
Abstract
Despite recent advances in therapy, multiple myeloma, the second most common hematologic tumor in the Western world, is still incurable. Identification of substances that display a wide range of tumor-killing activities and target cancer-specific pathways constitute a basis for the development of novel therapies. In this study, we investigate the cytotoxic effect of the natural substance cnicin in multiple myeloma. Cnicin treatment reveals potent antiproliferative effects and induces cell death in cell lines and primary myeloma cells even in the presence of survival cytokines and the tumor microenvironment. Other cell lines of hematopoietic origin also succumb to cell death whereas stromal cells and endothelial cells are unaffected. We show that activation of caspases, accumulation of reactive oxygen species and downregulation of nuclear factor kappa-light-chain-enhancer of activated B cell contribute to the cytotoxic effects of cnicin. Microarray analysis reveals downregulation of Pim-2, a serine/threonine kinase. We provide evidence that Pim-2 constitutes a new survival kinase for myeloma cells in vitro and is highly expressed in malignant but not in normal plasma cells in vivo. Combining cnicin with current standard or experimental therapeutics leads to enhanced cell death. Thus, our data indicate that cnicin induces myeloma cell death via several pathways and reveals Pim-2 as a novel target. These findings provide a rational for further evaluation of cnicin as a new anti-tumor drug and underline the potential of sesquiterpene lactones in tumor therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma is a B-cell malignancy characterized by clonal expansion of plasma cells in the bone marrow. Myeloma cells have a low proliferation index and accumulation of malignant cells is rather due to resistance against apoptosis than to excessive growth. Expansion and survival are fueled by activation of survival pathways including nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) [1], AKT [2], and p38 [3], and probably others, as yet unidentified ones. In this regard, new substances which display a broad spectrum of antitumor activity and uncover novel targets could essentially contribute to therapeutic success in this still incurable malignancy.
Natural substances already constitute the base of many commonly used chemotherapeutics (reviewed in [4]) and are still a rich source for the discovery of novel antitumor drugs. Sesquiterpene lactones are one class of natural compounds that display cytotoxic activities and different components purified from various sources have been found to efficiently kill cancer cells [5–13]. Depending on the source of the isolated compounds and the tumor entities tested, the related mechanisms of sesquiterpene lactone-induced apoptosis are multifaceted, including inhibition of NF-κB, accumulation of reactive oxygen species (ROS), cell cycle arrest, induction of endoplasmic reticulum stress, and activation of caspase 8. Targeting multiple pathways is a prerequisite for efficient induction of myeloma cell death and sesquiterpene lactones might therefore constitute potential therapeutic substances in the fight against multiple myeloma. The sesquiterpene lactone cnicin shows antibacterial and antineoplastic activities in cancer cell lines [14] and exerts anti-inflammatory effects [15]. However, its cytotoxic effect on myeloma cells and the underlying pathways have not been investigated so far.
Expression of the serine/threonine kinase Pim-2 has been shown to support proliferation in normal B-cell development [16] and deregulation of this protein could also play a role in the survival of malignant B-cells. Intriguingly, hyperactivation of Pim-2 has already been correlated with disease progression in B-CLL and non-Hodgkin lymphoma [17]. In multiple myeloma, enhanced mRNA levels of Pim-2 were shown as part of a disease-specific signature [18] and, just recently, overexpression of Pim-2 protein has been discussed as an anti-apoptotic mechanism in myeloma cells [19].
Pim-2 is a transcriptionally regulated mediator of cytokine-dependent survival signals [20] and in normal adult tissue expression is limited to peripheral blood cells [21]. It confers rapamycin resistance to regulatory T cells and primary hematopoietic cells and investigations assign Pim-2 to a parallel signaling pathway from the PI3K/AKT/mTOR pathway [22, 23]. Pim-2 is also known as a proto-oncogene that enables v-abl transformation [24] and supports c-myc-driven malignancies partially by stabilizing the oncogene [25, 26]. In animal models, overexpression of Pim-2 predisposes to tumor formation, especially in lymphomas [27]. In addition, it has been shown to affect clinical outcome of lymphoma therapy [28].
These characteristics suggest that additionally targeting Pim-2 could be a relevant approach in myeloma. In this study, we investigated the potential of cnicin as a new antimyeloma drug. We could show that cnicin potently kills myeloma cells via different pathways and we identified Pim-2 as a novel-specific target of this sesquiterpene lactone.
Materials and methods
Reagents
Cnicin (Roth, Karlsruhe, Germany) and parthenolide (Sigma Aldrich, Vienna, Austria) were dissolved in methanol and ethanol as 10 mM stock solution, respectively, and stored at −20°C. IGF-I, IL-6, and Pan-caspase inhibitor Q-VD-OPh were purchased from R&D systems (Abbingdon, UK), melphalan was obtained from GlaxoSmithKline Austria (Vienna, Austria), and bortezomib from Janssen-Cilag (Vienna, Austria). AKT-Inhibitor VIII, N-acetylcysteine, and dithiothreitol were purchased from Sigma Aldrich.
Cell culture
Myeloma cell lines RPMI-8226, U266, and NCI-H929 as well as stromal cell line HS-5 were obtained from American Type Culture Collection (Rockville, MD, USA), and OPM-2 and LP-1 from DSMZ (Braunschweig, Germany). MM.1R and MM.1S were kindly provided by Dr. N.L. Krett [29] (R.H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, USA). Cell lines of other hematopoietic origin as well as colon cancer cell lines were obtained from DSMZ (Braunschweig, Germany). Cell lines were grown in RPMI-1640 medium (Life Technologies, Paisley, UK) supplemented with 10% fetal calf serum (FCS; PAA, Linz, Austria), l-glutamine 100 μg/ml, and penicillin–streptomycin 100 U/ml, with exceptions mentioned below. For MM.1 cells, fungizone 1.25 μg/ml (Fischer Scientific Austria, Vienna) was added. SUP-B15 cells were cultured in McCoy’s 5A media supplemented with 20% FCS. Human bone marrow endothelial cell line HBMEC-28 was kindly provided by Dr. C.E. van der Schoot [30] (CLB, Amsterdam, The Netherlands). Cells were cultured in M199 media (PAA) supplemented with 25 mM HEPES (MERCK, Darmstadt, Germany), 10% human serum (Biowhittaker, Verviers, Belgium), 10% fetal calf serum (PAA), bFGF 1 ng/ml (Clonetics Corporation, San Diego, CA, USA), heparin 5 U/ml (Biochrom Ltd, Cambridge, UK), l-glutamine 0.3 mg/ml, penicillin–streptomycin 100 U/ml, and geneticin 100 μg/ml (all Fischer Scientific Austria, Vienna) in fibronectin-coated (1 μg/cm2) tissue flasks. Human umbilical vein endothelial cells (HUVEC) were kindly provided by Dr. David Bernhard (Medical University Innsbruck) and cultured in EBM media (Cell Systems, Kirkland, WA, USA) supplemented with growth factors (EGM-MV kit; Lonza Ltd, Basel, Switzerland) in gelatin-coated (0.2%) tissue flasks. All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2, periodically fingerprinted and tested for mycoplasma negativity.
Preparation of PBMC, patient myeloma cells and bystander cells
Peripheral blood mononuclear cells (PBMC) from healthy donors as well as bone marrow mononuclear cells from patient samples were utilized after obtaining informed consent at the University Hospital Salzburg. Cells were subjected to Ficoll separation (Ficoll-Paque®) and further incubated with RPMI-1640 media plus supplements as indicated. Cells were treated as specified and stained with cell-type specific antibodies (CD4, CD8, CD19, CD38, CD45; all from Becton Dickinson, Heidelberg, Germany) and AnnexinV-APC for analysis on a FACS Calibur (Becton Dickinson) or on a FC500 (Beckman Coulter) and further analyzed using CellQuest software (Becton Dickinson). Patients’ myeloma cells were identified by gating on the CD38high/CD45low-neg fraction (patient 1–3 MM), bone marrow bystander cells were defined as the myeloma-cell negative fraction (patient 1–3 BM) and lymphocytes were analyzed gating on CD45++ (Pat 1–3 CD45++).
Proliferation and apoptosis assay
Proliferation was measured by 3H-thymidine incorporation assays. Myeloma cells (5 × 103cells/well) were seeded into a 96-well plate with/without the addition of cytokines and cnicin as described and 1 μCi of 3H-thymidine was added to each well for the last 16 h of a 72-h incubation period. Cells were harvested onto glass fiber filters, scintillation liquid was added and radioactivity was measured using a Scintillation counter (Beckman Coulter, model LS-6500). Experiments were repeated at least three times in quadruplicates and statistical analyses were performed utilizing paired Student’s t test.
Induction of apoptosis was measured by flow cytometry. Briefly, 0.5 × 106 cells/ml were incubated for 24 and 48 h with or without cnicin (1–20 μM). Appropriate solvent controls were always included and never showed any effects. Experiments were repeated at least three times in duplicates. The extent of cell survival was calculated as percentage of AnnexinV/propidium iodide negative cells compared to control. The same procedure was used to analyze the impact of pan-caspase inhibitor Q-VD-OPH (20 μM), N-acetylcysteine (1–10 mM) and dithiothreitol (100–500 μM), parthenolide (1–4 μM), and the effect of cnicin in combination with melphalan, bortezomib, and AKT-Inhibitor VIII. Calculation of synergism (defined as combination index (CI) lower or equal to 0.9), antagonism (CI >1.1) or additive effects (0.9–1.1) of the combined treatments was performed employing Calcusyn software (Biosoft, Cambridge, UK).
Coculture assay
NCI-H929 (50,000 cells/well) cells and HS-5 stromal cells (30,000cells/well) were either grown alone or in combination in 96-wells and cnicin was added at the indicated concentrations for 48 h. Expression of CD10 (Dako, clone SS2/36, FITC labeled) on stromal cells was utilized to distinguish between the two cell types and survival was measured by AnnexinV-APC/PI staining in flow cytometry analysis.
Cell cycle analysis
For cell cycle studies, cells were treated with/without different concentrations of cnicin for 48 h, collected, fixed in ice-cold ethanol, and stained with propidium iodide. The DNA content was measured by flow cytometry on a FACS Calibur (Becton Dickinson) and data were analyzed using CellQuest software.
Ex ovo chicken chorioallantoic membrane assay
The chorioallantoic membrane (CAM) assay used in this study was performed as previously described [31] with some modifications. Briefly, fertilized white leghorn chicken eggs (SPF eggs) were purchased (Charles River, Kiesslegg, Germany) and incubated at 37°C with 80% humidity (model BSS 160, Grumbach Incubators, Asslar, Germany) for 3 days. At day 3, eggs were opened and transferred to plastic weighting boats. Ex ovo cultures were covered with a square Petri dish and incubated at 37°C and 80% humidity for 6 days. In the meantime, onplants containing tumor xenografts were generated. Therefore, native, nonpepsinized, type I rat tail collagen (BD Bioscience) was neutralized with 0.1 N NaOH solution and mixed with 10x DMEM medium (Fischer Scientific Austria, Vienna) on ice. Then, 3 × 105 NCI-H929, MM.1S, and U266 cells, respectively, were added to 30 μl of this solution, and dropped into a petri dish covered with sterile parafilm. After 45 min at 37°C, collagen-onplants were applied to the chicken chorioallantoic membrane (five onplants/CAM) with the addition of cnicin (163 nM per CAM, n = 10) or methanol (0.0025% per CAM) as a solvent control (n = 10) and incubated for 5 days. Prior to this treatment, different dose of cnicin were tested for toxicity on the embryo. Xenotransplants were microscopically analyzed (Olympus SZX10, Olympus E410, Vienna, Austria). For histological analysis, onplants were excised, fixed in 4% paraformaldehyde solution, processed for paraffin sectioning and stained with Hemalaun solution (Merck, Darmstadt, Germany). To determine the tumor area the ImageJ program (NIH, USA) was used.
Western blot analysis
Cells were either lysed directly using sodium dodecyl sulfate (SDS) loading buffer or using cell lysis buffer (Cell Signaling, Frankfurt, Germany) with the addition of 1 mM PMSF. Proteins were separated by SDS page and transferred onto nitrocellulose membranes (Millipore Corporation, Hertfordshire, UK). Primary antibodies against human caspase 8, caspase 9, poly(ADP)ribose polymerase (PARP), Pim-2, p-AKT (all from Cell Signaling), caspase 3 (Axxora, Lörrach, Germany), p-p38 (Santa Cruz) and actin (Neomarkers, Fremont, USA), were used for detection on immunoblots. Complexes with appropriate horseradish peroxidase (HRP)-linked secondary antibodies (Cell Signaling) were detected using chemiluminescence reagents (LumiGLO, Cell Signaling) followed by exposure to X-ray films.
Confocal imaging and measurement of mitochondrial ROS
For reactive oxygen species (ROS) measurements, NCI-H929 or MM.1S cells (1 × 106/ml) were plated in Lab-Tek chambered coverglass (Nunc, Rochester, NY, USA) and incubated with MitoTracker Red CM-H2XRos (0.2 μM, Invitrogen Molecular Probes, Eugene, OR, USA) for 30 min added directly to the cell culture medium. The digital images were taken using the microlens-enhanced Nipkow disk-based confocal system UltraVIEW RS (Perkin Elmer, Wellesey MA, USA) mounted on an Olympus IX-70 inverse microscope with a 40× water immersion objective (Olympus, Nagano, Japan). Images were acquired with the UltraVIEW RS software (Perkin Elmer). MitoTracker Red CM-H2XRos fluorescence was excited at 568 nm.
NF-κB ELISA
The amount of DNA-bound and thus activated NF-κB in nuclear extracts of myeloma cell lines NCI-H929, OPM-2, and MM.1S was quantified by ELISA using the TransAM NF-κB p65 Transcription Factor Assay kit (Active Motif North America, Carlsbad, CA, USA). Briefly, cells stimulated or not with tumor necrosis alpha (TNF-α; 10 ng/ml) for 3 h were concurrently treated with cnicin (5 μM) or left untreated. Nuclear cell extracts were prepared utilizing a Nuclear Cell Extraction kit (Active Motif) according to instructions. Twenty microgram protein/well were then incubated in 96-well plates coated with immobilized oligonucleotides containing the consensus binding side for the activated p65 subunit of NF-κB. Competitive addition of oligonucleotides carrying wild-type or mutated sequences to TNF-α stimulated extracts were utilized as controls for specific binding. Detection was enabled by the addition of primary antibody which recognizes an epitope that is accessible only when NF-κB is bound to its target. Addition of HRP-conjugated secondary antibody and appropriate substrate permitted quantification which was obtained at 450 nm with a reference wavelength of 655 nm using the Benchmark Microplate Reader (Bio-Rad, Vienna, Austria).
Microarray hybridization
Total RNA from cell lines NCI-H929, U266, and OPM-2 were isolated 6 h after incubation with or without 2 μM (NCI-H929) and 5 μM cnicin (U266 and OPM-2), respectively, using the RNAeasy Kit (Qiagen, Hilden, Germany). Microarray production and hybridization procedures were described previously [32]. Briefly, human oligonucleotides were used consisting of 33,072 features corresponding to 29,550 different genes. All hybridizations were repeated with reversed dye assignment (dye swap) and tiff-files were generated using the Genepix 4100A scanner (Molecular Devices, Sunnyvale, CA, USA).
Real-time PCR
RNA from all cell lines was isolated after 6 and 24 h of cnicin treatment (5 μM, except for NCI-H929, 2 μM), respectively, using TRIzol reagent according to the manufacturer’s instructions (Sigma-Aldrich). Complementary DNA probes were generated using random hexamer primers and employing SuperscriptTMII Reverse Transcriptase (Invitrogen, Austria). Real-time PCR was performed using a SYBR-Green PCR kit (Qiagen) and analysis was performed on an iCycler (iQ5; BioRad, Vienna, Austria). Expression of human Pim-2 and beclin-1 was normalized against the amount of 18S RNA and HPRT, respectively. Primers were selected using the Primer Express software (Applied Biosystems) and sequences were as follows: huPim-2 forward: CCCTACACTGACTTTGATGG, huPim-2 reverse: ACACCATGTCATAGAGGA; 18S forward: CCATTCGAACGTCTGCCCTAT, 18S reverse: TCACCCGTGGTCACCATG; Hprt forw TTTGGGCCACTGGCTGATCTATCT; Hprt rev: ATGACCAGTCAACAGGGGAC; beclin-1 for: TGTGGTGAAGACAGCTAGTG; beclin-1 rev: GTCATCACCCTGTTGATTAGAG.
Silencing studies using small interfering RNAs (siRNA)
A pool of four predesigned siRNAs for hPIM-2 and control nontargeting siRNA (NTC) were purchased from DHARMACON RNA Technologies Inc. (Lafayette, CO, USA) selecting the ON-TARGETplus Smartpool duplexes as the RNAi-specific technology platform. NCI-H929 and MM.1S cells, respectively, were transfected using the Nucleofector II device (Amaxa Biosystems). Cell survival was quantified by Annexin V/PI-staining after 48 h and at the same time protein was harvested to confirm the efficiency of Pim-2 knockdown by immunoblotting. Control-siRNA and Pim-2 knockeddown cells were additionally treated with different concentrations of cnicin for 48 h to delineate additive effects.
Immunohistochemical detection of Pim-2
This study was performed in accordance with the local research ethics committee. Briefly, immunohistochemical analyses were performed on paraffin-embedded serial sections (5 μm) derived from bone marrow of myeloma patients (n = 10; five males, five females; mean age, 64.2 ± 15). Additionally, bone marrow sections from healthy controls (n = 10; five males, five females, mean age, 69.42 ± 12.45 years) and patients with monoclonal gammopathy of unknown significance (MGUS) (n = 11; four males, seven females; mean age, 68.3 ± 3.7 years) were analyzed. Antigen retrieval was performed by heat induced epitope retrieval in pH 9 retrieval buffer (Dako, Glostrup, Denmark) at 95°C for 60 min. After blocking endogenous peroxidase (Dako), sections were incubated with antibody against hPim-2 (1:100, Sigma-Aldrich) and signals were detected using the EnVision Detection System (Dako). Visualization of all sections was performed using diaminobenzidine as the chromogen substrate (Roche Molecular Biochemicals, Mannheim, Germany) according to the manufacturer’s instructions. Nuclei were counterstained with hemalaun (Roth, Karlsruhe, Germany). Signals were digitalised using the ImageAccess Enterprise5 software (Imagic Bildverarbeitung, Glattbrugg, Switzerland). Percentage of positive cells was assessed by calculating the average of positive cells in five high power fields (patient samples) or by analyzing 50–100 plasma cells (controls, MGUS). ANOVA analysis was performed on this data set and equality of the data was tested according to Kruskal–Wallis.
Results
Cnicin blocks proliferation and induces apoptosis of myeloma cells even in the presence of components of the bone marrow microenvironment
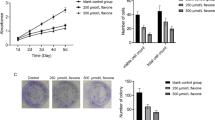
We first analyzed the effect of cnicin treatment on the proliferation of myeloma cells and found a dose-dependent blockade in all cell lines (Fig. 1a, 72 h; p < 0.05 for all measured concentrations except for U266 1 μM). As shown for NCI-H929 cell line, even the addition of myeloma growth factors such as IL-6, IGF-I, or the combination of both could not prevent this blockade (Fig. 1b).

Impact of cnicin on proliferation and survival of cells. a Proliferation was measured by 3H thymidine incorporation assay after 72 h in six different myeloma cell lines (n = 4, p < 0.05 for all, except U266 (1–2 μM cnicin) and RPMI (1 μM); paired student’s t test), and b in NCI-H929 cell line ± the addition of IL-6 and/or IGF-I. c Survival of myeloma cell lines treated with increasing concentrations of cnicin for 48 h were measured by flow cytometry analysis after AnnexinV/PI staining (n > 3). Cell survival (defined as AnnV/PI negativity) is displayed as percentage of control. d Impact of cnicin treatment on coculture of HS-5 stromal cells and NCI-H929 myeloma cells (48 h) was investigated by flow cytometry analysis as above. One representative example out of three is shown. e Endothelial cell line HBMEC-28 (HB-28), primary HUVECs, and stromal cell line HS-5 as well as different cell fractions (CD19+ B-cells, CD4+ and CD8+ T-cells) from PBMC of healthy donors (n = 4) were treated and analyzed as above (48 h). f Colon carcinoma cell lines Lovo and HRT18, T-ALL cell lines CEM-C7 and Jurkat, B cell precursor leukemia cell line Nalm-6, B-ALL cell lines SD-1 and SUP-B15, erythroleukemia cell line HEL and AML cell line HL-60 were also treated with cnicin for 48 h. Cell survival as percentage of control is presented
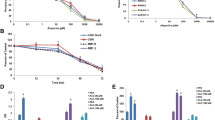
The effect of cnicin on cell viability was then measured by AnnexinV/propidium iodide staining after 48 h using flow cytometry analysis (see suppl. Fig. 1a for a representative analysis of NCI-H929 cells after 48 h). All tested myeloma cell lines readily succumb to cell death (p < 0.005 for concentrations ≥5 μM for NCI-H929 and MM.1S, and for ≥ 10 μM for all other cell lines) and IC50 values varied between 3 and 13 μM cnicin (Fig. 1c). Time–course experiments utilizing NCI-H929 revealed that apoptosis was time- and dose-dependent and was rapidly induced starting within 6 h (suppl. Fig. 1b). Coculturing NCI-H929 cells with HS-5 cells showed that stromal support attenuated the extent but could not totally prevent cnicin-induced tumor cell death (Fig. 1d).
Previously, treatment with a sesquiterpene lactone has been shown to arrest ovarian cancer cells at G2/M cell cycle phase [33] which could be the reason for the above described cell death. We did not find an accumulation of cells in a specific cell cycle phase testing cnicin at lower drug dose (1–2 μM, 48 h). Treatment using 5 μM cnicin, however, led to the accumulation of cells in subG1 phase, revealing a direct induction of cell death without prior cell cycle arrest in NCI-H929 cells (suppl. Fig. 1c). Similar data was obtained for OPM-2 and U266 cells using 10 μM cnicin (data not shown).
Recently, the sesquiterpene lactone parthenolide has been described to efficiently kill cancer cells including myeloma cells [12, 13]. We thus further compared the extent of apoptosis induced by either cnicin or parthenolide in our system and found both substances to be equally effective in vitro (suppl. Fig. 1d).
Cnicin preferentially kills myeloma cells and cells of other hematologic tumors
Cells of other origin such as endothelial cells (cell line HBMEC-28 and primary HUVEC from 2 different donors) were largely resistant to treatment as was the stromal cell line HS-5 (Fig. 1e). Testing PBMC from four healthy donors and analyzing the different immune cell subsets revealed that B-cells (CD19+) were more susceptible to cnicin-induced apoptosis (IC50 = 4.5 μM) than CD4+ T-cells (IC50 = 14.7 μM) and CD8+ T-cells (IC50 = 11.2 μM; Fig. 1e). This differential susceptibility was even more pronounced at shorter treatment periods (24 h; suppl. Fig. 2a). We also tested cell lines of other hematologic tumors as well as colon cancer cell lines (Fig. 1f). Whereas colon cancer cells (Lovo, HRT-18) were not affected by cnicin treatment, cancer cell lines representing T-ALL (CEM-C7, Jurkat), B-ALL (SD-1, SUP-B15), AML (HL-60), and erythroleukemia cells (HEL) were also efficiently killed. IC50 values varied between 10 and 14 μM except for Nalm-6 cells, a human precursor B cell leukemia cell line (IC50 = 3.4 μM).
In order to test cnicin treatment on primary myeloma cells in the context of a tumor-supporting microenvironment, mononuclear cells from bone marrow samples from three myeloma patients were treated with cnicin and further analyzed as above. We found that myeloma cells (gated as CD38++/CD45low-neg cells; patient 1–3 MM) preferentially died whereas bone marrow bystander cells (patient 1–3 BM, gated on all cells except MM) and lymphocytes (gated as CD45++) were generally less affected by the treatment (Fig. 2a). For all patient samples, viability of untreated bone marrow cells after 48 h in vitro cultures was higher than 70%.

Effect of cnicin on primary bone marrow cells and on survival of tumor xenografts. a Bone marrow mononuclear cells from myeloma patients were treated with cnicin (48 h) at indicated dosis and survival of myeloma cells (CD38++/CD45low; patients 1–3 MM, black circles), bystander cells (patients 1–3 BM, white squares) and CD45++ lymphocytes (patients 1–3, CD45++, white triangles) were analyzed as above. b Xenotransplants of NCI, MM.1S, and U266 cells, respectively, treated with methanol (control) and cnicin were digitalized (left pictures). Paraffin sections of these xenotransplants (right pictures) were stained with haemalaun and the extent of tumor area defined by plasma cell morphology was measured (n = 12, p < 0.0001; one representative example for each treatment is shown). Black bars indicate 500 μM
Cnicin efficiently kills myeloma cells in an ex ovo CAM assay
Originally developed to investigate angiogenesis, the CAM assay evolved to an accepted and reliable in vivo model to replace animal experiments for testing different substances and chemotherapeutics [34–36]. Toxicity of cnicin for the embryo was tested (data not shown) and cancer xenografts were then treated with methanol (as a solvent control) or with cnicin at nonlethal concentrations and pictures were taken after 6 days growth. Cancer areas were visualized by staining paraffin sections of control and treated onplants with hemalaun (Fig. 2b) and by defining the plasma cell areas. Results were obtained by calculating the tumor area (NCI-H929, control: 1.21 ± 0.19, cnicin 0.20 ± 0.12, mm2 ± SD, p < 0.0001; MM.1S, control: 1.22 ± 0.27; cnicin 0.52 ± 0.11, p < 0.0001; U266, control 1.11 ± 0.24, cnicin 0.27 ± 0.08, p < 0.0001) and revealed that cnicin treatment-blocked tumor growth of all three cell lines.
Cnicin-induced cell death is partially caspase dependent, induces ROS and blocks NF-κB activation
Common pathways engaged by sesquiterpene lactones in different cancer cells include activation of caspase 8 and accumulation of ROS. As shown for NCI-H929 (Fig. 3) and MM.1S (suppl. Fig. 3), caspase activation by cnicin was confirmed by immunoblot, predominantly revealing down-regulation of full-length caspase 3 and concomitant appearance of cleaved caspases 3. Caspases 8 and 9 cleavage was also observed but to a rather low extent, and cleavage of PARP additionally confirmed caspase activation (Fig. 3a). Addition of the pan-caspase inhibitor Q-VD-OPH to cnicin-treated cells could partially restore cell viability (Fig. 3b). While flow cytometry data exclude the involvement of necrosis as an alternative death pathway, the contribution of autophagy could still play a role in this setting. We therefore analyzed cnicin-induced regulation of beclin-1, a main player in the formation of the autophagosome. As depicted in suppl. Fig. 2b, cnicin-induced upregulation of beclin-1 in NCI-H929 suggests that autophagy contributes to the observed effects.

Effects of cnicin-treatment on activity of caspases, ROS, and NF-κB. a NCI-H929 cells were exposed to cnicin (2 μM) for 3–12 h and cleavage of Caspases (Casp) 3, 8, 9 (FL full length protein, CL cleaved fragment) and PARP was analyzed. Actin served as loading control. b NCI-H929 cells were treated with indicated concentrations of cnicin for 24 and 48 h ± pan-caspase inhibitor Q-VD-OPH. Cell survival was measured by flow cytometry analysis (mean percentage ± SD, n = 3, *p < 0.05, Students’ paired t test). c Activation of ROS in NCI-H929 was analyzed by confocal microscopy after 1 h incubation with different concentrations of cnicin as indicated. ROS accumulation is visible as white spots within the cells. The effect of concomitant treatment with cnicin and ROS-inhibitors (NAC, 1 mM, dithiothreitol, 0.1 mM, n = 3) on cell survival was measured after 48 h as above. White bars indicate 10μm. d NF-κB p65 was detected by ELISA in nuclear extracts of three cell lines either untreated or treated with cnicin (3 h, 5 μM cnicin). In addition, the effect of TNF-α stimulation (10 ng/ml, 3 h) and concomitant cnicin-treatment on NF-κB binding was tested. Absorbance at 450 nm is shown in a representative experiment out of three
Next, activation of ROS was measured by confocal microscopy. As shown in Fig. 3c, already short (1 h) treatment of NCI-H929 cells with different concentrations of cnicin led to enhanced production of ROS indicating a potential involvement in the observed cell death. In line with this, concomitant treatment of cells with cnicin and the ROS scavengers N-acetylcysteine and dithiothreitol, respectively, could restore cell survival (Fig. 3c, graph). These results were confirmed using myeloma cell line MM.1S (see suppl. Fig 3c).
NF-κB constitutes a major myeloma survival pathway and blockade would also modulate microenvironmental support of the malignant cells in vivo. We thus investigated NF-κB activity in nuclear extracts of cells treated with cnicin, TNF-α (also serving as positive control), and combination of both using NCI-H929, OPM-2, and MM.1S cells. As shown in Fig. 3d, constitutive NF-κB activation as well as TNF-α-induced upregulation was blocked by cnicin.
Combinatorial treatment of cnicin and chemotherapeutic agents partially enhances apoptotic effects
Further analysis revealed that cnicin treatment also induced myeloma prosurvival pathways which might attenuate the anti-tumor effect observed. We found induction of low levels of phospho-AKT which was timely limited as well as phospho-p38 expression (NCI-H929, Fig. 4a, MM.1S, suppl. Fig. 3d). As a consequence, we investigated the impact of therapeutically combining a p-AKT inhibitor (AKT-inhibitor VIII; Akti) with cnicin. Similarly, we evaluated the effects of cnicin applied jointly with standard therapeutics such as melphalan and bortezomib. NCI-H929 cells, which were most sensitive to cnicin, and U266 cells, which were least sensitive to cnicin, were treated with different combinations of these substances and apoptosis was measured after 24 h (data not shown) and 48 h (Fig. 4b). The respective combination index (CI) of the treatment was calculated using Calcusyn software (Biosoft, Cambridge, UK; see suppl. Tab. 1a–c for detailed values). Combining cnicin with Akti and melphalan revealed synergism in both cell lines tested. Concomitant treatment with cnicin and bortezomib led to additive effects in U266 cells and was antagonistic in NCI-H929 cells.

Upregulation of survival pathways by cnicin and effects of combination treatments. a Expression of p-AKT and p-p38 in NCI-H929 cells was investigated by immunoblot at the indicated time points of cnicin treatment (2 μM; asterisks marks an unspecific binding of the p-p38 antibody, actin serves as loading control). b Combined treatment with cnicin and AKT-inhibitor VIII (akti), melpahalan (mel), and Bortezomib (btz), respectively, for 48 h, a representative combination is shown (dosis in micromolar for cni, mel, akti; nanogram per milliliter for btz) in NCI-H929 and U266 cell lines. Percentage of specific apoptosis shown as percentage of Ann ± PI- and Ann ± PI + cells is displayed. Combination index was calculated applying Calcusysn software. Complete values are summarized in supplemental figures
Microarray analysis reveals induction of genes involved in stress response and down-regulation of Pim-2 as an early response to cnicin treatment
In search for other pathways that might be specifically involved in cnicin-induced cell death, we further utilized genome-wide mRNA profiling of treated cells. At the short time of treatment and relatively low concentration used, no concomitant induction of apoptosis was observed in parallel flow cytometry analysis (data not shown). We found that only few genes (i.e., seven genes, see suppl. Tab. 2) were regulated greater than or equal to twofold at 6 h treatment concomitantly in all three cell lines. Genes encoding for stress response (Hspa1a; HSP-70), antioxidant defense (Hmox1; hemoxygenase-1), and cell cycle regulation (Cdkn1a; p21) were upregulated, probably representing the effort of the cells to cope with cytotoxic stress and we could identify these genes also in screens of other treatment schemes (data not shown). Therefore, we further focused on downregulated genes to identify specific cnicin response genes. From these, Pim-2, a known proto-oncogene, further attracted our interest. Pim-2 constitutes a known B-cell survival factor and has been implicated in the progression of B-CLL and non-Hodgkin lymphoma. Modulation of Pim-2 by cytotoxic treatment has not been shown before. We further confirmed and expanded data on cnicin-induced downregulation of Pim-2 by real-time PCR in several myeloma cell lines (Fig. 5a) and B-cells of healthy donors (suppl. Fig. 4a) and we could show downregulation of the described Pim-2 isoforms also on protein level (NCI Fig. 5b and MM.1S suppl. Fig 4b).

Expression analyses of cnicin target-gene Pim-2 and functional analysis. a Pim-2 mRNA levels of myeloma cell line ± cnicin treatment (2or 5 μM) for 6 h were analyzed by real-time PCR (% expression of control is shown). b Pim-2 protein is downregulated in response to cnicin treatment (2 μM; NCI-H929). c Comparison of spontaneous apoptosis rates of control siRNA (NTC) and Pim-2 siRNA-treated NCI-H929 cells 48 h past transfection (left graph; n = 5,% cell survival ± SD compared to control are displayed, p = 0.013, Students’ paired t test). Right blots efficacy of Pim-2 knockdown in NCI-H929 cells analyzed by Western blot (NTC nontargeting control, SM size marker). Actin is used as a loading control. d Cnicin-induced decrease of cell survival in control (NTC) and Pim-2 siRNA transfected cells, treated for 48 h with indicated dosis of cnicin. e Bone marrow sections of controls, MGUS patients, and myeloma patients were stained for Pim-2 expression (brown) and nuclei were counterstained with hemalaun (blue). One representative staining for each entity is shown. Bars indicate 10 μm, pictures are shown in 100× magnification. Percentages of Pim-2 positive cells for the different entities is summarized in the graph (***p < 0.0001; Kruskal–Wallis testing for equality)
Pim-2 expression contributes to survival of myeloma cell lines in vitro and expression correlates with myeloma disease in vivo
In order to dissect the role of Pim-2 downregulation in myeloma cells, we next transfected NCI-H929 and MM.1S cells, respectively, with NTC and Pim-2 specific (Pim-2) siRNA, respectively, and confirmed the knock-down by immunoblot after 48 h (Fig. 5c and suppl. Fig 4c, right blots). The concomitant measured decrease in cell survival in cells lacking Pim-2 confirmed a vital role for Pim-2 (Fig. 5c and suppl. Fig. 4c, left graph, n = 5, cell survival NCI: 66.5% ± 17.7% SD; p = 0.013, MM.1S: 78.9% ± 6.7% SD; p = 0.0029). Cnicin treatment further enhanced cell death of Pim-2-silenced cells, probably by involving above described pathways (Fig. 5d). Recently, Asano et al. [19] showed that Pim-2 expression in myeloma cells is upregulated via IL-6/Stat 3 and TNF-α/NF-κB contributing to enhanced cell survival. In contrast, constitutive expression of Pim-2 showed no correlation with NF-κB or Stat3 levels. Interestingly, we could show that cnicin counteracts both, constitutive as well as IL-6-induced expression of this survival kinase (see suppl. Fig. 4d).
We further investigated the in situ expression of Pim-2 in bone marrow samples from age-matched healthy donors (controls, n = 10), patients diagnosed with MGUS, (n = 11) and myeloma (n = 10). A very low percentage of plasma cells from controls (2 ± 1.4 SD; range, 0–5%,) showed Pim-2 expression. In MGUS patients, few plasma cells (7 ± 2.3 SD; range, 4–10%) stained positive for Pim-2. In myeloma cells, however, expression was observed in about 85% of the cells (88.7 ± 5.1 SD; range ,75–95%). Additionally, high staining intensity of Pim-2 was found only in plasma cells from myeloma patients (data not shown). In Fig. 5e, examples for Pim-2 staining of plasma cells from a healthy control, an MGUS patient, and a myeloma patient are shown. Mean percentages of Pim-2 positive cells are summarized for each entity in the attached graph (graph Fig. 5e, percentage of positive cells ±SD; ***p < 0.0001, Kruskal–Wallis test).
Discussion
In this study, we describe that cnicin, a plant natural product, efficiently kills myeloma cells even in the context of survival cytokines and bystander cells in vitro as well as in an in vivo model. It enhances cell death in certain combination treatments and thus displays a promising profile as an alternative antimyeloma drug.
The pathways involved in cnicin-induced cell death are multifaceted and partially match those assigned to sesquiterpene lactone-induced killing of various tumor cell entities in literature such as activation of ROS and blockade of NF-κB activity. However, we found no major role for activation of caspase 8 as well as cell cycle arrest as suggested previously. In contrast, combining cnicin with melphalan, an alkylating agent reveals synergistic cell death implicating that the DNA damage pathway is not essentially targeted by single treatment with cnicin. Combination of cnicin and bortezomib, however, was additive in one and antagonistic in the other cell line and we speculate that several similar pathways might be activated by these drugs (e.g., ROS activation, NF-κB downregulation, induction of stress response) abrogating additive/synergistic effects. Detailed further investigations will have to delineate the dimension of this finding in vivo.
While other members of this substance family have been shown to efficiently induce cell death in colon carcinoma cells and endothelial cells [37] cnicin showed no deleterious effects on these cell types. In contrast, we identified B cell survival kinase Pim-2 as a novel target of cnicin-induced cytotoxicity in myeloma cells. Also in line with the specific targeting of a survival kinase is our observation that cnicin induces beclin-1, a main player in the activation of autophagy.
Cnicin treatment also induced compensatory mechanisms at mRNA level as well as activation of known myeloma survival proteins. The observed phosphorylation of AKT and p-38 could be a consequence of enhanced ROS accumulation as has been shown in other malignancies [38, 39]. These rescue mechanisms are likely to interfere with the observed extent of cell death induction in our setting. In line with this, we could show that combining cnicin with an AKT inhibitor had synergistic and additive effects in the tested cell lines, respectively, probably by interfering with the observed p-AKT upregulation and thus modulating cell death kinetics. Interestingly, Pim-2, the novel target of cnicin, is known to regulate several targets in common with the AKT pathway (for a review, see [40]). Hyperactivation of AKT constitutes a therapeutic target in about 50% of myeloma patients [41] and it is tempting to speculate that Pim-2 signaling could be a compensatory mechanism in the other half of patients. Thus, cnicin would hold the potential to interfere with this suggested compensation, however, the in vivo impact of the timely limited p-AKT induction observed in vitro will have to be clarified beforehand.
Expression of Pim-2 and AKT have been shown to contribute to malignant transformation in mouse models of lymphoma partially via common targets [23]. But whereas AKT resistance in these models was easily reversed by the addition of rapamycin, Pim-2 resistance was refractory to this TORC-1 inhibition. Importantly, in human disease, Pim-2 expression has recently been shown to affect the clinical outcome of lymphoma therapy in retrospective analysis [28]. Chemoresistance is also a threat in multiple myeloma and we suggest that Pim-2 could also play an important role in multiple myeloma refractory disease. Thus, the cnicin-induced down-regulation of Pim-2 could be of vital future therapeutic interest.
Pim-2 regulation by cytotoxic treatment is a new discovery and might also be of interest for the treatment of other solid cancer entities displaying activation of this kinase such as prostate cancer [42]. In addition, Behan et al. [43] recently found that adipocytes prevented chemotherapy-induced apoptosis in ALL-patients partially due to upregulation of Pim-2.
Overexpression of Pim-2 in multiple myeloma has only recently been described for a small number of myeloma cell lines and primary myeloma cells [19] and the role of microenvironmental cytokines in the observed upregulation has been delineated. We here present data that cnicin can efficiently counteract constitutive as well as IL-6-induced Pim-2 expression underlining its possible relevance in vivo. We also confirmed and extended Pim-2 expression data and showed for the first time that Pim-2 protein expression is mostly limited to malignant plasma cells. This finding argues against a developmental role for this kinase at the level of plasma cells as has been suggested for other B-cell subtypes and further supports a specific role of Pim-2 in myeloma progression. In addition, myeloma cells revealed rather high staining intensity of Pim-2 compared to MGUS and controls suggesting massive protein accumulation. Interestingly, premalignant plasma cells from MGUS patients displayed a significantly higher percentage of Pim-2 positive cells than plasma cells from healthy donors and further investigations are needed to clarify a possible role of Pim-2 in the transformation of these cells.
Although cnicin is a promising novel drug several issues have to be clarified in the course of further development. In general, major pitfalls of sesquiterpene lactones are thought to constitute their lack of selectivity towards cancer cells in vitro and in vivo, their high toxicity in normal cells, and their low bioavailability. Nevertheless, they developed into lead compounds in clinical cancer trials (for reviews, see [44, 45]) since several compounds revealed targeting of cancer specific signaling pathways. Here, we suggest that cnicin might be added to this group by targeting the B cell survival factor Pim-2. In addition, although we observed relatively high toxicity in normal lymphocytes, especially in B cells, we also present data that further investigations utilizing shorter time periods contain the potential to define a useful therapeutic window for cnicin treatment. However, in vivo data is urgently needed to definitely clarify this aspect. As to bioavailability, data on sesquiterpene lactones is rare. It was shown that parthenolide given orally to patients could not be detected in the blood [46]. The low bioavailiability of parthenolide is reflected in its chemical structure which has several differencies compared to cnicin. We therefore calculated the octanol/water partition coefficient (XlogP3-AA) which is a readout of the expected water solubility of a given substance, for parthenolide and cnicin and found it to be 0.2 and 2.3, respectively. This reflects a more than 20-fold higher water solubility of cnicin. Thus, we suggest that this higher bioavailability of cnicin could contribute to reach physiologically relevant concentrations. However, experimental proof will have to be established.
In summary, from our data it appears that cnicin preferentially kills lymphatic cells (predominantly B-cell derived) and hits myeloma cells at several known pathways and also on a novel one, survival kinase Pim-2, each potentially lethal. Its efficacy in the tumor microenvironment as well as in an in vivo model highlights the potential of this sesquiterpene lactone to contribute to a better targeting of the heterogenous malignant clone in myeloma disease.
References
Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V et al (2002) NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem 277:16639–16647
Kawauchi K, Ogasawara T, Yasuyama M, Otsuka K, Yamada O (2009) The PI3K/Akt pathway as a target in the treatment of hematologic malignancies. Anticancer Agents Med Chem 9:550–559
Hideshima T, Akiyama M, Hayashi T, Richardson P, Schlossman R, Chauhan D, Anderson KC (2003) Targeting p38 MAPK inhibits multiple myeloma cell growth in the bone marrow milieu. Blood 101:703–705
Newman DJ, Cragg GM (2007) Natural products as sources of new drugs over the last 25 years. J Nat Prod 70:461–477
Bruno M, Rosselli S, Maggio A, Raccuglia RA, Bastow KF, Wu CC, Lee KH (2005) Cytotoxic activity of some natural and synthetic sesquiterpene lactones. Planta Med 71:1176–1178
Hung JY, Hsu YL, Ni WC, Tsai YM, Yang CJ, Kuo PL, Huang MS (2010) Oxidative and endoplasmic reticulum stress signaling are involved in dehydrocostuslactone-mediated apoptosis in human non-small cell lung cancer cells. Lung Cancer 68:355–365
Ichikawa H, Nair MS, Takada Y, Sheeja DB, Kumar MA, Oommen OV, Aggarwal BB (2006) Isodeoxyelephantopin, a novel sesquiterpene lactone, potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis through suppression of nuclear factor-kappaB (nf-kappaB) activation and nf-kappaB-regulated gene expression. Clin Cancer Res 12:5910–5918
Lee J, Hwangbo C, Lee JJ, Seo J, Lee JH (2010) The sesquiterpene lactone eupatolide sensitizes breast cancer cells to TRAIL through down-regulation of c-FLIP expression. Oncol Rep 23:229–237
Lesiak K, Koprowska K, Zalesna I, Nejc D, Duchler M, Czyz M (2010) Parthenolide, a sesquiterpene lactone from the medical herb feverfew, shows anticancer activity against human melanoma cells in vitro. Melanoma Res 20:21–34
Saroglou V, Karioti A, Demetzos C, Dimas K, Skaltsa H (2005) Sesquiterpene lactones from Centaurea spinosa and their antibacterial and cytotoxic activities. J Nat Prod 68:1404–1407
Steele AJ, Jones DT, Ganeshaguru K, Duke VM, Yogashangary BC, North JM, Lowdell MW, Kottaridis PD, Mehta AB, Prentice AG et al (2006) The sesquiterpene lactone parthenolide induces selective apoptosis of B-chronic lymphocytic leukemia cells in vitro. Leukemia 20:1073–1079
Suvannasankha A, Crean CD, Shanmugam R, Farag SS, Abonour R, Boswell HS, Nakshatri H (2008) Antimyeloma effects of a sesquiterpene lactone parthenolide. Clin Cancer Res 14:1814–1822
Wang W, Adachi M, Kawamura R, Sakamoto H, Hayashi T, Ishida T, Imai K, Shinomura Y (2006) Parthenolide-induced apoptosis in multiple myeloma cells involves reactive oxygen species generation and cell sensitivity depends on catalase activity. Apoptosis 11:2225–2235
Vanhaelen-Fastre R, Vanhaelen M (1976) Antibiotic and cytotoxic activity of cnicin and of its hydrolysis products. Chemical structure–biological activity relationship. Planta Med 29:179–189 (author’s transl)
Schneider G, Lachner I (1987) Analysis and action of cnicin. Planta Med 53:247–251
Woodland RT, Fox CJ, Schmidt MR, Hammerman PS, Opferman JT, Korsmeyer SJ, Hilbert DM, Thompson CB (2008) Multiple signaling pathways promote B lymphocyte stimulator dependent B-cell growth and survival. Blood 111:750–760
Cohen AM, Grinblat B, Bessler H, Kristt D, Kremer A, Schwartz A, Halperin M, Shalom S, Merkel D, Don J (2004) Increased expression of the hPim-2 gene in human chronic lymphocytic leukemia and non-Hodgkin lymphoma. Leuk Lymphoma 45:951–955
Claudio JO, Masih-Khan E, Tang H, Goncalves J, Voralia M, Li ZH, Nadeem V, Cukerman E, Francisco-Pabalan O, Liew CC et al (2002) A molecular compendium of genes expressed in multiple myeloma. Blood 100:2175–2186
Asano J, Nakano A, Oda A, Amou H, Hiasa M, Takeuchi K, Miki H, Nakamura S, Harada T, Fujii S et al (2011) The serine/threonine kinase Pim-2 is a novel anti-apoptotic mediator in myeloma cells. Leukemia 25:1182–1188
Fox CJ, Hammerman PS, Cinalli RM, Master SR, Chodosh LA, Thompson CB (2003) The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev 17:1841–1854
Amson R, Sigaux F, Przedborski S, Flandrin G, Givol D, Telerman A (1989) The human protooncogene product p33pim is expressed during fetal hematopoiesis and in diverse leukemias. Proc Natl Acad Sci U S A 86:8857–8861
Fox CJ, Hammerman PS, Thompson CB (2005) The Pim kinases control rapamycin-resistant T cell survival and activation. J Exp Med 201:259–266
Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB (2005) Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood 105:4477–4483
Chen JL, Limnander A, Rothman PB (2008) Pim-1 and Pim-2 kinases are required for efficient pre-B-cell transformation by v-Abl oncogene. Blood 111:1677–1685
Allen JD, Verhoeven E, Domen J, van der Valk M, Berns A (1997) Pim-2 transgene induces lymphoid tumors, exhibiting potent synergy with c-myc. Oncogene 15:1133–1141
Zhang Y, Wang Z, Li X, Magnuson NS (2008) Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 27:4809–4819
Breuer ML, Cuypers HT, Berns A (1989) Evidence for the involvement of pim-2, a new common proviral insertion site, in progression of lymphomas. EMBO J 8:743–748
Schatz JH, Oricchio E, Wolfe AL, Jiang M, Linkov I, Maragulia J, Shi W, Zhang Z, Rajasekhar VK, Pagano NC et al (2011) J Exp Med 208:1799–1807
Greenstein S, Krett NL, Kurosawa Y, Ma C, Chauhan D, Hideshima T, Anderson KC, Rosen ST (2003) Characterization of the MM.1 human multiple myeloma (MM) cell lines: a model system to elucidate the characteristics, behavior, and signaling of steroid-sensitive and -resistant MM cells. Exp Hematol 31:271–282
Rood PM, Calafat J, von dem Borne AE, Gerritsen WR, van der Schoot CE (2000) Immortalisation of human bone marrow endothelial cells: characterisation of new cell lines. Eur J Clin Invest 30:618–629
Deryugina EI, Quigley JP (2008) Chapter 2. Chick embryo chorioallantoic membrane models to quantify angiogenesis induced by inflammatory and tumor cells or purified effector molecules. Methods Enzymol 444:21–41
Kulterer B, Friedl G, Jandrositz A, Sanchez-Cabo F, Prokesch A, Paar C, Scheideler M, Windhager R, Preisegger KH, Trajanoski Z (2007) Gene expression profiling of human mesenchymal stem cells derived from bone marrow during expansion and osteoblast differentiation. BMC Genomics 8:70–76
Choi EJ, Ahn WS (2009) Antiproliferative effects of dehydrocostuslactone through cell cycle arrest and apoptosis in human ovarian cancer SK-OV-3 cells. Int J Mol Med 23:211–216
Kunzi-Rapp K, Genze F, Kufer R, Reich E, Hautmann RE, Gschwend JE (2001) Chorioallantoic membrane assay: vascularized 3-dimensional cell culture system for human prostate cancer cells as an animal substitute model. J Urol 166:1502–1507
Saw CL, Heng PW, Liew CV (2008) Chick chorioallantoic membrane as an in situ biological membrane for pharmaceutical formulation development: a review. Drug Dev Ind Pharm 34:1168–1177
Taizi M, Deutsch VR, Leitner A, Ohana A, Goldstein RS (2006) A novel and rapid in vivo system for testing therapeutics on human leukemias. Exp Hematol 34:1698–1708
Parada-Turska J, Paduch R, Majdan M, Kandefer.Szerszen M, Rzeski W (2007) Antiproliferative activity of parthenolide against three human cancer cell lines and umbilical vein endothelial cells. Pharmacol Rep 59:233–237
Zunino SJ, Storms DH, Ducore JM (2010) Parthenolide treatment activates stress signaling proteins in high-risk acute lymphoblastic leukemia cells with chromosomal translocation t(4;11). Int J Oncol 37:1307–1313
Clerkin JS, Naughton R, Quiney C, Cotter TG (2008) Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett 266:30–36
Amaravadi R, Thompson CB (2005) The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest 115:2618–2624
Zollinger A, Stuhmer T, Chatterjee M, Gattenlohner S, Haralambieva E, Muller-Hermelink HK, Andrulis M, Greiner A, Wesemeier C, Rath JC et al (2008) Combined functional and molecular analysis of tumor cell signaling defines 2 distinct myeloma subgroups: Akt-dependent and Akt-independent multiple myeloma. Blood 112:3403–3411
Dai H, Li R, Wheeler T, Diaz DV, Frolov A, Tahir S, Agoulnik I, Thompson T, Rowley D, Ayala G (2005) Pim-2 upregulation: biological implications associated with disease progression and perinueral invasion in prostate cancer. Prostate 65:276–286
Behan JW, Yun JP, Proektor MP, Ehsanipour EA, Arutyunyan A, Moses AS, Avramis VI, Louie SG, Butturini A, Heisterkamp N et al (2009) Adipocytes impair leukemia treatment in mice. Cancer Res 69:7867–7874
Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N (2010) What made sesquiterpene lactones reach clinical trials? Drug Discov Today 15:668–678
Merfort I (2011) Perspectives on sesquiterpene actones in inflammation and cancer. Curr Drug Targets 12:1560–1573
Curry EA 3rd, Murry DJ, Yoder C, Fife K, Armstrong V, Nakshatri H, O’Connell M, Sweeney CJ (2004) Invest New Drugs 22:299–305
Acknowledgments
The authors would like to thank Katrin Janke and Claudia Zavadil for their excellent technical assistance. This project was supported by grants of the Austrian Cancer Society/Tirol (to KJ, MO, and JP), the SFB021 (to RG), and the Klinische Malignom- und Zytokinforschung Innsbruck-Salzburg gemeinnützige GmbH.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
DOC 35 kb
Rights and permissions
About this article
Cite this article
Jöhrer, K., Obkircher, M., Neureiter, D. et al. Antimyeloma activity of the sesquiterpene lactone cnicin: impact on Pim-2 kinase as a novel therapeutic target. J Mol Med 90, 681–693 (2012). https://doi.org/10.1007/s00109-011-0848-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-011-0848-x