fashion [16]. In some contexts, however, Stat3β promotes gene expression through interactions with transcription factors such as c-Jun [17, 19]. Mice in which Stat3β is selectively ablated exhibit impaired recovery from endotoxic shock. In such mice a subset of endotoxin-inducible genes is chronically overexpressed and hyperresponsive to induction, consistent with a role for Stat3β as a global suppressor of systemic inflammation [18].
Because Stat3β exerts its suppressive effects largely on non-classical APR genes, the role of Stat3β in protection against the consequences of sustained systemic inflammation has remained unclear. We have now tested this role by examining the effect of Stat3β deficiency on the development of atherosclerosis in the apoE-deficient mouse model. In mice doubly deficient in apoE and Stat3β, atherosclerotic plaque formation is accelerated relative to mice deficient in apoE alone. This effect is accompanied by increased representation of pro-inflammatory TH17 lymphocytes among aortic-infiltrating T cells, consistent with the dependence of TH17 differentiation on Stat3 and the ability of Stat3β to oppose Stat3 function. Our observations indicate that Stat3β is protective against atherogenesis in a mouse model and suggest that Stat3β exerts its protective effect, at least in part, by opposing production of TH17 cells.
Materials and methods
Animals
C57BL/6 J apoE−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The Stat3β− allele [18] was backcrossed onto a C57BL/6 J background for nine generations and bred with C57BL/6 J apoE−/− mice; apoE−/−Stat3β+/+ and apoE−/−Stat3β−/− mice were obtained by interbreeding apoE−/−Stat3β+/− animals. Mice were housed under pathogen-free conditions. Upon weaning at 3 weeks mice were maintained on normal (5.67 % fat, 0 % cholesterol; Teklad #7012) or high fat (20 % fat, 1.5 % cholesterol, 0.5 % sodium cholate; Teklad #96354) diets. Animals were maintained in accordance with the guidelines of the Johns Hopkins Animal Care and Use Committee.
Measurement of triglyceride and cholesterol levels
Mice were fasted overnight and sacrificed. Blood was drawn from the inferior vena cava. Total serum cholesterol and triglyceride levels were measured using commercial reagents (Thermo Electron).
Measurement of atherosclerotic lesions
For en face measurement of aortic lesions, mice were euthanized and aortas were perfused with ice-cold PBS. Aortas were excised from the ascending aorta to the iliac bifurcation and fixed in 4 % paraformaldehyde. After removal of adventitial and adipose tissue, aortas were incised longitudinally, stained with Sudan IV and pinned to wax plates. Images were captured with a Zeiss AxioCam camera connected to a Zeiss Stemi 2000-C dissection microscope. The contour of the aorta was defined by manual tracing. Plaque contours were defined by an automated procedure in which a representative plaque was sampled to define a color threshold which was then applied to the en face aortic image to define the margins of plaques satisfying the threshold criterion. Total aortic area and plaque areas were determined using ImageJ (http://imagej.nih.gov/ij/) by summing the pixels within the aortic contour and plaque contours, respectively, and scaling to metric area.
For measurement of aortic root lesions, hearts were sectioned perpendicular to the axis of the ascending aorta; upper halves were embedded in OCT and frozen at −80 C. Serial sections (10 μm) were stained with Oil Red O (PolyScientific) and counterstained with hematoxylin. Images were captured with a Zeiss AxioCam camera connected to a Zeiss Axioskop2. Total lesion area was quantified by manual tracing of intimal lesions in four aortic root sections spaced at 100 μm intervals.
Preparation of primary mouse peritoneal macrophages
Resting peritoneal macrophages were harvested by peritoneal lavage with Dulbecco’s modified Eagle medium supplemented with L-glutamine, penicillin, streptomycin and 10 % heat-inactivated FBS. Peritoneal cells were plated at 37 °C in the presence of 5 % CO2 in culture medium overnight and non-adherent cells were removed.
Quantitative RT-PCR
Total RNA was reverse transcribed using random hexamer primers. The resulting cDNA was quantified by real-time PCR in the presence of forward and reverse primers at 150 nM each and SYBR Green (BioRad) in an iCycler thermal cycler (BioRad). Relative gene expression was determined by ∆∆Ct approximation. The expression level of each gene (represented as the Ct value) was first normalized to that of a reference gene (HPRT1) [∆Ct = Ct(gene of interest) − Ct(HPRT1)]. The linear fold difference in expression of a given gene between two samples was then determined by taking the difference between the corresponding ∆Ct values (∆∆Ct) and computing 2−(∆∆Ct).
Oligonucleotides
Sequences of forward and reverse PCR primers are provided below.
-
Stat3α: forward, 5′-GCGCTTCAGCGAGAGCAGCAAAG-3′
-
Reverse, 5′-CATCGGCAGGTCAATGGTATTGC-3′
-
Stat3β: forward, 5′-GCGCTTCAGCGAGAGCAGCAAAG-3′
-
reverse, 5′-GTTATTTCCAAACTGCATCAATGAATGG-3′
-
HPRT1: forward, 5′-CAGTCCCAGCGTCGTGATTA-3′
-
reverse, 5′-CATGACATCTCGAGCAAGTCTTTC-3′
-
Thy1: forward, 5′-CAACTTCACCACCAAGGAATG-3′
-
reverse, 5′-TCTGAACCAGCAGGCTTATG-3′
-
RORγt: forward, 5′-CCGCTGAGAGGGCTTCAC-3′
-
reverse, 5′-TGCAGGAGTAGGCCACATTACA-3′
-
F4/80: forward, 5′-AGGCTTTGTCTTGAATGGCT-3′
-
reverse, 5′-GCCCTCCTCCACTAGATTCA-3′
Immunohistochemistry
Frozen heart sections, embedded in OCT, were fixed in acetone, rehydrated in PBS and treated with Protein Block (Dako) for 15 min. For detection of macrophages, sections were incubated with rat anti-mouse MOMA-2 antibody (AbD Serotec) at 2.5 μg/ml for 12 h at 4 °C followed by incubation with rhodamine conjugated goat anti-rat IgG (Jackson ImmunoResearch) at 15 μg/ml for 1 h at room temperature.
Immunological detection of Stat3 protein
Aortic smooth muscle cells (SMCs) were prepared as described [20]. Whole cell lysates from SMCs and aorta were fractionated by SDS polyacrylamide gel electrophoresis and analyzed by immunoblotting with an anti-Stat3 antibody (Cell Signaling Technology).
Analysis of gene expression in peritoneal macrophages
After starvation for 24 h in DMEM containing 0.5 % FBS, peritoneal macrophages were stimulated in the presence or absence of IL6 (40 ng/ml) and sIL6R (50 ng/ml) for 12 h. RNA was harvested (Rneasy, Qiagen) and reverse transcription was performed using a mixture of random hexamer and oligo-dT primers. A PCR array (Qiagen) was used to assess expression of 84 genes associated with atherosclerosis. The list of genes analyzed is available online at http://www.sabiosciences.com/rt_pcr_product/HTML/PAMM-038Z.html. Data analysis was performed using ΔΔCt-based fold-change calculations. Expression was normalized to that of HPRT1.
Transcripts exhibiting differential signals in the screening assay above were retested. Peritoneal macrophages were harvested from four to seven mice, plated for 12 h, washed with PBS and serum starved for 24 h in DMEM supplemented with 0.5 % FBS. Macrophages were then incubated for 12 h in the presence or absence of IL6 (40 ng/ml) and sIL6R (50 ng/ml). RNA was harvested and reverse transcription was primed with random hexamers. The resulting cDNA was amplified with Sybr Green master mix (BioRad) with 150 nM of gene-specific primers. Data analysis was performed using ΔΔCt-based fold-change calculations. Expression was normalized to that of HPRT1. Primer sequences are as follows:
-
HPRT1: forward, 5′-CAGTCCCAGCGTCGTGATTA-3′
-
reverse, 5′-CATGACATCTCGAGCAAGTCTTTC-3′
-
IL1β: forward, 5′-CACTACAGGCTCCGAGATGA-3′
-
reverse, 5′-TTTGTCGTTGCTTGGTTCTC-3′
-
SerpinB2: forward, 5′-CCATAGTTCTCCTCGGTGCT-3′
-
reverse, 5′-GCCACTGAAGTTCTCTGGGT-3′
-
IFNγ: forward, 5′-AGCTCTTCCTCATGGCTGTT-3′
-
reverse, 5′-TTTGCCAGTTCCTCCAGATA-3′
-
VCAM: forward, 5′-AGAACCCAGGTGGAGGTCTA-3′
-
reverse, 5′-ATCTCCAGATGGTCAAAGGG-3′
-
SerpinE1: forward, 5′-TGGTGAAACAGGTGGACTTC-3′
-
reverse, 5′-CCCTTGGCCAGTAAGTCATT-3′
-
MMP3: forward, 5′-GGAGATGCTCACTTTGACGA-3′
-
reverse, 5′-TGAGCAGCAACCAGGAATAG-3′
-
CCL5: forward, 5′-TCTTGCAGTCGTGTTTGTCA-3′
-
reverse, 5′-CCACTTCTTCTCTGGGTTGG-3′
Assay of cytokine production by CD4 T cells
Splenocytes were plated at 2 × 106/ml in the presence or absence of stimuli as defined below. At 20 h cells were treated with brefeldinA (Becton, Dickinson). After 4 h cells were stained for CD4, fixed and permeabilized in the presence of antibodies against IFNγ and IL17, tagged with APC and PE, respectively. Expression of IFNγ and IL17 in the CD4-gated population was detected by flow cytometry.
Some assays were performed in round-bottomed wells in the presence of soluble anti-CD3 (1 μg/ml), anti-CD3 (1 μg/ml) and anti-CD28 (1 μg/ml), anti-CD3 (1 μg/ml) and IL6 (40 ng/ml), or IL6 alone (40 ng/ml). Other assays were performed in flat-bottomed wells in the presence of plate-bound anti-CD3 (1 μg/ml), anti-CD3 (1 μg/ml) and soluble anti-CD28 (1 μg/ml), anti-CD3 (1 μg/ml) and IL6 (40 ng/ml), or IL6 alone (40 ng/ml). As a positive control splenocytes were incubated for 4 h with PMA (20 ng/ml) and ionomycin (2 μg/ml) in the presence of brefeldin A and assayed as above.
Proliferation assay
Splenocytes were loaded with 5 μM CFSE for 5 min in the presence of PBS supplemented with 5 % PBS. CFSE-loaded cells were stimulated as above. At 5 days after stimulation CFSE dilution was assessed in the CD4+, 7AAD− population.
Apoptosis assay
At 24 h after stimulation splenocytes were stained with anti-CD4, annexinV and 7AAD. AnnexinV and 7AAD were assayed in the CD4-gated population.
Statistical analysis
Except where indicated, statistical significance was determined by the two-tailed Student’s t test. Differences were considered significant if P ≤ 0.05.
Results
Derivation of apoE−/−Stat3β−/− mice
Stat3β, which is encoded by an alternatively spliced mRNA isoform, lacks 55 amino acid residues found at the carboxyl terminus of Stat3α, including the transactivation domain (Fig. 1a). Stat3β-deficient mice were previously generated by targeted mutation of the alternative splice acceptor site [18]. Evidence that Stat3β is a negative modulator of systemic inflammation [18] led us to predict that Stat3β deficiency might exacerbate disease phenotypes associated with chronic inflammation.

Absence of Stat3β transcript and protein in apoE−/−Stat3β−/− mice. a Generation of Stat3 isoforms by alternative splicing. Middle line, exons 22 through 24 of the Stat3 locus (filled boxes). Alternative splicing patterns generate mRNA encoding Stat3α (above) or Stat3β (below). Filled boxes translated regions, open boxes untranslated regions. Top and bottom, Stat3α and Stat3β protein isoforms, respectively. DBD DNA-binding domain, SH2 SH2 domain, TAD transactivation domain, Y Tyr705, S Ser727. b Absence of Stat3β mRNA and protein in Stat3β−/− mice. Total RNA and protein were prepared from aorta, peritoneal macrophages and aortic smooth muscle cells (SMC) from apoE−/−Stat3β+/+ and apoE−/−Stat3β−/− mice. Stat3α (top panel) and Stat3β (middle panel) transcripts were detected by isoform-specific RT-PCR. Stat3α and Stat3β proteins (bottom panel) were assayed by immunoblotting with a pan-specific anti-Stat3 antibody. Positions of Stat3α and Stat3β are indicated by arrows. a Whole cell lysate of NIH3T3 cells transfected with plasmids encoding Stat3α and Stat3β (NIH3T3 + Stat3α,β) was used as a positive control
We chose to test this in the apoE-deficient mouse model for atherosclerosis. The Stat3β allele was backcrossed onto a C57BL/6 background and then introduced into the apoE-deficient, C57BL/6 strain. Stat3α and Stat3β mRNA isoforms were assayed in aorta, aortic smooth muscle cells and peritoneal macrophages of apoE−/−Stat3β+/+ and apoE−/−Stat3β−/− mice. Stat3β mRNA was undetectable in samples from apoE−/−Stat3β −/− mice, while Stat3α mRNA was present at similar amounts in samples from mice of both genotypes. Similarly, Stat3β protein was selectively absent from tissues of animals doubly deficient in apoE and Stat3β (Fig. 1b).
Association of Stat3β deficiency with increased atherosclerotic plaque area in the aortic trunks of apoE-deficient female mice
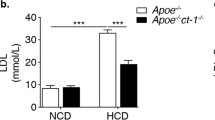
After weaning at 3 weeks, apoE−/−Stat3β+/+ and apoE−/−Stat3β−/− mice were maintained on a normal diet for 10 or 20 weeks. At 13 weeks of age the absence of Stat3β had no apparent effect on serum cholesterol, triglycerides or weight in male or female mice maintained on a normal diet (Table 1). By 23 weeks of age, female apoE−/−Stat3β−/− mice exhibited a decrease in weight and a small increase in serum triglycerides, relative to apoE−/−Stat3β+/+ controls (Table 2). These differences were not observed in 23-week-old male mice (Table 2).
We next examined the effect of Stat3β deficiency on atherosclerotic plaque development. Aortic trunks, extending from the ascending aorta to the iliac bifurcation, were harvested at 13 or 23 weeks from apoE−/−Stat3β+/+ and apoE−/−Stat3β−/− mice, maintained on normal diet. Aortas were mounted en face and plaque surface area was measured. At 13 weeks of age, apoE−/−Stat3β+/+ and apoE−/−Stat3β−/− female mice did not differ significantly with respect to atherosclerotic plaque area (Fig. S1, A–D). At 23 weeks, apoE−/−Stat3β−/− females had smaller aortic areas (49.32 ± 2.15 mm2) than their apoE−/−Stat3β+/+ counterparts (54.53 ± 5.01 mm2) (Fig. 2a, b), consistent with the difference between these groups with respect to weight. Total aortic plaque area was substantially increased in apoE−/−Stat3β−/− mice (1.17 ± 0.49 mm2) relative to apoE−/−Stat3β+/+ animals (0.58 ± 0.35 mm2) (Fig. 2c). This difference was even more striking when total plaque area was normalized to total aortic area (2.40 ± 1.06 % and 1.07 ± 0.65 % for apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice, respectively) (Fig. 2d).

Association of Stat3β deficiency with enhanced atherogenesis in 23-week-old apoE−/− female mice that had been maintained for 20 weeks on normal diet. a Representative en face aortic preparations, stained with Sudan IV, from apoE−/−Stat3β+/+ (left) or apoE−/−Stat3β−/− (right) female mice. b Total luminal surface area of aortas from apoE−/−Stat3β+/+ or apoE−/−Stat3β−/− mice. c Total plaque area of aortas from apoE−/−Stat3β+/+ or apoE−/−Stat3β−/− mice. d Total plaque area, normalized to total luminal surface area, of aortas from apoE−/−Stat3β+/+ or apoE−/−Stat3β−/− mice. e Mean aortic root lesion area from 23-week-old apoE−/−Stat3β+/+ (left) or apoE−/−Stat3β−/− (right) female mice maintained for 20 weeks on normal diet. For each mouse, lesion areas were determined for four serial sections, each 100 μm apart; the average of these values represents the mean atherosclerotic lesion area. f Representative aortic root sections, stained with Oil red O, from apoE−/−Stat3β+/+ (left) or apoE−/−Stat3β−/− (right) mice. For b–e, circles represent values obtained for individual mice; for each group, the mean and S.D. are indicated by long and short horizontal bars, respectively. *P < 0.05, **P < 0.01
Association of Stat3β deficiency with increased atherosclerotic plaque area is maintained in advanced aortic root lesions
Atherosclerotic plaque does not appear synchronously along the aortic trunk, but tends to form first at sites of high curvature, such as branching points and the aortic root [21–23]. To determine whether the pro-atherogenic effect of Stat3β deficiency was also evident in these more advanced lesions, we assessed plaque development in the aortic root. Hearts of 13- or 23-week-old female mice that had been maintained on a normal diet were harvested, frozen, and sectioned. Atherosclerotic plaque was detected by staining with Oil red O and lesion areas were quantified by digital image analysis. At 13 weeks, atherosclerotic lesions in the aortic root were of similar size in apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ females (Fig. S1E, F). By 23 weeks, however, atherosclerotic lesions in the aortic root were significantly larger in apoE−/−Stat3β−/− females than in their apoE−/−Stat3β+/+ counterparts (Fig. 2e, f), indicating that the exacerbation of atherosclerotic plaque formation by Stat3β deficiency is evident even at more advanced stages of plaque development.
Atheroprotection by Stat3β under high fat dietary conditions
The development of atherosclerotic plaque in murine models is sensitive to dietary differences, developing more rapidly in animals maintained on a high fat diet [21, 24]. We maintained cohorts of apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ female mice on a diet containing 20 % fat, 1.5 % cholesterol and 0.5 % sodium cholate for 4 weeks after weaning at 3 weeks and total aortic plaque area was then measured. Like 23-week-old apoE−/−Stat3β−/− females, 7-week-old apoE−/−Stat3β−/− females that had been maintained on a high fat diet for 4 weeks had smaller aortic areas than apoE−/−Stat3β+/+ controls (42.18 ± 2.45 mm2 compared to 45.62 ± 3.13 mm2) (Fig. 3a, b). Moreover, total aortic plaque area in 7-week-old apoE−/−Stat3β−/− females maintained on high fat diet was significantly increased (0.512 ± 0.32 mm2) relative to that of apoE−/−Stat3β+/+ control mice (0.29 ± 0.096 mm2) (Fig. 3c, d). These observations suggested that in female, apoE-deficient mice, Stat3β is also atheroprotective under high fat dietary conditions.

Association of Stat3β deficiency with increased plaque burden in 7-week-old apoE−/− female mice that had been maintained for 4 weeks on high fat diet. a Representative en face aortic preparations, stained with Sudan IV, from apoE−/−Stat3β+/+ (left) or apoE−/−Stat3β−/− (right) female mice. b Total luminal surface area of aortas from apoE−/−Stat3β+/+ or apoE−/−Stat3β−/− mice. c Total plaque area of aortas from apoE−/−Stat3β+/+ or apoE−/−Stat3β−/− mice. d Total plaque area, normalized to total luminal surface area, of aortas from apoE−/−Stat3β+/+ or apoE−/−Stat3β−/− mice. For b–d, circles represent values obtained for individual mice; for each group, the mean and S.D. are indicated by long and short horizontal bars, respectively. *P < 0.05, **P < 0.01
Exacerbation of plaque development by Stat3β deficiency is unmasked in male mice by high fat diet
Unlike their female counterparts, 23-week-old male apoE−/−Stat3β−/− mice that had been maintained on a normal diet for 20 weeks did not exhibit an increase in total aortic plaque area, relative to apoE−/−Stat3β+/+ controls (Fig. 4). This was of interest because among apoE-deficient mice, males exhibit smaller atherosclerotic lesions than females, although this difference decreases with age [25]. We therefore reasoned that an effect of Stat3β deficiency on atherosclerotic plaque development in male mice might be unmasked under conditions of accelerated atherogenesis, such as maintenance on a high fat diet. Male apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice were maintained on a high fat diet for 10 weeks after weaning. No significant differences in weight, serum triglycerides or cholesterol were observed between the two genotypes (Table 1). Among 13-week-old mice maintained on a 10-week high fat diet, both genotypes exhibited increased aortic plaque area, relative to animals maintained on a normal diet for 20 weeks. The apoE−/−Stat3β−/− males exhibited a significantly larger total plaque area (7.10 ± 3.57 mm2) than their apoE−/−Stat3β+/+ counterparts (4.13 ± 1.49 mm2) (Fig. 5), indicating that Stat3β is protective against atherosclerosis in both females and males.

No significant increase in plaque burden of male apoE−/−Stat3β−/− mice maintained on normal diet for 20 weeks. a Representative en face aortic preparations, stained with Sudan IV, from apoE−/−Stat3β+/+ (left) or apoE−/−Stat3β−/− (right) male mice. b Total luminal surface area of aortas from apoE−/−Stat3β+/+ or apoE−/−Stat3β−/− males. c Total plaque area of aortas from apoE−/−Stat3β+/+ or apoE−/−Stat3β−/− males. d Total plaque area, normalized to total luminal surface area, of aortas from apoE−/−Stat3β+/+ or apoE−/−Stat3β−/− males. For b–d, circles represent values obtained for individual mice; for each group, the mean and S.D. are indicated by long and short horizontal bars, respectively

Association of Stat3β deficiency with increased plaque burden in 13-week-old apoE−/− male mice that had been maintained for 10 weeks on high fat diet. a Representative en face aortic preparations, stained with Sudan IV, from apoE−/−Stat3β+/+ (left) or apoE−/−Stat3β−/− (right) male mice maintained on high fat diet for 10 weeks. Total luminal surface area (b), total plaque area (c) and total normalized plaque area (d) are displayed as in Fig. 4. *P < 0.05
No increased representation of macrophages in lesions of Stat3β-deficient mice
Macrophages and their derivatives, foam cells, are a major component of atherosclerotic plaques. Because Stat3 is implicated in the recruitment of macrophages to an inflammatory microenvironment [26], we asked whether macrophages were overrepresented in atherosclerotic plaque or aortic infiltrates from apoE−/−Stat3β−/− mice, relative to apoE−/−Stat3β+/+ controls. Aortic root sections from 23-week-old female mice, maintained on normal diet, were immunohistochemically stained with an antibody to the monocyte- and macrophage-specific antigen MOMA-2 [27]. For each section, the total plaque area and the area stained by MOMA-2 were measured. The ratio of MOMA-2-positive area to total plaque area did not differ significantly between apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice (Fig. S3). To obtain an independent assessment of macrophage infiltration in aortas of apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice we developed a quantitative assay based on the detection of transcripts for F4/80, a macrophage-restricted receptor [28]. In reconstitution experiments in which macrophage RNA was combined in varying amounts with RNA from fibroblastoid cells, the PCR signal cycle threshold (Ct) for a particular sample was linearly related to the log percentage of macrophage RNA present over a range of 0.01 through 100 % (Fig. S4A). Next, whole aortic RNA from 23-week-old wild-type or apoE−/−Stat3β+/+ mice, maintained on normal diet, was assayed for transcripts encoding F4/80 and these values were normalized to those obtained for transcripts encoding HPRT1. The relative amounts of F4/80 RNA were four- to sixfold greater in samples from apoE−/−Stat3β+/+ animals than in those from wild-type mice (Fig. S4B), consistent with the association of macrophage-containing atherosclerotic lesions with apoE deficiency. Finally, we quantified F4/80 transcripts, normalized to transcripts for HPRT1, in aortic RNA samples from apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ littermate pairs. No systematic difference in the normalized expression levels was observed (Fig. S5). Taken together these observations suggest that while apoE deficiency is associated with a relative increase in the number of macrophages in aortic infiltrates, this number is not further increased when apoE deficiency is combined with Stat3β deficiency.
Expression of inflammatory markers in resting and IL6-stimulated peritoneal macrophages
Although the available evidence indicated that apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice did not differ with respect to the number of macrophages present in atherosclerotic lesions, it remained possible that macrophages from Stat3β-deficient animals were hyperresponsive to inflammatory stimuli. We previously observed that activated macrophages from Stat3β-deficient and wild-type littermates produce similar amounts of TNFα, IL-1β, IL-6, and IL-10 in response to LPS [18]. We proceeded to ask whether stimulation with IL-6, which signals through Stat3, elicits differential expression of inflammatory markers in macrophages from apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice.
Pooled peritoneal macrophages from four male mice of the apoE−/−Stat3β−/− or apoE−/−Stat3β+/+ genotype were assayed for expression of transcripts encoding 84 inflammatory markers before and after stimulation with IL-6 for 12 h. In this initial screen, several transcripts, including those encoding IFN-γ, IL-1β, VCAM-1, SerpinB2 and SerpinE1, exhibited modest overaccumulation (greater than twofold) in resting macrophages from apoE−/−Stat3β−/− mice relative to apoE−/−Stat3β+/+ controls; one of these transcripts, IL-1β, showed an apparent overinduction of about twofold in the Stat3β-deficient animals (data not shown). Nonetheless, when resting or IL6-stimulated macrophages from individual apoE−/−Stat3β−/− or apoE−/−Stat3β+/+ mice were assayed for accumulation of these transcripts, no significant difference between the two genotypes was observed (data not shown). We were therefore unable to detect a robust difference between peritoneal macrophages from apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice with respect to the resting levels of inflammatory transcripts or their induction by IL-6.
Increase in IL17 production by CD4+splenocytes from apoE−/−Stat3β−/− mice
The presence of T lymphocytes in atherosclerotic lesions, albeit in fewer numbers than macrophages, has suggested one or more roles in atherogenesis. CD4+ splenocytes from apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice did not differ with respect to proliferation or apoptosis in response to anti-CD3, anti-CD3 and anti-CD28, or anti-CD3 and IL6 (data not shown). As the helper T cell subset TH17 is associated with inflammation [29] and TH17 cells have been implicated in atherogenesis [30, 31], we assessed CD4+ splenocytes for production of IL17 and IFNγ in response to anti-CD3, anti-CD3 and anti-CD28, or anti-CD3 and IL6. In the presence of soluble anti-CD3 and anti-CD28, CD4+ splenocytes from apoE−/−Stat3β−/− mice produced significantly more IL17 than those from apoE−/−Stat3β+/+ littermates (Fig. S6). In the presence of soluble anti-CD3 or soluble anti-CD3 plus IL6 the numbers of IL17-producing CD4+ splenocytes were also greater in samples from apoE−/−Stat3β−/− mice, but these differences were not statistically significant. No consistent difference was seen between the two genotypes with respect to production of IFNγ in samples treated with soluble antibody (Fig. S6), or with respect to either cytokine in samples treated with plate-bound antibody (data not shown).
Increase in RORγt expression in ApoE−/−Stat3β−/− aortas compared to that of ApoE−/−
In the mouse, differentiation to the TH17 subset is promoted by the transcription factor RORγt, whose induction by cytokines such as IL-6 is dependent on Stat3 [32–34]. The role of Stat3 in driving TH17 differentiation and the negative modulatory function of Stat3β suggested that RORγt positive TH17 cells might be overrepresented in atherosclerotic lesions of Stat3β-deficient mice.
We therefore determined the relative abundance of TH17 cells in aortic T cell infiltrates from, 23-week-old apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice that had been maintained on normal diet for 20 weeks. To minimize the effects of uncontrolled environmental stimuli we analyzed paired littermates of differing genotype, as others have done [35, 36]. We used expression of the nuclear receptor RORγt as a marker for TH17 cells. While RORγt is also expressed by other cell types in the thymus and in secondary lymphoid organs, outside of these tissues it is highly specific for TH17 cells and can therefore be used as a surrogate marker for this effector subset. Aortic RNA was assayed for the presence of transcripts encoding the pan T cell marker Thy1 or RORγt by real-time PCR as we performed for quantification of F4/80. Comparison of 12 apoE−/−Stat3β−/− mice with paired apoE−/−Stat3β+/+ littermates using the Wilcoxon paired rank order test revealed a significantly higher proportion of RORγt transcripts, when normalized to Thy1 transcript levels, in the apoE−/−Stat3β−/− animals, relative to their apoE−/−Stat3β+/+ counterparts (P = 0.0122) (Fig. 6). These observations suggest that selective ablation of Stat3β is associated with increased aortic infiltration by TH17 cells in apoE-deficient mice.

Increased representation of TH17-specific transcripts in aortas from apoE−/−Stat3β−/− mice. Total aortic RNA from paired apoE−/−Stat3β+/+ and apoE−/−Stat3β−/− littermates was assayed by quantitative PCR for the presence of transcripts encoding the TH17-specific marker RORγt or the pan T cell marker Thy1. The ratio of RORγt transcripts to Thy1 transcripts in apoE−/−Stat3β+/+ (filled squares) or apoE−/−Stat3β−/− (filled triangles) mice is indicated; lines connect the values obtained from paired littermates. Statistical significance was determined by the paired Wilcoxon rank test
Discussion
Stat3β acts predominantly as a suppressor of the hepatic acute phase response to LPS [18]. Moreover, the ability of Stat3β to oppose the induction of TNF and IL-6 by Stat3α [37] is consistent with a role for Stat3β as a negative mediator of systemic inflammation. These findings have suggested that Stat3β might in some settings protect against pathological effects of chronic inflammation, a prediction which we have tested here. Consistent with hypothesis, we observed that in the apoE-deficient mouse, selective ablation of Stat3β exacerbates the formation of atherosclerotic plaque. To our knowledge, these results provide the first evidence that Stat3β can mitigate the course of an inflammatory disease.
At 23 weeks, the average weight of female apoE−/−Stat3β−/− mice was lower than that of apoE−/−Stat3β+/+ control animals. Several Stat3 activators, including leptin, effect a decrease in weight [38]. Accordingly, ablation of Stat3 in mature adipocytes is associated with increased weight [39]. The relative decrease in the weight of apoE−/−Stat3β−/− mice is therefore consistent with the ability of Stat3β to oppose the actions of Stat3α. The association of Stat3β deficiency with lower body weight is not necessarily related to its association with increased atherogenesis. Weight loss as a result of dietary restriction is associated with diminished plaque formation [40], while weight loss resulting from administration of leptin is associated with enhanced atherogenesis [41]. The effects of Stat3β deficiency described in the present study are consistent with the divergent effects of leptin, a Stat3 activator, on atherogenesis and body weight.
In humans, elevated triglyceride is associated with an increased risk of CAD; upon adjustment for high-density lipoprotein, which is inversely related to triglyceride, a significant association does not persist [42]. Nonetheless, triglyceride levels may be a synergistic risk factor for CAD in humans [43] and in several mouse models, triglyceride levels are positively correlated with atherogenesis [44]. At 23 weeks, an increase in serum triglyceride levels was observed in apoE−/−Stat3β−/− female mice maintained on normal diet, relative to apoE−/−Stat3β+/+ controls. The relationship between triglyceride levels and atherosclerosis in our cohorts, however, is equivocal, because in male apoE−/−Stat3β−/− and apoE−/−Stat3β+/+ mice maintained on a high fat diet we observed no significant difference in triglyceride levels, despite a significant enhancement of atherogenesis in the Stat3β-deficient group.
Because the Stat3β and Stat3α coding sequences overlap, it was not feasible to undertake selective conditional ablation of Stat3β to identify cell types responsible for the pro-atherogenic phenotype. Using reciprocal bone marrow transfer, we attempted to determine whether the pro-atherogenic effects of Stat3β deficiency could be attributed to cells of hematopoietic or non-hematopoietic origin; all donor-recipient pairs, however, exhibited elevated plaque burdens, consistent with the ability of irradiation to accelerate atherogenesis [45].
Th17 cells constitute a specific T helper subset with strong pro-inflammatory capacity [29]. The nuclear receptor RORγt, whose expression is positively regulated by Stat3, plays a central role in the differentiation of TH17 cells: RORγt is required for induction of IL-17 in response to TGF-β and IL-6, and in RORγt-deficient mice TH17 cells are absent from the lamina propria, where they constitutively reside in wild-type animals [46]. In the mouse, ablation of Stat3 signaling impairs induction of RORγt and blocks differentiation of TH17 cells [47, 48]. We observed a relative increase in the number of IL17-producing CD4+ splenocytes in apoE−/−Stat3β−/− mice. Moreover, the abundance of RORγt transcripts, relative to Thy1, was significantly greater in aortic infiltrates from apoE−/−Stat3β−/− mice than in infiltrates from paired apoE−/−Stat3β+/+ littermates. This observation strongly suggests that TH17 cells are overrepresented in aortic T cell infiltrates of apoE−/−Stat3β−/− mice. The relative increase in infiltrating TH17 cells is consistent with a mechanism in which Stat3β-deficiency permits Stat3α to function unopposed, thereby promoting Th17 differentiation and mobilization.
Although several lines of evidence suggest that TH17 cells, and IL-17 in particular, promote atherogenesis [30, 31, 49–51], in a discordant study ablation of the Stat3 inhibitor SOCS3 was associated with elevated production of IL-17 and IL-10 as well as increased atherosclerotic plaque size [52]. Interpretation of this result, however, is not straightforward, as IL-10 can suppress atherogenesis [53]. Our observations are consistent with a pro-atherogenic role for IL-17, as they suggest that a skewing toward TH17 differentiation may contribute to the enhanced atherogenesis observed in Stat3β-deficient animals. Nonetheless, Stat3 is a global regulator of systemic inflammation and in that setting Stat3β deficiency is known to affect expression of more than 100 genes [15, 18]. Thus, it seems likely that the effect of Stat3β deficiency on atherosclerotic plaque development reflects the dysregulation of multiple transcriptional targets, of which those regulating TH17 differentiation may be a subset.
References
Hansson GK, Libby P (2006) The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol 6:508–519
Ridker PM, Rifai N, Stampfer MJ, Hennekens CH (2000) Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 101:1767–1772
Koenig W, Sund M, Frohlich M, Fischer HG, Lowel H, Doring A, Hutchinson WL, Pepys MB (1999) C-Reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation 99:237–242
Speidl WS, Graf S, Hornykewycz S, Nikfardjam M, Niessner A, Zorn G, Wojta J, Huber K (2002) High-sensitivity C-reactive protein in the prediction of coronary events in patients with premature coronary artery disease. Am Heart J 144:449–455
Ridker PM, Hennekens CH, Buring JE, Rifai N (2000) C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 342:836–843
Ridker PM, Hennekens CH, Roitman-Johnson B, Stampfer MJ, Allen J (1998) Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet 351:88–92
Schonbeck U, Varo N, Libby P, Buring J, Ridker PM (2001) Soluble CD40L and cardiovascular risk in women. Circulation 104:2266–2268
Asanuma Y, Oeser A, Shintani AK, Turner E, Olsen N, Fazio S, Linton MF, Raggi P, Stein CM (2003) Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med 349:2407–2415
Riikola A, Sipila K, Kahonen M, Jula A, Nieminen MS, Moilanen L, Kesaniemi YA, Lehtimaki T, Hulkkonen J (2009) Interleukin-6 promoter polymorphism and cardiovascular risk factors: the Health 2000 Survey. Atherosclerosis 207:466–470
Blankenberg S, Barbaux S, Tiret L (2003) Adhesion molecules and atherosclerosis. Atherosclerosis 170:191–203
Breslow JL (1996) Mouse models of atherosclerosis. Science 272:685–688
Moore KJ, Tabas I (2011) Macrophages in the pathogenesis of atherosclerosis. Cell 145:341–355
Bjorkbacka H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, Lee MA, Means T, Halmen K, Luster AD, Golenbock DT et al (2004) Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med 10:416–421
Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M (2003) Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 23:656–660
Desiderio S, Yoo JY (2003) A genome-wide analysis of the acute-phase response and its regulation by Stat3beta. Ann N Y Acad Sci 987:280–284
Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JA, Lammers JW, Koenderman L, de Groot RP (1996) STAT3beta, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J Biol Chem 271:13221–13227
Schaefer TS, Sanders LK, Nathans D (1995) Cooperative transcriptional activity of Jun and Stat3 beta, a short form of Stat3. Proc Natl Acad Sci U S A 92:9097–9101
Yoo JY, Huso DL, Nathans D, Desiderio S (2002) Specific ablation of Stat3beta distorts the pattern of Stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell 108:331–344
Schaefer TS, Sanders LK, Park OK, Nathans D (1997) Functional differences between Stat3alpha and Stat3beta. Mol Cell Biol 17:5307–5316
Ray JL, Leach R, Herbert JM, Benson M (2001) Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci 23:185–188
Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R (1994) ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 14:133–140
Reddick RL, Zhang SH, Maeda N (1994) Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb 14:141–147
Tangirala RK, Rubin EM, Palinski W (1995) Quantitation of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J Lipid Res 36:2320–2328
Ma Y, Wang W, Zhang J, Lu Y, Wu W, Yan H, Wang Y (2012) Hyperlipidemia and atherosclerotic lesion development in LDLR-deficient mice on a long-term high-fat diet. PLoS One 7:e35835
Caligiuri G, Nicoletti A, Zhou X, Tornberg I, Hansson GK (1999) Effects of sex and age on atherosclerosis and autoimmunity in apoE-deficient mice. Atherosclerosis 145:301–308
Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, Yoshimura A, Reindl W, Sipos B, Akira S et al (2011) Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 19:456–469
Kraal G, Rep M, Janse M (1987) Macrophages in T and B cell compartments and other tissue macrophages recognized by monoclonal antibody MOMA-2. An immunohistochemical study. Scand J Immunol 26:653–661
Austyn JM, Gordon S (1981) F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol 11:805–815
Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 Cells. Annu Rev Immunol 27:485–517
Gao Q, Jiang Y, Ma T, Zhu F, Gao F, Zhang P, Guo C, Wang Q, Wang X, Ma C et al (2010) A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J Immunol 185:5820–5827
Smith E, Prasad KM, Butcher M, Dobrian A, Kolls JK, Ley K, Galkina E (2010) Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation 121:1746–1755
Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, Takahashi H, Sun HW, Kanno Y, Powrie F et al (2010) Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 32:605–615
Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR (2007) IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8:967–974
Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS et al (2008) Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29:44–56
Avagyan S, Aguilo F, Kamezaki K, Snoeck HW (2011) Quantitative trait mapping reveals a regulatory axis involving peroxisome proliferator-activated receptors, PRDM16, transforming growth factor-beta2 and FLT3 in hematopoiesis. Blood 118:6078–6086
Chang X, Gao JX, Jiang Q, Wen J, Seifers N, Su L, Godfrey VL, Zuo T, Zheng P, Liu Y (2005) The Scurfy mutation of FoxP3 in the thymus stroma leads to defective thymopoiesis. J Exp Med 202:1141–1151
Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, Murray-Tait V, Chiarle R, Poli V (2004) The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol 5:401–409
Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E et al (2003) STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 421:856–859
Cernkovich ER, Deng J, Bond MC, Combs TP, Harp JB (2008) Adipose-specific disruption of signal transducer and activator of transcription 3 increases body weight and adiposity. Endocrinology 149:1581–1590
Lyngdorf LG, Gregersen S, Daugherty A, Falk E (2003) Paradoxical reduction of atherosclerosis in apoE-deficient mice with obesity-related type 2 diabetes. Cardiovasc Res 59:854–862
Chiba T, Shinozaki S, Nakazawa T, Kawakami A, Ai M, Kaneko E, Kitagawa M, Kondo K, Chait A, Shimokado K (2008) Leptin deficiency suppresses progression of atherosclerosis in apoE-deficient mice. Atherosclerosis 196:68–75
Austin MA (1989) Plasma triglyceride as a risk factor for coronary heart disease. The epidemiologic evidence and beyond. Am J Epidemiol 129:249–259
Gotto AM Jr (1998) Triglyceride as a risk factor for coronary artery disease. Am J Cardiol 82:22Q–25Q
Zadelaar S, Kleemann R, Verschuren L, de Vries-Van, der Weij J, van der Hoorn J, Princen HM, Kooistra T (2007) Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol 27:1706–1721
Stewart FA, Heeneman S, Te Poele J, Kruse J, Russell NS, Gijbels M, Daemen M (2006) Ionizing radiation accelerates the development of atherosclerotic lesions in ApoE−/− mice and predisposes to an inflammatory plaque phenotype prone to hemorrhage. Am J Pathol 168:649–658
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH et al (2007) Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol 179:4313–4317
Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C (2007) STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 282:9358–9363
Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, Wang Y, Bockler D, Katus HA, Dengler TJ (2009) Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol 183:8167–8175
van Es T, van Puijvelde GH, Ramos OH, Segers FM, Joosten LA, van den Berg WB, Michon IM, de Vos P, van Berkel TJ, Kuiper J (2009) Attenuated atherosclerosis upon IL-17R signaling disruption in LDLr deficient mice. Biochem Biophys Res Commun 388:261–265
Butcher MJ, Gjurich BN, Phillips T, Galkina EV (2012) The IL-17A/IL-17RA axis plays a proatherogenic role via the regulation of aortic myeloid cell recruitment. Circ Res 110:675–687
Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, Esposito B, Perez N, Yasukawa H, Van Snick J et al (2009) Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med 206:2067–2077
Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF, Soubrier F, Esposito B, Duez H, Fievet C et al (1999) Protective role of interleukin-10 in atherosclerosis. Circ Res 85:e17–e24
Acknowledgements and disclosure statement
We are grateful to Dominic Dordai for expert technical support and Karen Fox-Talbot for preparation and staining of aortic root samples. The work was supported by National Institutes of Health grant HL073971 and by a gift to the Johns Hopkins Institute for Cell Engineering. J.L. was supported in part by National Institute of General Medical Sciences training grant 5T32GM008752. The authors have no commercial or other associations that would pose a conflict of interest in connection with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 21735 kb)
Rights and permissions
About this article
Cite this article
Lee, J., Baldwin, W.M., Lee, CY. et al. Stat3β mitigates development of atherosclerosis in apolipoprotein E-deficient mice. J Mol Med 91, 965–976 (2013). https://doi.org/10.1007/s00109-013-1013-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-013-1013-5