
Abstract
Increasing data suggest that the initiation, relapse, and progression of human cancers are driven by specific cell populations within an individual tumor. However, inconsistencies have emerged in precisely defining phenotypic markers that can reliably identify these “cancer stem cells” in nearly every human malignancy studied to date. Multiple myeloma, one of the first tumors postulated to be driven by a rare population of cancer stem cells, is no exception. Similar to other diseases, controversy surrounds the exact phenotype and biology of multiple myeloma cells with the capacity for clonogenic growth. Here, we review the studies that have led to these controversies and discuss potential reasons for these disparate findings. Moreover, we speculate how these inconsistencies may be resolved through studies by integrating advancements in both myeloma and stem cell biology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple myeloma is characterized by the clonal expansion of malignant plasma cells that results in anemia, renal insufficiency, and bone disease [1]. Although plasma cells phenotypically characterize the disease, recent studies have suggested that these cells lack significant proliferative capacity and instead, arise from clonogenic cells that resemble memory B cells [2–5]. However, these results are far from conclusive as other reports suggest that some or all malignant plasma cells have tumorigenic and self-renewal properties [6, 7]. The precise reasons for these disparate experimental findings are unclear, but they bring to light recurring issues regarding cancer stem cells in several human diseases, namely, inconsistencies between reports that describe their phenotypes. For example, differing stem cell phenotypes have been reported in acute lymphocytic leukemia and colorectal, pancreatic, bladder, brain, and breast cancers [8–22]. In this review, we describe the experimental approaches that have been used to identify tumorigenic cells in multiple myeloma, discuss potential factors that may contribute to the conflicting data and speculate on how advancements in the general understanding of myeloma may ultimately resolve these controversies.
Phenotypic identification of cancer stem cells
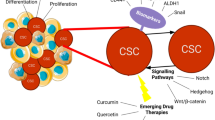
Cancer stem cells have been identified in many human cancers, and several general approaches have been employed to identify markers that distinguish them from bulk tumor cells. In myeloid leukemias and brain tumor, two of the earliest cancers examined, the surface antigens expressed by normal hematopoietic and neural stem cells were found to enrich for clonogenic tumor cells [19, 23–25]. These findings have suggested that human cancers may arise from normal stem cells and retain a cellular hierarchy with self-renewing cancer stem cells giving rise to differentiated cells that ultimately make up the majority of the tumor but lack significant long-term proliferative capacity. Furthermore, cancer stem cells may be isolated using markers expressed by their normal counterparts.
This approach has not been useful in most solid tumors since few surface antigens that mark normal stem cells are known. In these tissues, normal stem cells can be identified by their histological location, such as bulge cells in the skin or within crypts in the gastrointestinal tract or by intracellular antigens that cannot be used to isolate viable cells. Instead, the identification of surface antigens expressed by solid tumor stem cells has been somewhat empiric, such as the use of CD44 and CD24 in breast cancer [21]. These antigens were previously noted to be expressed in breast cancer and hypothesized to play a role in cell motility, metastasis, and disease progression [26, 27]. However, little existing data suggested that they would be differentially expressed by breast cancer stem cells. Nevertheless, this pioneering study by Al Hajj et al. was the first to prospectively identify human solid tumor stem cells, and CD44 expression has been subsequently found to enrich for clonogenic cells in colorectal, pancreatic, and head and neck squamous cell carcinomas [14, 15, 28].
Although no universal stem cell antigen currently exists, several properties common to normal stem cells from different tissues may also be used to identify tumorigenic cells. Normal stem cells are highly resistant to toxic injury because of multiple cellular processes that include high expression of membrane-bound drug transporters and intracellular detoxification enzymes [29, 30]. Tumor regrowth following treatment suggests that clonogenic cells are resistant to therapy, and flow cytometric assays based on these drug resistance mechanisms, such as the side population assay and measurement of intracellular aldehyde dehydrogenase (ALDH) activity have been able to identify clonogenic cells in several cancers [5, 31–33].
Functional assessment of cancer stem cell activity
Following the identification of putative cancer stem cells, it is necessary to evaluate their functional properties. In vitro, the examination of colony formation in semisolid media, such as soft-agar or methylcellulose, has long been used to evaluate clonogenic growth potential. Since colony formation using either of these assays may arise from either stem cells or self-limited progenitors, long-term proliferative potential may serve as a surrogate for self-renewal. Therefore, the ability to form secondary colonies or spheres through serial rounds of replating may aid in distinguishing functionally primitive populations [4].
Since these in vitro methods may not account for cell-extrinsic factors that influence cancer stem cell function, in vivo assays have emerged as the gold-standard to evaluate tumorigenic potential. The development of immunodeficient strains of mice have provided a means of overcoming xenografting barriers and allows the growth of human tumor cells to be assessed. NOD/SCID mice that lack B and T cells have been utilized in both the initial studies identifying clonogenic stem cells in acute myeloid leukemia and subsequently, in many other human cancers [23, 24]. The phenotypic analysis of formed tumors allows the differentiation capacity of injected cells to be assessed. Moreover, self-renewal potential may be demonstrated through serial transplantation.
Although NOD/SCID mice are most frequently used to demonstrate in vivo clonogenic growth potential, recent studies have suggested that significant xenografting barriers persist in these animals that may skew the engraftment capacity of different tumor cell populations. For example, initial studies in acute myeloid leukemia found that CD34+CD38neg tumor cells could engraft NOD/SCID mice. However, a recent report suggested that CD34+CD38+ leukemic cells could also be transplanted if NOD/SCID mice were further treated with an antibody against natural killer cells in addition to the radiation normally used for conditioning [34]. Another study using a more severely immunodeficient mouse strain (NOD/SCID/IL2γreceptorko) that also lacks natural killer cells found that a high proportion of human melanoma cells were capable of engraftment unlike previous reports using NOD/SCID mice [35–37]. Furthermore, no specific phenotypic population of engrafting cells could be identified suggesting that at least in melanoma, the relationship between distinct phenotypes and functional capacities may not be firmly linked.
It is likely that some of the differences between reports describing the phenotype of putative cancer stem cells may reflect the distinct requirements for cellular growth and expansion within each of these assay systems. In the following sections, we will focus on the identification of clonogenic cells in multiple myeloma and the potential role that the various assays utilized to study their functional properties may have played in discrepancies regarding their phenotype.
Phenotypic heterogeneity in multiple myeloma
Following antigen exposure, normal naïve B cells with immunoglobulin V(D)J gene rearrangements engage in germinal center reactions where they undergo class switch recombination and somatic hypermutation. The immunoglobulin gene sequences in multiple myeloma plasma cells are somatically hypermutated and remain constant throughout the clinical course suggesting that the disease arises from a postgerminal center B cell [38, 39]. Several studies have found that multiple myeloma patients harbor phenotypic B cells expressing the immunoglobulin gene sequence and idiotype unique to the individual myeloma clone [40–44]. Therefore, multiple myeloma may arise from these clonotypic B cells and recapitulate aspects of normal plasma cell development. The ability to induce differentiation of clonotypic B cells into plasma cells in vitro provides support for this theory [45, 46]. These findings imply that clonotypic B cells may be involved in the human disease process but offer no definitive proof that B cells represent the proliferating tumor compartment.
Clonogenic growth of multiple myeloma
The clonogenic potential in multiple myeloma has been examined using both in vitro and in vivo assays, and a comparison of the merits of these approaches is summarized in Fig. 1. The first successful in vitro system capable of growing human myeloma colonies was described by Hamburger and Salmon and later used soft agar along with a feeder of either human erythrocytes or mouse spleen cells [47]. This system suggested that the clonogenic frequency of clinical myeloma specimens was 0.001% to 0.1% of all tumor cells. These results were confirmed in our studies utilizing methylcellulose supplemented with lymphocyte conditioned media as a source of growth factors [4]. Furthermore, we found that myeloma plasma cells characterized by surface expression of CD138 were incapable of significant clonogenic growth, but that CD138neg cells expressing typical B cell surface antigens produced tumor colonies that could be serially passaged. More recently, a novel in vitro 3D stromal culture system has been developed that recapitulates both cellular and extracellular features of the bone marrow, and tumor growth in this assay also appears to arise from clonotypic B cells [48].

A comparison of in vitro and in vivo cancer stem cell functional assays
The first in vivo assay capable of supporting the growth of human myeloma was developed by Yaccoby and Epstein using a SCID mouse implanted with human fetal bone fragments to create a humanized microenvironment (SCID-hu) [6, 7]. Using this model, mature CD38++CD45neg plasma cells generated disease that included circulating M protein, hypercalcemia, and resorption of the human bone fragment. In contrast, CD38negCD45pos peripheral blood B cells were unable to engraft suggesting that only mature plasma cells are clonogenic. In a subsequent study, Pilarski et al. found that G-CSF mobilized peripheral blood specimens resulted in lytic bone lesions and malignant plasma cells in the bone marrow following intracardiac or direct intraosseous injection into NOD/SCID mice [2]. Since the injected cells did not contain phenotypic plasma cells, the authors concluded that clonotypic B cells were responsible for tumor formation in this model. The same group subsequently demonstrated that clonotypic B cells isolated from an advanced myeloma patient could generate disease in NOD/SCID mice [3]. Similarly, we recently found that CD138+ plasma cells failed to engraft NOD/SCID mice following tail vein injection [4, 5]. However, peripheral blood cells lacking CD138 and expressing the memory B cell markers CD19 and CD27 were able to serially engraft mice and give rise to clonotypic CD138+ plasma cells functionally capable of producing circulating M protein. Therefore, these studies suggest that clonotypic B cells, rather than CD138+ plasma cells, are clonogenic in vivo.
It is likely that differences between functional assays contribute to the discrepancies in reported cancer stem cell phenotypes. Although the capacity for self-renewal may be cell intrinsic, external factors within the stem cell niche also regulate this process [49]. Little data exists regarding the role or existence of the niche in regulating cancer stem cells, but the tumor microenvironment has emerged as a major focus of myeloma biology and serves as a therapeutic target in the disease [50]. Therefore, differences in extrinsic factors within each clonogenic assay may contribute to the ability of specific cell types to home, survive, and proliferate in vivo. For instance, the human fetal bone fragments used in the SCID-hu mice have the ability to support plasma cells but may lack factors required for the growth of human B cells. On the other hand, the bone marrow in NOD/SCID mice may not initially support mature plasma cells, but the engraftment of B cells may induce changes that subsequently allow plasma cell survival. The site of injection may also play a role in tumorigenic potential, and a variety of methods (e.g., intravenous, intracardiac, or intraosseous) have been used to assess myeloma growth, and it is possible that differences in cell trafficking also play a role in determining which cells can engraft. Other experimental differences that may contribute to discrepancies in the stem cell phenotype include distinct methods used to isolate specific cell populations, such as positive or negative selection or the derivation of tumor cells from the blood or bone marrow.
Like most human cancers, multiple myeloma displays a wide clinical biology that may also contribute to the reported differences in stem cell phenotypes. An individual patient’s stage of disease and previous therapy may impact the biology of tumorigenic cells. Moreover, several recurrent genetic alterations have been described in multiple myeloma [51], and it is possible that multiple myeloma represents a number of biologically distinct diseases each containing different initiating cells. For example, the t(4;14) chromosomal translocations may carry an especially poor prognosis, and it is possible that tumorigenic cells in these cases significantly differs from those carrying other genetic abnormalities.
Future directions in multiple myeloma stem cell research
The most definitive identification of myeloma stem cells would involve assessing the tumorigenic potential of candidate cell populations though syngeneic transplantation studies. Obviously, human studies of this nature cannot be carried out, but several mouse models of myeloma have been generated and may be useful in evaluating the clonogenic potential of specific cell populations. Several animal models displaying plasmacytosis have been derived from the aberrant expression of c-Myc. In one model, the coexpression of Bcl-XL and c-Myc in B cells results in polyclonal plasma cell expansion that later progresses to monoclonal plasmablastic malignancies [52]. In a separate model, c-Myc overexpression in postgerminal center B cells results in plasma cell expansion, serum monoclonal immunoglobulin, and deposition of the M protein in renal glomeruli [53]. Overexpression of the transcription factor Xbp-1, which is required for the differentiation of B cells into plasma cells in B cells, also results in the plasmacytosis and lytic bone lesions [54]. Despite recapitulation of some aspects of human disease, deregulation of c-Myc is thought to be a late event in human myeloma and specific genetic lesions that result in the overexpression of Xbp-1 have not been described. Therefore, the true fidelity of these models is largely unknown. As the genetic events associated with myeloma are better understood, it is possible that these will provide the basis for even better models to study myeloma stem cell biology. For example, recurrent chromosomal translocations that bring genes such as CYCLIN D1, FGFR3, MMSET, and c-MAF under the regulation of immunoglobulin enhancer elements are thought to represent disease initiating events since these can be found in monoclonal gammopathy of unknown significance (MGUS) [51], and transgenic models that include these abnormalities may be able to better recapitulate the full spectrum of human disease.
A major question in cancer stem cell biology remains as to what role they play in disease progression. In chronic myeloid leukemia, studies have suggested that the transition from chronic phase to blast crisis is mediated by changes in Wnt/β-catenin signaling [55]. Interestingly, these changes appear to occur not within the originating cell responsible for chronic phase, but rather within a phenotypically more mature progenitor. Thus, it is possible that cancer progression is driven by genetic or epigenetic events that confer self-renewal to more rapidly proliferating but previously self-limited progenitor compartments and that a “shift” in the clinically relevant stem cell occurs. Disease progression in multiple myeloma has been associated with several recurrent genetic events including amplifications of chromosome 1q, mutations in RAS and inactivation of p53 [56–59]. Therefore, it is possible that these lesions can be used to mark or track specific cellular compartments and provide evidence for their involvement during disease progression.
Conclusions
The true nature and phenotype of the cancer stem cell in multiple myeloma remains unclear and controversial. However, improvements in understanding the capacities and limitations of the clonogenic assays used to assess their functional properties, development of novel animal models of the disease, and incorporation of the growing understanding of myeloma biology may provide insights into the true nature of the cell responsible for clonogenic growth. The precise identification of the cancer stem cell in multiple myeloma may allow for development of novel therapeutic strategies that inhibit tumor regrowth, delay clinical relapse, and improve long-term outcome such as overall survival. Moreover, definite understanding of how or if the myeloma stem cell may be biologically distinct when driven by specific genetic lesions or over the course of human disease is likely to be highly relevant for many other cancers.
References
Kyle RA, Rajkumar SV (2004) Multiple myeloma. N Engl J Med 351:1860–1873
Pilarski LM, Hipperson G, Seeberger K, Pruski E, Coupland RW, Belch AR (2000) Myeloma progenitors in the blood of patients with aggressive or minimal disease: engraftment and self-renewal of primary human myeloma in the bone marrow of NOD SCID mice. Blood 95:1056–1065
Pilarski LM, Seeberger K, Coupland RW, Eshpeter A, Keats JJ, Taylor BJ, Belch AR (2002) Leukemic B cells clonally identical to myeloma plasma cells are myelomagenic in NOD/SCID mice. Exp Hematol 30:221–228
Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, Tanhehco Y, Smith BD, Civin CI, Jones RJ (2004) Characterization of clonogenic multiple myeloma cells. Blood 103:2332–2336
Matsui W, Wang Q, Barber JP, Brennan S, Smith BD, Borrello I, McNiece I, Lin L, Ambinder RF, Peacock C, Watkins DN, Huff CA, Jones RJ (2008) Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res 68:190–197
Yaccoby S, Barlogie B, Epstein J (1998) Primary myeloma cells growing in SCID-hu mice: a model for studying the biology and treatment of myeloma and its manifestations. Blood 92:2908–2913
Yaccoby S, Epstein J (1999) The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood 94:3576–3582
Cox CV, Evely RS, Oakhill A, Pamphilon DH, Goulden NJ, Blair A (2004) Characterization of acute lymphoblastic leukemia progenitor cells. Blood 104:2919–2925
George AA, Franklin J, Kerkof K, Shah AJ, Price M, Tsark E, Bockstoce D, Yao D, Hart N, Carcich S, Parkman R, Crooks GM, Weinberg K (2001) Detection of leukemic cells in the CD34+CD38{-} bone marrow progenitor population in children with acute lymphoblastic leukemia. Blood 97:3925
Hotfilder M, Rottgers S, Rosemann A, Jurgens H, Harbott J, Vormoor J (2002) Immature CD34+CD19- progenitor/stem cells in TEL/AML1-positive acute lymphoblastic leukemia are genetically and functionally normal. Blood 100:640–646
Hotfilder M, Rottgers S, Rosemann A, Schrauder A, Schrappe M, Pieters R, Jurgens H, Harbott J, Vormoor J (2005) Leukemic Stem Cells in Childhood High-Risk ALL/t(9;22) and t(4;11) Are Present in Primitive Lymphoid-Restricted CD34+CD19- Cells. Cancer Res 65:1442–1449
Wang L, O'Leary H, Fortney J, Gibson LF (2007) Ph+/VE-cadherin+ identifies a stem cell like population of acute lymphoblastic leukemia sustained by bone marrow niche cells. Blood 110:3334–3344
O'Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445:106–110
Dalerba P, Dylla SJ, Ik P, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, Shelton AA, Parmiani G, Castelli C, Clarke MF (2007) Phenotypic characterization of human colorectal cancer stem cells. Proceedings of the National Academy of Sciences 104:10158–10163
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM (2007) Identification of pancreatic cancer stem cells. Cancer Res 67:1030–1037
Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C (2007) Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 1:313–323
He X, Marchionni L, Hansel DE, Wayne Yu, Sood A, Yang J, Parmigiani G, Matsui W, Berman DM (2009) Differentiation of a Highly Tumorigenic Basal Cell Compartment in Urothelial Carcinoma. Stem Cells 27:1487–1495
Chan K, Espinosa I, Kim J, Ailles L, Zahilay G, Gill H, Presti J, van de Rijn M, Beachy P, Shortliffe L, Weissman I (2008) Molecular profiling reveals heterogeneity of active self-renewal pathways in bladder cancer stem cells. AACR Meeting Abstracts 2008:4998
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432:396–401
Read T-A, Fogarty MP, Markant SL, McLendon RE, Wei Z, Ellison DW, Febbo PG, Wechsler-Reya RJ (2009) Identification of CD15 as a Marker for Tumor-Propagating Cells in a Mouse Model of Medulloblastoma. Cancer Cell 15:135–147
Al Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 100:3983–3988
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1:555–567
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang TC-CJ, Minden M, Paterson B, Caligiuri MA, Dick JE (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367:645–648
Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3:730–737
Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI (2003) Cancerous stem cells can arise from pediatric brain tumors. Proceedings of the National Academy of Sciences 100:15178–15183
Aigner S, Ramos C, Hafezi-Moghadam A, Lawrence M, Friederichs J, Altevogt P, Ley K (1998) CD24 mediates rolling of breast carcinoma cells on P-selectin. FASEB J 12:1241–1251
Joensuu H, Klemi PJ, Toikkanen S, Jalkanen S (1993) Glycoprotein CD44 expression and its association with survival in breast cancer. Am J Pathol 143:867–874
Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE (2007) Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proceedings of the National Academy of Sciences 104:973–978
Goodell MA, Rosenzweig M, Kim H, Marks DF, DeMaria M, Paradis G, Grupp SA, Sieff CA, Mulligan RC, Johnson RP (1997) Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med 3:1337–1345
Hess DA, Meyerrose TE, Wirthlin L, Craft TP, Herrbrich PE, Creer MH, Nolta JA (2004) Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood 104:1648–1655
Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J, Gurney AL (2008) Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS ONE 3:e2428
Wicha MS, Liu S, Dontu G (2006) Cancer stem cells: an old idea—a paradigm shift. Cancer Res 66:1883–1890
Jones RJ, Gocke CD, Kasamon YL, Miller CB, Perkins B, Barber JP, Vala MS, Gerber JM, Gellert LL, Siedner M, Lemas MV, Brennan S, Ambinder RF, Matsui W (2009) Circulating clonotypic B cells in classic Hodgkin lymphoma. Blood 113:5920–5926. doi:10.1182/blood-2008-11-189688
Taussig DC, Miraki-Moud F, njos-Afonso F, Pearce DJ, Allen K, Ridler C, Lillington D, Oakervee H, Cavenagh J, Agrawal SG, Lister TA, Gribben JG, Bonnet D (2008) Anti-CD38 antibody-mediated clearance of human repopulating cells masks the heterogeneity of leukemia-initiating cells. Blood 112:568–575
Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder DE, Herlyn M (2005) A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res 65:9328–9337
Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C, Fuhlbrigge RC, Kupper TS, Sayegh MH, Frank MH (2008) Identification of cells initiating human melanomas. Nature 451:345–349
Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ (2008) Efficient tumour formation by single human melanoma cells. Nature 456:593–598
Bakkus MH, Heirman C, Van Riet I, Van Camp B, Thielemans K (1992) Evidence that multiple myeloma Ig heavy chain VDJ genes contain somatic mutations but show no intraclonal variation. Blood 80:2326–2335
Sahota SS, Leo R, Hamblin TJ, Stevenson FK (1997) Myeloma VL and VH gene sequences reveal a complementary imprint of antigen selection in tumor cells. Blood 89:219–226
Bakkus MH, Van RI, Van Camp B, Thielemans K (1994) Evidence that the clonogenic cell in multiple myeloma originates from a pre-switched but somatically mutated B cell. Br J Haematol 87:68–74
Billadeau D, Ahmann G, Greipp P, Van Ness B (1993) The bone marrow of multiple myeloma patients contains B cell populations at different stages of differentiation that are clonally related to the malignant plasma cell. J Exp Med 178:1023–1031
Bergsagel PL, Smith AM, Szczepek A, Mant MJ, Belch AR, Pilarski LM (1995) In multiple myeloma, clonotypic B lymphocytes are detectable among CD19+ peripheral blood cells expressing CD38, CD56, and monotypic Ig light chain. Blood 85:436–447
Chen BJ, Epstein J (1996) Circulating clonal lymphocytes in myeloma constitute a minor subpopulation of B cells. Blood 87:1972–1976
Rasmussen T, Kastrup J, Knudsen LM, Johnsen HE (1999) High numbers of clonal CD19+ cells in the peripheral blood of a patient with multiple myeloma. Br J Haematol 105:265–267
Caligaris-Cappio F, Bergui L, Tesio L, Pizzolo G, Malavasi F, Chilosi M, Campana D, van CB, Janossy G (1985) Identification of malignant plasma cell precursors in the bone marrow of multiple myeloma. J Clin Invest 76:1243–1251
Bergui L, Schena M, Gaidano G, Riva M, Caligaris-Cappio F (1989) Interleukin 3 and interleukin 6 synergistically promote the proliferation and differentiation of malignant plasma cell precursors in multiple myeloma. J ExpMed 170:613–618
Hamburger AW, Salmon SE (1977) Primary bioassay of human tumor stem cells. Science 197:461–463
Kirshner J, Thulien KJ, Martin LD, Debes Marun C, Reiman T, Belch AR, Pilarski LM (2008) A unique three-dimensional model for evaluating the impact of therapy on multiple myeloma. Blood 112:2935–2945. doi:10.1182/blood-2008-02-142430
Scadden DT (2006) The stem-cell niche as an entity of action. Nature 441:1075–1079
Mitsiades CS, Mitsiades NS, Richardson PG, Munshi NC, Anderson KC (2007) Multiple myeloma: a prototypic disease model for the characterization and therapeutic targeting of interactions between tumor cells and their local microenvironment. J Cell Biochem 101:950–968
Chng WJ, Glebov O, Bergsagel PL, Kuehl WM (2007) Genetic events in the pathogenesis of multiple myeloma. Best Practice & Research Clinical Haematology 20:571–596
Boylan KLM, Gosse MA, Staggs SE, Janz S, Grindle S, Kansas GS, Van Ness BG (2007) A transgenic mouse model of plasma cell malignancy shows phenotypic, cytogenetic, and gene expression heterogeneity similar to human multiple myeloma. Cancer Res 67:4069–4078
Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, Valdez R, Palmer SE, Haas SS, Stewart AK, Fonseca R, Kremer R, Cattoretti G, Bergsagel PL (2008) AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 13:167–180
Carrasco DR, Sukhdeo K, Protopopova M, Sinha R, Enos M, Carrasco DE, Zheng M, Mani M, Henderson J, Pinkus GS, Munshi N, Horner J, Ivanova EV, Protopopov A, Anderson KC, Tonon G, DePinho RA (2007) The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 11:349–360
Jamieson CHM, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, Sawyers CL, Weissman IL (2004) Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351:657–667
Hanamura I, Stewart JP, Huang Y, Zhan F, Santra M, Sawyer JR, Hollmig K, Zangarri M, Pineda-Roman M, van Rhee F, Cavallo F, Burington B, Crowley J, Tricot G, Barlogie B, Shaughnessy JD Jr (2006) Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood 108:1724–1732
Fonseca R, Van Wier S, Chng W, Ketterling R, Lacy M, Dispenzieri A, Bergsagel P, Rajkumar S, Greipp P, Litzow M (2006) Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia 20:2034–2040
Drach J, Ackermann J, Fritz E, Kromer E, Schuster R, Gisslinger H, DeSantis M, Zojer N, Fiegl M, Roka S, Schuster J, Heinz R, Ludwig H, Huber H (1998) Presence of a p53 gene deletion in patients with multiple myeloma predicts for short survival after conventional-dose chemotherapy. Blood 92:802–809
Portier M, Moles JP, Mazars GR, Jeanteur P, Bataille R, Klein B, Theillet C (1992) p53 and RAS gene mutations in multiple myeloma. Oncogene 7:2539–2543
Acknowledgments
Supported in part by National Institutes of Health grants R01CA127574 and K23CA107040, the Multiple Myeloma Research Foundation, the International Myeloma Foundation, and the Sidney Kimmel Foundation for Cancer Research. William Matsui is a scholar of the Leukemia and Lymphoma Society.
Conflicting interests
We have no conflicting interests related to this review article or the studies discussed in the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brennan, S.K., Matsui, W. Cancer stem cells: controversies in multiple myeloma. J Mol Med 87, 1079–1085 (2009). https://doi.org/10.1007/s00109-009-0531-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-009-0531-7