
Abstract
Activation of transcription factors nuclear factor-κB (NF-κB) and signal transducer and activator of transcription 3 (STAT3) is frequently observed in prostate cancer and has been linked with tumor cell proliferation, invasion, metastasis, and angiogenesis. In this study, we investigated the effect of ursolic acid (UA) on NF-κB and STAT3 signaling pathways in both androgen-independent (DU145) and androgen-dependent (LNCaP) prostate cancer cell lines and also prospectively tested the hypothesis of NF-κB and STAT3 inhibition using a virtual predictive functional proteomics tumor pathway technology platform. We found that UA inhibited constitutive and TNF-α-induced activation of NF-κB in DU145 and LNCaP cells in a dose-dependent manner. The suppression was mediated through the inhibition of constitutive and TNF-α-induced IκB kinase (IKK) activation, phosphorylation of IκBα and p65 and NF-κB-dependent reporter activity. Furthermore, UA suppressed both constitutive and inducible STAT3 activation in prostate cancer cells concomitant with suppression of activation of upstream kinases (Src and JAK2) and STAT3-dependent reporter gene activity. UA also downregulated the expression of various NF-κB and STAT3 regulated gene products involved in proliferation, survival, and angiogenesis and induced apoptosis in both cells lines as evidenced by DNA fragmentation and annexin V staining. In vivo, UA (200 mg/kg b.w.) treated for 6 weeks inhibited the growth of DU145 cells in nude mice without any significant effect on body weight. Overall, our results from experimental and predictive studies suggest that UA mediates its anti-tumor effects through suppression of NF-κB and STAT3 pathways in prostate cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prostate cancer is one of the leading causes of cancers among men worldwide [1]. Early in their development, prostate tumors require androgen stimulation for growth and survival [2]. Following remission, however, tumors frequently recur in an androgen-independent form refractory to current treatment modalities [2]. Studies in the last few years have revealed that chronic inflammation plays a pivotal role in carcinogenesis of the prostate [3]. Several inflammatory signaling cascades including nuclear factor-κB (NF-κB), signal transducer and activator of transcription (STAT3), serine/threonine protein kinase B (AKT), and cyclooxygenase (COX-2) have been linked with different stages of prostate cancer progression and are reported to regulate prostate cancer proliferation, survival, invasion, metastasis, and angiogenesis [4, 5]. Thus, agents that can suppress these inflammatory markers have potential in both the prevention and treatment of prostate cancer.
Ursolic acid (UA) (3β-hydroxy-urs-12-en-28-oic-acid) is a pentacyclic triterpenoid belonging to the cyclosqualenenoid family [6]. UA has been previously reported to suppress the proliferation of a variety of tumor cells, induce apoptosis [7–10], and inhibit tumor promotion, metastasis, and angiogenesis [11]. Although few studies on the apoptotic effects of UA on prostate cancer cells have been previously reported [7, 12], its detailed mechanism of action on prostate cancer cells and in vivo effects on prostate cancer growth has not been explored before.
Numerous studies support the critical role of NF-κB and STAT3 in prostate tumor cell survival, metastasis, and angiogenesis [13]; hence, we hypothesized that UA mediates its effects through suppression of both NF-κB and STAT3 pathways. In addition, the effect of UA on prostate cancer cells was also tested in a virtual predictive tumor cell platform [14]. The virtual epithelial tumor cell platform on which the predictive NF-κB and STAT3 inhibition studies were conducted is a comprehensive integration of the pathways representing the key cancer cell functions like proliferation, apoptosis, angiogenesis, metastasis, and tumor micro-environment including tumor-associated inflammation [14]. We further investigated the anti-cancer effects of UA on proliferation, apoptosis, as well as NF-κB and STAT3 regulated gene products in vitro and growth of prostate cancer in vivo. We found that UA is a potent inhibitor of constitutive and inducible NF-κB and STAT3 activation in prostate cancer cells and also significantly suppressed the growth of prostate cancer xenografts in vivo.
Materials and methods
Reagents
UA (98% pure) was purchased from Guangxi Changzhou Natural Products Development Company Ltd. (China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Tris, glycine, NaCl, SDS, curcumin, and BSA were purchased from Sigma-Aldrich (St. Louis, MO, USA). UA was dissolved in dimethyl sulfoxide (DMSO) as a 100 mM stock solution and stored at 4°C. RPMI 1640, fetal bovine serum (FBS), 0.4% trypan blue vital stain, and antibiotic–antimycotic mixture were obtained from Invitrogen (Carlsbad, CA, USA). Antibodies against phospho-STAT3 (Tyr 705), phospho-AKT (Ser 473), STAT3, IκBα, Bcl-2, Bcl-xL, survivin, XIAP, VEGF, PARP, AKT, annexin V-FITC assay kit, PTEN siRNA, c-Src, and scrambled control siRNA were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to phospho-specific Src (Tyr 416), Src, phospho-specific JAK2 (Tyr 1007/1008) and JAK2, phospho-specific p65 (Ser 536) and p65, phospho-specific IκBα (Ser 32), phospho-specific IKKα/β (Ser 180/Ser 181), IKKα, phospho-specific PDK1 (Ser 241), and PDK1 were purchased from Cell Signaling Technology (Beverly, MA, USA). Goat anti-rabbit–horseradish peroxidase (HRP) conjugate and goat anti-mouse HRP were purchased from Sigma-Aldrich. Bacteria-derived recombinant human TNF-α and IL-6 was purchased from ProSpec-Tany TechnoGene Ltd. (Rehovot, Israel). Death Detection ELISAPLUS DNA fragmentation kit was purchased from Roche Diagnostics (Mannheim, Germany). Bradford reagent was purchased from Bio-Rad (Hercules, CA, USA). Nuclear extract kit and DNA binding kit was obtained from Active Motif (Carlsbad, CA, USA). Lipofectamine was purchased from Invitrogen. Luciferase assay kit and β-galactosidase assay kit were purchased from Promega (Madison, WI, USA).
Cell lines
Human androgen-independent DU145 and androgen-dependent LNCaP prostate cancer cell lines were kindly provided by Prof. Shazib Pervaiz of our university. Cells were cultured in Rosewell Park Memorial Institute (RPMI 1640) medium containing 1× antibiotic–antimycotic solution with 10% FBS.
Cell proliferation assay (MTT)
The anti-proliferative effect of UA against prostate cancer cells was determined by the MTT dye uptake method as described previously [15].
Apoptosis detection by ELISA
Apoptosis of cells was determined as previously described [16] using Cell Death Detection ELISAPLUS kit according to the manufacturer’s protocol.
Annexin V assay
Apoptosis of cells was determined as previously described [16] using annexin V-FITC kit according to the manufacturer’s protocol.
Western blotting
Vehicle or UA treated whole-cell extracts were lysed in lysis buffer [20 mM Tris (pH 7.4), 250 mM NaCl, 2 mM EDTA (pH 8.0), 0.1% Triton X-100, 0.01 mg/ml aprotinin, 0.005 mg/ml leupeptin, 1 mM PMSF, and 4 mM NaVO4]. Lysates were then spun at 13,000 rpm for 10 min to remove insoluble material and resolved on a 10% SDS gel. After electrophoresis, the proteins were electrotransferred to a nitrocellulose membrane, blocked with 5% non-fat milk, and probed with antibodies of interest overnight at 4°C. The blot was washed, exposed to HRP-conjugated secondary antibodies for 1 h, and finally examined by chemiluminescence (ECL; GE Healthcare, Little Chalfont, Buckinghamshire, UK). Densitometric analysis of the scanned blots was performed using Image J software and the results are expressed as fold change relative to control.
NF-κB DNA binding assay
To determine NF-κB activation, we performed DNA binding assay using TransAM NF-κB Kit according to the manufacturer’s instructions and as previously described [17].
NF-κB and STAT3 luciferase reporter assay
The effect of UA on constitutive and TNF-α-induced NF-κB-dependent reporter gene transcription in prostate cancer cells was determined as described previously [18]. The STAT3-responsive elements linked to a luciferase reporter gene were transfected with wild-type or dominant-negative STAT3-Y705F (STAT3F) as described previously [19]. Luciferase activity was measured with a Tecan (Durham, NC, USA) plate reader and normalized to β-galactosidase activity. All luciferase experiments were done in triplicate and repeated twice.
siRNA transfection assay
DU145 cells were plated in 96-well plates and allowed to adhere for 24 h. On the day of transfection, 4 μl lipofectamine obtained from Invitrogen was added to 50 nM PTEN siRNA in a final volume of 100 μl of culture medium. After 48 h of transfection, cells were treated with UA (50 μM) for 24, 48, or 72 h. The anti-proliferative effect of UA against prostate cancer cells was determined by MTT assay as described previously [20]. DU145 cells were transfected with 50 nM Src siRNA or scrambled siRNA. The transfected cells were then treated with UA for 48 h and apoptosis was analyzed with (BD FACSCalibur; BD Biosciences, USA) as described previously [20].
In silico analysis
The Cellworks tumor cell platform provides a dynamic and transparent view of human cellular physiology at the proteomics abstraction level [14]. The virtual tumor cell platform consists of a dynamic and kinetic representation of the signaling pathways essential to tumor physiology at the bio-molecular level. Signaling pathways for different cancer phenotypes comprise 75 major signaling networks with more than 3,900 intracellular interacting molecules [14]. The baseline used for the study was an androgen-independent system with PTEN and p53 mutations. The following studies were conducted in disease state and the biomarker trends were evaluated as percentage change from disease values. Individual NF-κB and STAT3 inhibition and a combination of the NF-κB and STAT3 inhibition were tested. The results for the above studies individually or in combination were analyzed across known biomarker trends for UA and were compared with existing literature. The purpose of the above study was to determine the most probable mechanism of action of UA and to compare the virtual hypothesis testing with empirical data.
Prostate cancer xenograft mouse model
Xenograft studies were performed as previously described [21]. All mice were weighed before start of experiment. Animal experiments were conducted in accordance with Singapore NACLAR guidelines (Law as of November 2004) for laboratory animal use and care. Briefly, 4-week-old athymic balb/c nude male mice (Biopolis, Singapore) weighing 20 g were randomized into the following treatment and control group (n = 6). DU145 cells were s.c. injected (5 × 106 cells/mouse) to each mice. The control group was treated with 0.1% DMSO and treatment group mice received i.p. 200 mg/kg UA twice a week. The treatment was continued for 6 weeks from the date of randomization. The mice body weight and tumor sizes were recorded twice every week and the tumor size was determined by Vernier caliper and calculated using the formula \( \left[ {{\text{length}} \times {{\left( {{\text{width}}} \right)}^2}} \right]/2 \). At the end of 6 weeks, mice were sacrificed by a lethal dose of i.p. phentobarbital (40 mg/kg b.w.), blood was collected for measurement of UA, and tumor volume was measured and weighed.
Detection of UA in serum sample
Bioavailability of UA in mice serum was determined using LC–MS/MS as previously described with modifications [22]. The analyte was quantified with using single ion monitoring in MS multiple reactions monitoring mode (MRM, m/z 455.0 → 455.0). All samples were prepared in triplicate and analyzed in three separate analytical runs. Standard curves were constructed using weighted (1/x 2) linear least square regression analysis of the observed peak areas of UA. The unknown sample concentrations were calculated from the linear regression equation of the peak areas against concentrations of the calibration curve. The serum concentration of UA was calculated using Analyst software 1.4.2 (Applied Biosystems, USA).
Immunohistochemical analysis of tumor samples
Immunohistochemistry was performed using LSAB kit (Dako, Carpinteria, CA, USA) according to manufacturer’s instructions and as described previously [23]. Images were captured using Olympus BX51 microscope (magnification, ×20). Positive cells (brown) were quantitated using the Image-Pro plus 6.0 software package (Media Cybernetics, Inc.).
Statistical analysis
Statistical analysis was performed by Student’s t test. Probability (p) values less than 0.05 were considered statistically significant.
Results
Predictive proteomics analysis for NF-κB and STAT3 activities
In this study, predictive analysis was performed using the virtual tumor cell platform (Fig. 1a) which provides a comprehensive analysis to determine the primary target of UA in the prostate cancer cells. NF-κB activity was inhibited by 80% in a growth-factor over-expressed virtual tumor cell aligned to DU145. Figure 1b (left panel) shows that knocking down STAT3 activity by 80% had a very minimal impact on NF-κB activity. Knocking down NF-κB by 80% shows a reduction in phosphorylated JAK2 and Src (Fig. 1b, right panel). All of the key kinases such as ERK, p38, JNK, IKK, and AKT show a reduction with NF-κB inhibition and a minimal reduction with STAT3 inhibition excepting AKT where a minimal increase was observed (Fig. 1c, left panel). The key survival markers Bcl-2, Bcl-xL, survivin (BIRC5), and XIAP levels were reduced with NF-κB and STAT3 inhibition. Most of these markers were inhibited in the range of 35% to 50% with NF-κB inhibition; in fact, only 10% to 20% reduction was observed with STAT3 inhibition (Fig. 1c, right panel). Analyzing the increased predictive trends seen with the apoptotic markers caspases 3 and cleaved PARP1 (Fig. 1d, left panel) indicates an increase in the apoptotic phenotype. These predictions therefore support the hypothesis that UA’s effects on the proliferative and apoptotic phenotypes is through inhibition of both NF-κB and STAT3 activity. Furthermore, the predictive data shows a greater inhibition in metastatic (CXCR4) and angiogenic (VEGF) gene products with NF-κB inhibition compared to STAT3 where a relatively minimal inhibition is observed (Fig. 1d, right panel). The fact that NF-κB inhibition caused a significant reduction in STAT3 activity (around 45%) leads to the conclusion that UA mediates its effects by inhibiting both of these molecules (Fig. 1b, right panel).

Predictive in silico virtual tumor platform. a Overview of the predictive virtual tumor cell platform. b Left panel the percentage reduction in NF-κB. NF-κB shows only a minimal reduction with 80% inhibition of STAT3 activity. b Right panel the percentage inhibition of STAT3, p-JAK2 and p-Src levels with NF-κB and STAT3 activity inhibition by 80%. As seen experimentally, active Src shows the maximum reduction of about 60% as seen with NF-κB inhibition and a 20% reduction with STAT3 inhibition. Reduction of JAK2 was 10% with STAT3 inhibition but a higher reduction in JAK2 (40%) is seen with NF-κB inhibition. This indicates that NF-κB could be the more probabilistic pathway inhibited by UA. c Left panel the percentage reductions of key kinases, ERK, p38, JNK, IKK, and AKT. JNK shows a 38% reduction with NF-κB but only a minimal 3% reduction with STAT3 inhibition, NF-κB mediated inhibition was 16%, 30%, 23%, and 8% for p-ERK, p-p38, IKK, and AKT, respectively, while 5%, 2%, and 2% for p-ERK, p-p38, and IKK, respectively, with STAT3 inhibition and AKT shows a minimal increase. c Right panel the percentage reduction in key survival markers, Bcl-2, Bcl-xL, XIAP, and survivin following NF-κB and STAT3 inhibition. Bcl-xL shows a 50% reduction with NF-κB as compared to 18% reduction with STAT3 inhibition. The inhibition on Bcl-2, XIAP, and survivin was 42%, 45%, and 36%, respectively, with NF-κB inhibition and 16%, 10%, and 20%, respectively, with STAT3 inhibition. d Left panel the percentage increase in caspase-3 and cleaved PARP with NF-κB and STAT3 activity inhibition and the increasing trend of these markers supports the increase in apoptotic endpoint seen experimentally. d Right panel the percentage reductions for CXCR4 and VEGF with NF-κB and STAT3 inhibition. CXCR4 and VEGF show a 70% and 32% reduction, respectively, with NF-κB inhibition while VEGF and CXCR4 shows only minimal reduction with STAT3 inhibition
UA inhibits proliferation of prostate cancer cell lines
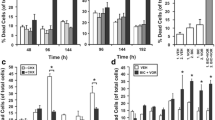
The structure of UA is shown in Fig. 2a. We first examined the anti-proliferative effects of UA using 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) method. The results of MTT assay indicated that treatment with 0, 10, 20, 25, and 50 μM UA for different time intervals inhibited cell proliferation in a dose- and time-dependent manner. Exposure to 50 μM UA for 72 h exhibited 45% cell growth inhibition in DU145 cells (Fig. 2b, left panel) as compared to a stronger 70% inhibition in LNCaP cells (Fig. 2b, right panel).

UA inhibits proliferation and induces apoptosis of prostate cancer cells. a The chemical structure of UA. b DU145 and LNCaP (5 × 105/ml) were plated in triplicate, treated with indicated concentrations of UA, and then subjected to MTT assay after 24, 48, or 72 h. Cell viability was analyzed by MTT method as described in “Materials and methods”. c DU145 and LNCaP were treated with 50 μM UA for the indicated times, apoptosis was determined by degree of DNA fragmentation in the cytoplasm of cells treated with UA. The assay was performed using DNA fragmentation ELISA kit as per manufacturer instructions (Roche). d DU145 and LNCaP (1 × 106/ml) were treated with 50 μM UA for the indicated times, after which the cells were washed, fixed, stained with annexin V and PI, and analyzed for apoptotic cells by flow cytometry. *p < 0.05 when compared to untreated group
UA induces apoptosis in prostate cancer cell lines
DNA fragmentation, a characteristic hallmark of apoptosis, was evaluated using cell death detection ELISA kit as described previously [16]. UA produced significant 12- and 18-fold increase in DNA fragmentation in DU145 (Fig. 2c, left panel) and LNCaP cells (Fig. 2c, right panel), respectively, following treatment for 72 h as compared to vehicle-treated control. Consistent with these results, when apoptosis was examined by annexin V staining, 35.4% of DU145 (Fig. 2d, left panel) and 47.8% of LNCaP cells (Fig. 2d, right panel) were found to be annexin V positive as compared to vehicle-treated control cells, thereby indicating that UA is a potent inducer of apoptosis in prostate cancer cells, although its apoptotic effects were less in DU145 as compared to LNCaP cells.
UA inhibits TNF-α-induced IκBα and p65 phosphorylation in LNCaP cells
Because IκBα phosphorylation is required for activation of NF-κB [23], we determined whether inhibition of TNF-α induced NF-κB activation in LNCaP cells by UA was due to inhibition of IκBα phosphorylation. We found that TNF-α induced IκBα phosphorylation was suppressed in a dose-dependent manner in UA-pretreated LNCaP cells (Fig. 3a, top panel). We next investigated the effect of UA on TNF-α-induced phosphorylation of p65 because phosphorylation is also required for its transcriptional activity [24]. LNCaP cells were further pre-incubated with different concentrations of UA and then stimulated with TNF-α. Nuclear extracts were prepared and tested for p65 phosphorylation using western blot analysis. As shown in Fig. 3a (middle panel), UA inhibited TNF-α induced p65 phosphorylation in a dose-dependent manner in LNCaP cells, with maximum inhibition seen at 50 μM. These results indicate that UA can modulate TNF-α-inducible IκBα and p65 phosphorylation and supports the predictive data related to NF-κB inhibition as shown in Fig. 1b (left panel).

a Top panel effect of UA on phosphorylation of IκBα. LNCaP cells (5 × 105/ml) were pretreated with different concentrations of UA for 4 h and then stimulated with TNF-α (1 nM) for 1 h. Cytoplasmic extracts were prepared and analyzed by western blotting using phospho-specific anti-IκBα (Ser32/36) and IκBα antibodies. The results shown are representative of three independent experiments. Equal protein loading was evaluated by β-actin. a Middle panel UA inhibits phosphorylation and nuclear translocation of p65. Nuclear extracts were prepared and analyzed for NF-κB activity. LNCaP cells (5 × 105/ml) were pretreated with UA for 4 h and then treated with TNF-α (1 nM) for 1 h. The results shown are representative of three independent experiments. For loading control of nuclear protein, the membrane was blotted with anti-PARP antibody. a Bottom panel effect of UA on the activation of IKK by TNF-α. LNCaP cells (5 × 105/ml) were pretreated with UA for 4 h and then treated with TNF-α (1 nM) for 15 min. Whole-cell extracts were prepared and probed for phospho-specific IKKα/β antibodies. The results shown are representative of three independent experiments. Equal protein loading was evaluated by IKKα. a Top right panel UA inhibits constitutive phosphorylation of AKT. LNCaP cells (5 × 105/ml) were treated with different concentrations of UA for 4 h. Whole-cell extracts were prepared and probed for phospho-specific AKT. The results shown are representative of three independent experiments. Equal protein loading was evaluated by antibodies specific for AKT. a Bottom right panel UA inhibits constitutive phosphorylation of PDK1. LNCaP cells (5 × 105/ml) were treated with different concentrations of UA for 4 h. Whole-cell extracts were prepared and probed for phospho-specific PDK1. The results shown are representative of three independent experiments. Equal protein loading was evaluated by antibodies specific for PDK1. b Effect of PTEN knockdown on DU145 cell proliferation. DU145 cells (5 × 105/ml) were transfected with either PTEN siRNA or scrambled siRNA (50 nM). After 48 h, cells were treated with 50 μM UA for 24, 48, and 72 h and MTT assay was performed to determine cell proliferation.*p < 0.05; **p < 0.001when compared to untreated group. c Effect of UA on TNF-α-inducible NF-κB DNA binding activity. LNCaP cells (5 × 105/ml) were pretreated with different concentrations of UA for 4 h and then treated with TNF-α (1 nM) for 1 h. Nuclear extracts were prepared, 20 μg of the nuclear extract protein was taken for DNA binding assay as described in “Materials and methods”. d UA inhibits TNF-α-induced reporter gene expression. LNCaP cells (1 × 105/ml) were transfected with NF-κB-luciferase and β-galactosidase reporter plasmid using lipofectamine, incubated for 24 h, and then treated with 10, 25, or 50 μM UA for 4 h or 50 μM curcumin for 4 h and then treated with TNF-α (1 nM) for 24 h. Cells were lysed in reporter lysis buffer and analyzed for luciferase activity and normalized with β-galactosidase activity. Results are expressed as fold activity over the activity of vector control. *p < 0.05
UA inhibits constitutive IκBα and p65 phosphorylation in DU145 cells
We further tested the effect of UA on IκBα phosphorylation in DU145 cells, which express constitutively active NF-κB [18]. Treatment with various concentrations of UA suppressed constitutive IκBα phosphorylation in a dose-dependent manner (Fig. 4a, top panel). We also examined the effect of UA on p65 phosphorylation in DU145 cells and found that exposure to different concentrations of UA suppressed constitutive p65 activation in these cells in a dose-dependent manner (Fig. 4a, middle panel).

a Top panel effect of UA on constitutive phosphorylation of IκBα. DU145 cells (5 × 105/ml) were pretreated with different concentrations of UA for 4 h. Cytoplasmic extracts were prepared and analyzed by western blotting using phospho-specific anti-IκBα (Ser32/36) and IκBα antibodies. The results shown are representative of three independent experiments. Equal protein loading was evaluated by β-actin. a Middle panel UA inhibits constitutive phosphorylation and nuclear translocation of p65. Nuclear extracts were prepared and analyzed for NF-κB activity. DU145 cells (5 × 105/ml) were pretreated with UA for 4 h. The results shown are representative of three independent experiments. For loading control of nuclear protein, the membrane was blotted with anti-PARP antibody. a Bottom panel effect of UA on the constitutive activation of IKKα/β. DU145 cells (5 × 105/ml) were pretreated with UA. Whole-cell extracts were prepared and probed for phospho-specific IKKα/β. The results shown are representative of three independent experiments. Equal protein loading was evaluated by IKKα. b Effect of UA on constitutive NF-κB DNA binding activity. DU145 cells (5 × 105/ml) were pretreated with different concentrations of UA for 4 h. Nuclear extracts were prepared, 20 μg of the nuclear extract protein was taken for DNA binding assay as described in “Materials and methods”. c UA inhibits constitutive activation of reporter gene expression. DU145 cells (1 × 105/ml) were transfected with NF-κB-luciferase and β-galactosidase reporter plasmid using lipofectamine, incubated for 24 h, and then treated with 10, 25, or 50 μM UA for 4 h. Cells were lysed in reporter lysis buffer and analyzed for luciferase activity and normalized with β-galactosidase activity. Results are expressed as fold activity over the activity of vector control. *p < 0.05; **p < 0.001
UA inhibits TNF-α-induced and constitutive activation of IKK in LNCaP and DU145 prostate cancer cell lines
It has been shown that UA inhibits the phosphorylation and degradation of IκBα; we next tested the effect of UA on TNF-α-induced and constitutive IKK activation, which is required for phosphorylation of IκBα [25]. LNCaP cells were pre-incubated with different concentrations of UA and then stimulated with TNF-α. Whole-cell extracts were prepared and tested for IKK phosphorylation using western blot analysis. As shown in Fig. 3a (bottom panel), UA inhibited TNF-α-induced IKK activation with maximum inhibition seen at 50 μM. We also examined the effect of UA on IKK activation in DU145 cells and found that treatment with various concentrations of UA suppressed constitutive IKK activation in these cells in a dose-dependent manner (Fig. 4a, bottom panel). However, UA was found to have a very minimal effect on expression of IKKα in both cell lines.
UA inhibits AKT activation in LNCaP cells with minimal effect on PDK1 phosphorylation
It has been reported that AKT and PDK1 can activate IKK [26]. Thus, it is possible that UA suppresses TNF-α-induced IKK activation in LNCaP cells through suppression of either of these upstream kinases. We found that UA treatment suppressed activation of AKT in a dose-dependent manner with a minimal effect on PDK1 phosphorylation (Fig. 3a, right panel). However, neither constitutive nor TNF-α-inducible phospho-AKT and phospho-PDK1 levels were detected in androgen-independent DU145 cell line (data not shown), most probably due to the fact that DU145 cells express PTEN, which is a negative regulator of PI3K/AKT signaling cascade, whereas LNCaP cells are PTEN null [27]. Interestingly, we also observed that the knockdown of PTEN gene by siRNA sensitized DU145 cells to anti-proliferative effects of UA as evidenced by more than 80% decrease in cell proliferation post-72 h after PTEN siRNA transfection (Fig. 3b). Overall, these results thus indicate that UA-induced inhibition of TNF-α-induced IKK activation in LNCaP cells may be mediated through the suppression of AKT activation.
UA inhibits TNF-α-induced and constitutive NF-κB DNA binding activity in prostate cancer cell lines
Since TNF-α-induced NF-κB activation cascade is well characterized [28], we next investigated the effect of UA on TNF-α induced NF-κB activity in LNCaP cells. Cells were pre-incubated with different concentrations of UA and then stimulated with TNF-α. Nuclear extracts were prepared and tested for NF-κB activity by DNA binding assay ELISA kit (ActiveMotif, Carlsbad, CA, USA). As shown in Fig. 3c, UA inhibited TNF-α-induced NF-κB DNA binding activity in a dose-dependent manner, with maximum inhibition seen at 50 μM. At 50 μM UA, a 2.1-fold decrease in DNA binding activity was observed compared to TNF-α-induced NF-κB activation. We also tested the effect of UA on NF-κB DNA binding activity in DU145 and found that treatment with various concentrations of UA suppressed constitutive NF-κB activity in a dose-dependent manner (Fig. 4b). These results indicate that UA can modulate both inducible and constitutive NF-κB activation in prostate cancer cells.
UA suppresses TNF-α-induced and constitutive NF-κB-dependent reporter gene expression
Although we observed by NF-κB DNA binding assay that UA inhibits NF-κB activation, DNA binding alone does not always correlate with NF-κB-dependent gene transcription, indicating that there are additional regulatory steps [29]. To determine the effects of UA on TNF-α-induced NF-κB-dependent reporter gene expression, we transiently transfected LNCaP cells with NF-κB regulate luciferase reporter construct and co-transfected with β-galactosidase vector. A 5-fold increase in luciferase activity was observed after stimulation with TNF-α (Fig. 3d). When the cells were pretreated with UA, TNF-α-induced NF-κB-dependent luciferase expression was inhibited in a dose-dependent manner at 50 μM concentration, which was comparable to curcumin, a potent NF-κB blocker (Fig. 3d). To determine the effects of UA on constitutive NF-κB-dependent reporter gene expression in DU145 cells, transfection was done as described above followed by treatment with UA for 4 h. In the presence of UA, NF-κB-dependent luciferase expression was inhibited in a dose-dependent manner with maximum inhibition at 50 μM (Fig. 4c). These results demonstrate that UA can inhibit both TNF-α-inducible and constitutive NF-κB-dependent reporter gene expression.
UA inhibits constitutive STAT3 phosphorylation in DU145 cells
The ability of UA to modulate constitutive STAT3 activation in DU145 cells was investigated. DU145 cells were incubated with different concentrations of UA for 4 h. As shown in Fig. 5a (left panel), UA inhibited the constitutive activation of STAT3 in DU145 cells in a dose-dependent manner, with maximum inhibition occurring at around 50 μM. As shown in Fig. 5a (right panel), the inhibition was time dependent, with maximum inhibition occurring at around 4–6 h with no effect on the expression of STAT3 protein. This data supports the predictive data on STAT3 inhibition as shown in Fig. 1b (right panel).

Effect of UA on constitutive STAT3 phosphorylation. a Left panel UA suppresses phospho-STAT3 levels in a dose-dependent manner. DU145 cells (5 × 105/ml) were treated with the indicated concentrations of UA for indicated dose. Whole-cell extracts were prepared, 30 μg of protein was resolved on 10% SDS–PAGE gel, electrotransferred onto nitrocellulose membranes, and probed with antibodies specific for phospho-STAT3. Equal protein loading was evaluated by STAT3. The results shown are representative of three independent experiments. a Right panel UA suppresses constitutive phospho-STAT3 levels in a time-dependent manner. DU145 cells (5 × 105/ml) were treated with the 50 μM UA for the indicated times and probed for phospho-STAT3 as described in “Materials and methods”. The same blots were stripped and reprobed with STAT3 antibody to verify equal protein loading. The results shown are representative of three independent experiments. b Left panel UA inhibits constitutively active phospho-Src in a time-dependent manner. DU145 cells (5 × 105/ml) were treated with 50 μM UA for the indicated times. Whole-cell extracts were probed for phospho-Src, as described in “Materials and methods”. The same blots were stripped and reprobed with Src antibody to verify equal protein loading. b Right panel effect of Src knockdown on DU145 apoptosis. DU145 (5 × 105/ml) cells were transfected with either Src siRNA or scrambled siRNA (50 nM). After 48 h, cells were treated with 50 μM UA for 72 h and analyzed for apoptosis (Sub-G1 accumulation). c UA suppresses constitutively active phospho-JAK2. DU145 cells (5 × 105/ml) were treated with the 50 μM UA for the indicated times. Whole-cell extracts were probed for phospho-JAK2, as described in “Materials and methods”. The same blots were stripped and reprobed with JAK2 antibody to verify equal protein loading. d Left panel UA suppresses inducible phospho-STAT3 levels in a time-dependent manner. LNCaP cells (5 × 105/ml) were treated with 50 μM UA for the indicated times. Whole-cell extracts were prepared and probed for phospho-STAT3. The same blots were stripped and reprobed with STAT3 antibody to verify equal protein loading. The results shown are representative of three independent experiments. d Right panel UA suppresses inducible phospho-JAK2 levels in a time-dependent manner. LNCaP cells (5 × 105/ml) were treated with the indicated 50 μM UA for the indicated times. Whole-cell extracts were prepared and probed for phospho-JAK2. The same blots were stripped and reprobed with JAK2 antibody to verify equal protein loading. e Left panel UA inhibits constitutive activation of STAT3-luciferase reporter gene expression. DU145 cells (1 × 105/ml) were transfected with STAT3-luciferase (STAT3-Luc) and β-galactosidase reporter plasmid, incubated for 24 h, and treated with 10, 25, or 50 μM UA for 6 h. Whole-cell extracts were then prepared in reporter lysis buffer and analyzed for luciferase activity. e Right panel UA inhibits inducible STAT3 in LNCaP cells. LNCaP cells (1 × 105/ml) were transfected with STAT3-luciferase (STAT3-Luc) and β-galactosidase reporter plasmid, incubated for 24 h, and treated with 10, 25, or 50 μM UA for 6 h and then stimulated with IL-6 (10 ng/ml) for 24 h. Whole-cell extracts were then prepared in reporter lysis buffer and analyzed for luciferase activity. The results shown are normalized to β-galactosidase activity and representative of three independent experiments. *p < 0.05
UA suppresses constitutive activation of Src and JAK2 kinases
STAT3 has also been reported to be activated by soluble tyrosine kinases of the Src and JAK family [4]. Hence, we determined the effect of UA on constitutive activation of Src and JAK2 kinase in DU145 cells. We found that UA suppressed the constitutive phosphorylation of Src kinase (Fig. 5b, left panel) and JAK2 (Fig. 5c) in a time-dependent manner. The level of non-phosphorylated Src remained unchanged under the same experimental conditions. These experimental results correlate with predictive data shown in Fig. 1b (right panel). Moreover, we observed that deletion of Src substantially reduced the apoptotic effects of UA as evidenced by a decrease in sub-G1 population in DU145 cells (Fig. 5b, right panel), thereby indicating a critical role of Src in UA-induced apoptosis.
UA represses IL-6-inducible STAT3 and JAK2 phosphorylation in LNCaP cells
IL-6 has been reported to induce STAT3 and JAK2 phosphorylation [5]. We next determined whether UA could inhibit IL-6-induced STAT3 and JAK2 phosphorylation in LNCaP cells. We found that IL-6-induced STAT3 phosphorylation was suppressed by UA in a time-dependent manner. Exposure of cells to UA for 6 h was sufficient to completely suppress IL-6-induced STAT3 and JAK2 phosphorylation (Fig. 5d, left and right panels). UA did not have any effect on non-phosphorylated STAT3 and JAK2 proteins.
UA suppresses constitutive and IL-6-induced STAT-3 reporter gene expression
Our above results clearly indicated that UA inhibited the phosphorylation of STAT3. We next determined whether UA affects STAT3-dependent gene transcription. When DU145 cells were transiently transfected with the pSTAT3-Luc construct, STAT3-mediated luciferase gene expression was increased and treatment with different concentrations of UA suppressed STAT3 reporter activity in a dose-dependent manner (Fig. 5e, left panel).
We next determined whether UA affects IL-6-inducible STAT3-dependent gene transcription. When LNCaP cells transiently transfected with the pSTAT3-Luc construct were stimulated with IL-6, STAT3-mediated luciferase gene expression was increased. When the cells were pretreated with UA, IL-6-induced STAT3 activity was inhibited in a dose-dependent manner (Fig. 5e, right panel).
UA downregulates the expression of NF-κB and STAT3-dependent gene products involved in survival and angiogenesis
It has been reported that NF-κB and STAT3 regulate the expression of the anti-apoptotic proteins Bcl-2, Bcl-xL, XIAP and survivin, and angiogenic (VEGF) gene products [13, 14]. We next investigated whether UA could modulate constitutive expression of these proteins. We found that UA blocked constitutive expression of these anti-apoptotic proteins in a time-dependent manner, with maximum inhibition occurring at 24 h (Fig. 6a). In addition, UA blocked constitutive expression of VEGF in a time-dependent manner, with maximum inhibition occurring at 24 h (Fig. 6a). These results correlate with predictive data shown in Fig. 1c (right panel) and Fig. 1d (right panel).

a Effect of UA on NF-κB and STAT3 regulated anti-apoptotic gene products. DU145 cells (5 × 105/ml) were treated with 50 μm UA for the indicated time, whole-cell extracts were prepared. Thirty micrograms of the protein was resolved on 10% gel and the separated proteins were electrotransferred to nitrocellulose membrane. The membrane was probed with antibodies specific for Bcl-2, Bcl-xL, XIAP, survivin, and VEGF. The same blots were stripped and reprobed with β-actin antibody to verify equal protein loading. b Left panel UA inhibits the prostate cancer growth in vivo. DU145 cells (5 × 106/ml) were injected into the left flank of mice, and the s.c. xenograft models were established as described in “Materials and methods”. The diameters were measured twice a week for 6 weeks using a Vernier caliper, and the tumor volume was calculated using the formula (L × W 2)/2 where W is the shortest diameter and L is the longest diameter (n = 6). Data is represented as mean tumor volume ± SE. *p < 0.05 for control group versus UA treatment group at the end of 6 weeks treatment. b Right panel effect of UA on body weight of mice. The mice were weighed twice a week for 6 weeks. The data is represented as mean body weight in grams ± SE. c Left panel mass spectrometric analysis of UA in mice serum. i Selected ion chromatograms of serum extracts from mice that had not received UA did not show any of the peak. ii Full scan spectrum showing the m/z peak with a molecular mass of 455 corresponds to UA. iii, iv Retention time was 3.52 min and was the same for both standard and sample. The chromatograms are representative of three independent experiments. For details of standard and sample preparation, refer to “Materials and methods”. c Right panel UA serum concentration was calculated using Analyst software 1.4.2 from the linear regression equation of the peak area ratio against the concentration of the calibration curve. The date is represented as average UA concentration (ng/ml) ± SE. d Immunohistochemical analysis of prostate tumor tissues for VEGF and caspase-3. Paraffin-embedded tumor tissue sections were processed as described in “Materials and methods”. The sections were then probed with antibodies specific for VEGF and caspase-3. The sections were then scored for percentage positive staining for the given biomarker. The photographs were taken at the magnification of ×200
UA inhibits the growth of prostate cancer in vivo
Our predictive proteomics and in vitro experiments so far have shown that UA is a potent inhibitor of NF-κB and STAT3 signaling cascades in prostate cancer cell lines. Next, we analyzed whether UA can inhibit the growth of prostate cancer DU145 xenograft in nude mice. Male nude mice were injected s.c. with DU145 cells and treated with UA (200 mg/kg b.w.) administered i.p. for 6 weeks (twice per week). We found that UA significantly suppressed the tumor growth in vivo following 6 weeks of treatment (Fig. 6b, left panel). At the end of experiment, insignificant change in body weight of UA-treated mice (Fig. 6b, right panel) was observed, thereby indicating that UA is a non-toxic compound. Immunohistochemical analysis of tumor tissue sections for VEGF (marker for angiogenesis) and caspase-3 (marker for apoptosis) showed substantial decrease in VEGF expression and an increase in caspase-3 expression in UA-treated mice as compared to control group (Fig. 6d, left and right panels). These results provide strong evidence of anti-angiogenic and pro-apoptotic effects of UA in prostate cancer.
Detection of UA in serum
We next determined the bioavailability of UA or its metabolites mice serum. MS analysis with ion-selected monitoring yielding molecular ions of m/z 455 corresponds to UA molecular mass. Serum samples were processed to isolate UA, analyzed by ion-selected monitoring in MS mode at the indicated m/z values (Fig. 6c, ii, left panel). The retention time for UA was 3.52 min (Fig. 6c, iii–iv, left panel). UA afforded peak with retention times identical to those of standard. Selected ion chromatograms of control serum extracts from mice that had not received UA did not show any peak (Fig. 6c, i, left panel). The average serum concentration in mice administered with UA was 515 ng/ml (Fig. 6c, right panel). UA does not generate any metabolites and thus no bio-transformed metabolites were detected in serum (Fig. 6c, left panel). UA detected in mice serum at the end of the study was far above the concentration required to exhibit its effects in vitro. The circulating concentration was sufficient to elicit significant biological effects as evidenced by inhibition of prostate tumor growth in nude mice.
Discussion
The aim of the present study was to elucidate the molecular mechanism(s) of anti-cancer effects of UA in prostate cancer cell lines and to investigate its effect on the growth of prostate cancer in vivo. We observed that UA suppressed both constitutive and TNF-α-induced NF-κB activation in prostate cancer cells as evidenced by suppression of NF-κB DNA binding activity. NF-κB inhibition was due to inhibition of IKK activation leading to suppression of IκBα phosphorylation and degradation, and phosphorylation and translocation of p65 and NF-κB dependent reporter activity. UA also exerted potent inhibitory effects on both constitutive and IL-6-inducible STAT3 activation in prostate cancer cells concomitant with suppression of upstream Src and JAK2 kinases. UA further downregulated the expression of various NF-κB and STAT3-regulated gene products involved in anti-apoptosis, metastasis, and angiogenesis. This hypothesis was also tested in a virtual predictive tumor cell platform with an NF-κB activity inhibition in the tumor cell generating more aligned biomarker trends as seen experimentally with UA effects on prostate cancer cells when compared to STAT3 inhibition alone. This downregulation led to the inhibition of proliferation, induction of apoptosis, and significant suppression of the growth of prostate cancer xenografts in vivo.
Although UA has been shown to inhibit constitutive NF-κB activation in different tumor cell lines [30], its effect on NF-κB signaling in prostate cancer cell lines has not been investigated before. Our results demonstrate for the first time that UA can indeed suppress both constitutive NF-κB activation in DU145 cells and TNF-α-induced NF-κB activation in LNCaP cells as also confirmed by the predictive studies. The inhibition of IKK activation by UA suggests that it abolishes NF-κB activation in prostate cancer cells through a suppression of IKK phosphorylation. Several kinases, such as MEKK1, MEKK3, PKC, glycogen synthase kinase-3β, TAK1, PDK1, and AKT [31], have been reported to function upstream of IKK. A recent study suggests that UA can induce apoptosis through suppression of AKT activation in androgen-independent PC-3 cells [12]. We also found that UA can suppress constitutive AKT activation in LNCaP cells, although it had a minimal effect on PDK1 phosphorylation. In addition, we observed that siRNA-targeted inhibition of PTEN significantly sensitized PTEN wild-type DU145 cells to the anti-proliferative effects of UA. These results are in line with a previously published report showing that liposomal encapsulated curcumin and resveratrol can prevent prostate cancer incidence in PTEN null mice [32]. Overall, these results thus indicate that UA-induced inhibition of IKK activation may be mediated through the suppression of AKT activation.
We further observed that UA could suppress both constitutive and inducible STAT3 activation in prostate cancer cells, and that these effects of UA on STAT3 phosphorylation correlated with the suppression of upstream protein tyrosine kinases c-Src and JAK2. Previous studies have indicated that Src and JAK2 kinase activities cooperate to mediate constitutive activation of STAT3 [33]. Our findings suggest that UA can block cooperation of Src and JAK2 involved in tyrosine phosphorylation of STAT3 and deletion of Src can substantially reduce the apoptotic effects of UA. We also observed that UA suppressed IL-6-induced reporter activity of STAT3. These results are consistent with another report in which UA was found to suppress activation of STAT3 and its regulated gene products in multiple myeloma cell lines [8].
STAT3 phosphorylation plays a critical role in proliferation and survival of tumor cells [4]. Several types of cancers have been shown to express constitutively active STAT3 [4, 33]. The suppression of constitutively active STAT3 in prostate cancer cells raises the possibility that this novel STAT3 inhibitor might also inhibit constitutively activated STAT3 in other types of cancer cells. Interestingly, a recent report indicated that STAT3 can prolong NF-κB nuclear retention through acetyltransferase p300-mediated RelA acetylation, thereby interfering with NF-κB nuclear export [34]. Thus, it is possible that suppression of NF-κB activation may mediate inhibition of STAT3 activation by UA. This assumption is supported by the predictive proteomics analysis that it is indeed NF-κB inhibition that mediates a 45% inhibition of STAT3. Pathak et al. have also reported that UA can induce the expression of a protein tyrosine phosphatase (PTP), SHP-1 expression in multiple myeloma cells [8]. Whether UA-induced inhibition of STAT3 activation also involves a protein tyrosine phosphatase (PTP) in prostate cancer needs additional investigation.
We also found that UA suppressed the expression of several NF-κB and STAT3-regulated genes, including anti-apoptotic gene products (Bcl-2, Bcl-xL, XIAP, and survivin) and angiogenic gene product (VEGF). The downregulation of the expression of Bcl-2, Bcl-xL, XIAP, and survivin induces apoptosis in prostate cancer cells as evidenced by annexin V staining and DNA fragmentation results. These observations are consistent with recent reports in which UA was demonstrated to induce apoptosis in prostate cancer cells through downregulation of Bcl-2 and activation of JNK [10]. The down modulation of VEGF expression as reported here may also explain the anti-angiogenic potential of this triterpene that requires further investigation.
Whether these in vitro observations with UA has any relevance to that in vivo was also investigated. Our results show for the first time that UA significantly suppressed prostate tumor growth in nude mice without any significant decrease in body weight. To the best of our knowledge, no prior studies with UA in xenograft prostate cancer models have been reported. We next determined the systemic bioavailability of UA in serum samples obtained from nude mice. UA was detected in all serum samples 24 h after last injection. Systemic bioavailability of UA was in nanogram range and metabolites of UA were not detected in the samples. These results indicate that UA is well absorbed in the mouse peritoneum and supports the role of UA as a potent compound for chemoprevention and therapy of prostate cancer. Overall, our predictive proteomics and experiment results clearly indicate that anti-proliferative and pro-apoptotic effects of UA in prostate cancer are mediated through suppression of multiple cell survival pathways and provide a sound basis for pursuing the use of UA further, either alone or in combination with existing therapy for the therapeutic intervention of prostate cancer.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ (2008) Cancer statistics, 2008. CA Cancer J Clin 58:71–96
Damber JE, Aus G (2008) Prostate cancer. Lancet 371:1710–1721
Vasto S, Carruba G, Candore G, Italiano E, Di Bona D, Caruso C (2008) Inflammation and prostate cancer. Future Oncol 4:637–645
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9:798–809
Grivennikov SI, Karin M (2010) Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 21:11–19
Liu J (1995) Pharmacology of oleanolic acid and ursolic acid. J Ethnopharmacol 49:57–68
Kassi E, Papoutsi Z, Pratsinis H, Aligiannis N, Manoussakis M, Moutsatsou P (2007) Ursolic acid, a naturally occurring triterpenoid, demonstrates anticancer activity on human prostate cancer cells. J Cancer Res Clin Oncol 133:493–500
Pathak AK, Bhutani M, Nair AS, Ahn KS, Chakraborty A, Kadara H, Guha S, Sethi G, Aggarwal BB (2007) Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol Cancer Res 5:943–955
Manu KA, Kuttan G (2008) Ursolic acid induces apoptosis by activating p53 and caspase-3 gene expressions and suppressing NF-kappaB mediated activation of bcl-2 in B16F-10 melanoma cells. Int Immunopharmacol 8:974–981
Kassi E, Sourlingas TG, Spiliotaki M, Papoutsi Z, Pratsinis H, Aligiannis N, Moutsatsou P (2009) Ursolic acid triggers apoptosis and Bcl-2 downregulation in MCF-7 breast cancer cells. Cancer Invest 27:723–733
Kiran MS, Viji RI, Sameer Kumar VB, Sudhakaran PR (2008) Modulation of angiogenic factors by ursolic acid. Biochem Biophys Res Commun 371:556–560
Zhang Y, Kong C, Zeng Y, Wang L, Li Z, Wang H, Xu C, Sun Y (2010) Ursolic acid induces PC-3 cell apoptosis via activation of JNK and inhibition of Akt pathways in vitro. Mol Carcinog 49:374–385
Li F, Sethi G (2010) Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta 1805:167–180
Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S, Ikeda H, Perrone G, Gorgun G, Patel K, Desai N, Sportelli P, Kapoor S, Vali S, Mukherjee S, Munshi NC, Anderson KC, Raje N (2010) Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther 9:963–975
Tan SM, Li F, Rajendran P, Kumar AP, Hui KM, Sethi G (2010) Identification of beta-escin as a novel inhibitor of signal transducer and activator of transcription 3/Janus-activated kinase 2 signaling pathway that suppresses proliferation and induces apoptosis in human hepatocellular carcinoma cells. J Pharmacol Exp Ther 334:285–293
Rabi T, Shukla S, Gupta S (2008) Betulinic acid suppresses constitutive and TNFalpha-induced NF-kappaB activation and induces apoptosis in human prostate carcinoma PC-3 cells. Mol Carcinog 47:964–973
Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, Remacle J (2001) Development of a sensitive multi-well colorimetric assay for active NFkappaB. Nucleic Acids Res 29:E21
Mukhopadhyay A, Bueso-Ramos C, Chatterjee D, Pantazis P, Aggarwal BB (2001) Curcumin downregulates cell survival mechanisms in human prostate cancer cell lines. Oncogene 20:7597–7609
Li F, Fernandez PP, Rajendran P, Hui KM, Sethi G (2010) Diosgenin, a steroidal saponin, inhibits STAT3 signaling pathway leading to suppression of proliferation and chemosensitization of human hepatocellular carcinoma cells. Cancer Lett 292:197–207
Li F, Rajendran P, Sethi G (2010) Thymoquinone inhibits proliferation, induces apoptosis and chemosensitizes human multiple myeloma cells through suppression of signal transducer and activator of transcription 3 activation pathway. Br J Pharmacol 161:541–554
Mickey DD, Stone KR, Wunderli H, Mickey GH, Vollmer RT, Paulson DF (1977) Heterotransplantation of a human prostatic adenocarcinoma cell line in nude mice. Cancer Res 37:4049–4058
Liao Q, Yang W, Jia Y, Chen X, Gao Q, Bi K (2005) LC–MS determination and pharmacokinetic studies of ursolic acid in rat plasma after administration of the traditional Chinese medicinal preparation Lu-Ying extract. Yakugaku Zasshi 125:509–515
Rajendran P, Ong TH, Chen L, Li F, Shanmugam M, Vali S, Abbasi T, Kapoor S, Sharma A, Kumar AP, Hui KM, Sethi G (2011) Suppression of signal transducer and activator of transcription 3 activation by butein inhibits growth of human hepatocellular carcinoma in vivo. Clin Cancer Res 17:1425–1439
Ghosh S, May MJ, Kopp EB (1998) NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 16:225–260
Sethi G, Sung B, Aggarwal BB (2008) TNF: a master switch for inflammation to cancer. Front Biosci 13:5094–5107
Tanaka H, Fujita N, Tsuruo T (2005) 3-Phosphoinositide-dependent protein kinase-1-mediated IkappaB kinase beta (IkkB) phosphorylation activates NF-kappaB signaling. J Biol Chem 280:40965–40973
Gustin JA, Maehama T, Dixon JE, Donner DB (2001) The PTEN tumor suppressor protein inhibits tumor necrosis factor-induced nuclear factor kappa B activity. J Biol Chem 276:27740–27744
Sethi G, Tergaonkar V (2009) Potential pharmacological control of the NF-kappaB pathway. Trends Pharmacol Sci 30:313–321
Ahn KS, Sethi G, Aggarwal BB (2007) Nuclear factor-kappa B: from clone to clinic. Curr Mol Med 7:619–637
Shishodia S, Majumdar S, Banerjee S, Aggarwal BB (2003) Ursolic acid inhibits nuclear factor-kappaB activation induced by carcinogenic agents through suppression of IkappaBalpha kinase and p65 phosphorylation: correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res 63:4375–4383
Sethi G, Ahn KS, Sandur SK, Lin X, Chaturvedi MM, Aggarwal BB (2006) Indirubin enhances tumor necrosis factor-induced apoptosis through modulation of nuclear factor-kappa B signaling pathway. J Biol Chem 281:23425–23435
Narayanan NK, Nargi D, Randolph C, Narayanan BA (2009) Liposome encapsulation of curcumin and resveratrol in combination reduces prostate cancer incidence in PTEN knockout mice. Int J Cancer 125:1–8
Garcia R, Bowman TL, Niu G, Yu H, Minton S, Muro-Cacho CA, Cox CE, Falcone R, Fairclough R, Parsons S, Laudano A, Gazit A, Levitzki A, Kraker A, Jove R (2001) Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 20:2499–2513
Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, Forman S, Jove R, Pardoll DM, Yu H (2009) Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 15:283–293
Acknowledgments
This work was supported by grants from National Medical Research Council of Singapore (Grant R-184-000-168-275, Grant R-184-000-157-214) to GS. APK was supported by grants from the National Medical Research Council of Singapore (Grant R-713-000-124-213) and Cancer Science Institute of Singapore, Experimental Therapeutics I Program (Grant R-713-001-011-271). KMH was supported by grant from the National Medical Research Council of Singapore, Biomedical Research Council of Singapore, and the Singapore Millennium Foundation.
Conflict of interests
No potential conflict of interest is declared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shanmugam, M.K., Rajendran, P., Li, F. et al. Ursolic acid inhibits multiple cell survival pathways leading to suppression of growth of prostate cancer xenograft in nude mice. J Mol Med 89, 713–727 (2011). https://doi.org/10.1007/s00109-011-0746-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-011-0746-2