
Abstract
Transferrin receptors (TfR) are overexpressed in brain tumors, but the pathological relevance has not been fully explored. Here, we show that TfR is an important downstream effector of ets transcription factors that promotes glioma proliferation and increases glioma-evoked neuronal death. TfR mediates iron accumulation and reactive oxygen formation and thereby enhanced proliferation in clonal human glioma lines, as shown by the following experiments: (1) downregulating TfR expression reduced proliferation in vitro and in vivo; (2) forced TfR expression in low-grade glioma accelerated proliferation to the level of high-grade glioma; (3) iron and oxidant chelators attenuated tumor proliferation in vitro and tumor size in vivo. TfR-induced oxidant accumulation modified cellular signaling by inactivating a protein tyrosine phosphatase (low-molecular-weight protein tyrosine phosphatase), activating mitogen-activated protein kinase and Akt and by inactivating p21/cdkn1a and pRB. Inactivation of these cell cycle regulators facilitated S-phase entry. Besides its effect on proliferation, TfR also boosted glutamate release, which caused N-methyl-d-aspartate-receptor-mediated reduction of neuron cell mass. Our results indicate that TfR promotes glioma progression by two mechanisms, an increase in proliferation rate and glutamate production, the latter mechanism providing space for the progressing tumor mass.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cells produce reactive oxidants as metabolic by-products and as messengers for specific cellular signaling pathways [1]. However, under pathological conditions, oxidants are often produced in excess and account for abnormal levels of proliferation, differentiation, migration, and apoptosis [2]. High oxidant levels may even oxidize proteins and lipids and thereby cause cellular transformation [3]. Consequently, many tumors—including gliomas—contain very high levels of reactive oxidants, which initiate tumor proliferation by an unknown mechanism [4].
Glioma is the most common primary brain cancer and represents a morphologically diverse group of tumors [5]. These cancers are rapidly proliferating and leave patients with a mean survival of only 12 months after diagnosis [6]. Gliomas most likely arise from transformed stem and progenitor cells of the brain after combined overactivation of the Ras/mitogen-activated protein kinase (MAPK) and Akt (PKB) pathways [7]. Together with frequently occurring mutations in tumor suppressors of the G1 cell cycle check point, the activated MAPK and Akt signaling maintains high proliferation levels and strong apoptosis resistance in gliomas. In order to progress, gliomas release high amounts of glutamate which induces excitotoxic neuronal cell death [8, 9]. The elimination of neurons is necessary for the tumor to grow since otherwise the progressing glioma would press the brain mass against the skull and the tumor size could be limited by the increasing intracranial pressure.
Gliomas are long known to overexpress transferrin receptors (TfR) and the extent of TfR expression positively correlates with tumor grading [10]. As in other neoplasms, TfR in gliomas induces increased intracellular iron accumulation [11], which can catalyze the generation of reactive oxidants via the “Fenton reaction” [1, 12]. Although in some tumors like breast cancer TfR has been discussed to fulfill growth-factor-like functions [13], the exact role of TfR overexpression in glioma is largely unexplored. How TfR overexpression in glioma is regulated is also not known. Under physiological conditions, transcription of TfR is controlled by binding of ets1 to the TfR promoter region [14, 15] but is also extensively regulated on the posttranscriptional level [16]. In glioma and many other cancers, ets transcription factors control a range of tumor-promoting pathways [17–19].
In our present study, we provide evidence that in gliomas TfR is an important downstream effector of ets transcription factors which induces excess oxidant production, promotes tumor growth, and causes N-methyl-d-aspartate (NMDA)-receptor-mediated reduction of neuronal cell mass.
Materials and methods
Cell cultures
U373 human glioblastoma, 132N1 low-malignancy human astrocytoma, and GL261 mouse glioblastoma cell lines were purchased from the National Cancer Institute, NCI-Frederick (MD, USA) and cultured in Dulbecco’s modified Eagle’s medium with 10% heat-inactivated fetal calf serum (Atlanta Biological), 4 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in 25-cm2 tissue culture flasks (Falcon model 3023, Becton Dickinson, Lincoln Park, NJ, USA). The medium was changed every 2 days and cells were passaged when the cell density reached confluency. Cell cultures were maintained at 37°C in humidified 5% CO2–95% air incubator (Heraeus, Hanau, Germany).
Cell lines
The cDNA-encoding EtsDN (Kind gift from H. Sato), which lacks a transcription activation domain and corresponds to amino acid residues 306–441 [20], was inserted into the pCEP4-IRES-EGFP expression plasmid (Invitrogen), allowing bicistronic expression of EtsDN and EGFP. Bulk U373 EtsDn cultures were obtained by stable transfection of that construct into U373 cells with Lipofectamine 2000 (Invitrogen, Carlsbad, USA). Transfected U373 cells were cultured in the presence of 800 μg/ml hygromycin B, and resistant cells were cloned. U373 wild-type cells were seeded at clonal density and then cultivated to give rise to a number of clonal U373-WT cultures. A tetracycline-inducible system was used to get single clones of etsDn-expressing cells of U373. EtsDn-IRES-EGFP was subcloned in to the pTRE-tight vector (kind gift from M. Gossen). Doxycycline (200 ng/ml) was used to induce the expression of the insert. U373-etsDN cultures were then grown from single clones of the stably transfected cells (as described for U373-WT cells).
Further, we subcloned the open reading frame for TfR (from pCD-TR1; a kind gift from L. Kuhn; [16]) into a cytomegalovirus (CMV)-promoter-containing vector (Rc-CMV; Invitrogen) and then transfected U373 cells constitutively expressing EstDN and WT 1321N1 cells with the TfR construct by using Lipofectamin-2000 Selection of cells stably expressing etsDN + TfR was achieved with hygromycin and G418 (0.6 mg/ml; Gibco, Gaithersburg, MD, USA). Finally, cultures were grown from single clones of U373-etsDN + TfR cells. All reagents were purchased from Sigma (Taufkirchen, Germany), unless otherwise specified.
Hippocampal cell culture
All animals were sacrificed and hippocampal cultures from E19 Wistar rats were prepared as previously described [21] and maintained in B27- and 1%-FCS-supplemented Neurobasal medium (GibcoBRL, Bethesda, MD, USA). The initial cell density was 45,000 per square centimeter. Tumor cells (2,800 per square centimeter) were added to the hippocampal cell culture 2 weeks after plating. After 24 h of tumor cell growth, hippocampal neurons were processed for immunocytochemistry. Neuron integrity was assessed according to previously published experimental parameters (dendrite length, dendritic swellings and varicosities, fragmentation, as described [22, 23]). In addition, cell nuclei were visualized using 4′,6-diamidino-2-phenylindole (DAPI)-containing vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA). Finally, the total number of microtubule-associated proteins 2 (MAP2)-positive neurons per view field was quantified. Data are presented as means ± SEM obtained from three independent experiments, each including analysis of at least two different coverslips and five to ten randomly chosen view fields.
Immunofluorescence
All stainings were performed on the cells on coverslips and the primary antibodies were applied overnight in the Tris-buffered saline buffer (100 mM Tris and 150 mM NaCl) containing 1% Tx-100 and 3% donkey serum. We used the following primary antibodies: rabbit anti p21 (Santa Cruz Biotechnology, USA), mouse anti-TfR (Dianova, Germany), goat anti-GFP (Acris Antibodies, Hiddenhausen, Germany), and microtubule-associated proteins (MAP) 2a and 2b (Sigma-Aldrich, Deisenhofen, Germany). Fluorescein-isothiocyanate- and Rhodamine-Red-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Confocal microscopy
All confocal microscopy was performed using a spectral confocal microscope (Leica TCS SP2). Appropriate gain and black level settings were determined on control tissues stained with secondary antibodies alone. Overview images were processed with Photoshop (Version CS, Adobe, San Jose, CA, USA) and colocalization images with Volocity (Version 2.6.1, Lexington, MA, USA).
Immunoprecipitation and Western blot analysis
The 106 cells were lysed for 20 min on ice in 500 μl of complete radioimmunoprecipitation assay lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 2 mM ethylene glycol tetraacetic acid, complete protease and phosphatase inhibitors). Lysates were clarified by centrifugation and immunoprecipitated for 4 h at 4°C with 1–2 μg of the specific antibodies. Immune complexes were collected on protein A Sepharose, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto nitrocellulose. Immunoblots were incubated in 3% bovine serum albumin, 10 mM Tris–HCl, pH 7.5, 1 mM ethylenediaminetetraacetic acid, and 0.1% Tween-20 for 1 h at room temperature, probed first with specific antibodies and then with secondary antibodies.
Oxyblot and detection of oxidized proteins
To identify carbonyl groups that are introduced into the amino acid side chain after oxidative modification of proteins, oxyblot analysis was performed. The derivative that is produced by reaction with 2,4-dinitrophenylhydrazine was immunodetected by an antibody specific to the attached dinitrophenol (DNP) moiety of proteins using a commercial kit (Chemicon, Temecula, CA, USA). Briefly, the electrophoresis was performed in the same way as described earlier [24], and the gels were transferred to a nitrocellulose membrane using a semidry transfer system (BioRad, Munich, Germany). Membranes were then blocked and incubated with a rabbit anti-DNP antibody (1:150; Chemicon) for 1 h at room temperature. The secondary antibody incubation was performed using horseradish-peroxidase-conjugated antirabbit immunoglobulin G (IgG; 1:300; Chemicon) for 1 h at room temperature. The immunoreactivity was visualized by enhanced chemiluminescence using commercial reagents (Roth, Germany). The kit used for the oxyblot analysis is sensitive to detect as little as 10 fmol of dinitrophenol residues.
Reporter assays
A 455-bp fragment of the human TfR promoter region [16] was cloned in front of luciferase gene and the resulting construct was transiently transfected into U373 wild-type glioma cells using an electroporator (Amaxa, Cologne, Germany). The activity of pTfR-Luc was compared with mutated TfR promoter constructs in which the Ets respectively the AP1, ATF, and CREB sites had been altered by site-directed mutagenesis (Strategene, La Jolla, CA, USA) with the following primers (from MWG Biotech, Ebersberg, Germany): m-EBS: fwd: 5′-CTCGCGAGCGTACGTGCCTCACATCGTGACGCACAGCCCCCCTGG-3′, rev: 5′-CCAGGGGGGCTGTGCGTCACGATGTGAGGCACGTACGCTCGCGAG-3′; mutated-AP1/Cre/EBS: fwd: 5′-CGTGCCTCACATCGTGTGGTGCAGCCCCCCTG-3′; rev: 5′-CAGGGGGGCTGCACCACACGATGTGAGCGACG; mutated AP1/Cre: fwd: 5′-CGTGCCTCAGGAAGTGTGGTGCAGCCCCCCTG-3′; rev: 5′-CAGGGGGGCTGCACCACACTTCCTGAGGCACG-3′.
The Dual Luciferase assay (Promega, Mannheim, Germany) was used according to the manufacturer’s instructions. Briefly, cells were seeded at 105 cells per 3.5-cm-diameter dish, incubated in medium with or without tet for 48 h, then washed with phosphate-buffered saline and agitated gently for at least 20 min with 500-μl passive lysis buffer. The cell lysate was cleared by centrifugation and 20 μl was added to 100 μl of luciferase assay reagent in a luminometer tube. The relative light units (RLU) were measured immediately in a Berthold-2000 luminometer. The average of ten RLU measurements taken at 2-s intervals was recorded.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (chIP) assays were carried out using a commercially available kit the chIP-IT™ Express (Active Motif) according to the manufacturer’s instructions. One 30-cm dish of 80% confluent wtU373 cells and U373 cells stably expressing etsDN or etsDN + TfR was used for one immunoprecipitation (IP) reaction. Cross-linking of DNA proteins was induced by an addition of formaldehyde (1% final concentration) directly to the culture medium for 10 min at 37°C. Cells were lysed and DNA in the supernatant was sheared by enzymatic digestion for 10 min at 37°C. An aliquot of the digested chromatin sample (“input”) was removed for polymerase chain reaction (PCR) analysis, and the remainder was used for IPs. Immunoprecipitation was performed with 1 μg of Ets1 (C-20) antibody (Santa Cruz Biotechnology) or normal goat IgG (Active Motif) that had been precoated onto Protein G-magnetic Sepharose beads, and reactions were incubated for 4 h with constant rotation at 4°C. Protein G beads were collected using magnetic stand and protein DNA complexes were eluted from the beads followed by a cross-link reversal step. DNA from each IP reaction (3 μl) was used for PCR with a primer pair to amplify the 110-bp region of interest of human TfR gene: forward primer, TGTAGGCTGCCCATTGTAAC; reverse primer, CAGAATTCCCACACATCCACT. Input or immunoprecipitated DNA was amplified by PCR (94°C, 20 s; 55°C, 30 s; and 72°C, 30 s) for 35 cycles. In parallel, chromatin was precipitated with positive control antibody (RNA polII) and negative control IgG from the chIP-IT™ control kit (Active Motif); then, input and immunoprecipitated DNA was amplified by 35-cycle PCR with human-specific positive GAPDH primers under recommended conditions, generating 166-bp products. All PCR products were resolved in 1.5% agarose gels.
Iron imaging
Iron measurements were performed using a previously established method [25]. Briefly, cells plated on glass coverslips were loaded with the fluorescent dye Phen Green SK (20 μM; Molecular Probes, Karlsruhe, Germany; 488-nm excitation and 505-nm emission) for 15 min at 37°C. Cells were then transferred to a microscope (Avioskop FS; Zeiss, Oberkochen, Germany) equipped with a perfusion chamber and superfused with Hepes buffer; 5 mM 1,10-phenantroline (Sigma) was used to chelate the free intracellular iron (Fe2+), which liberated the fluorescent dye from Fe2+ and thereby led to an increase in fluorescence. The difference in fluorescence intensity before and after iron chelation is a direct measure for the amount of free iron in a cell. Fluorescence changes were detected with a cooled charge-coupled device camera (SensiCam; Pico, Kelkheim, Germany) and images were analyzed with conventional software (Image Pro; Media Cybernetics, Silver Spring, MD, USA).
ROS measurement
Intracellular reactive oxygen species (ROS) production was assessed using 2′7′-dichlorodihydrofluorescin diacetate (DCFH-DA; Molecular Probes). DCFH-DA is oxidized by ROS within cells to the fluorescent product DCF. Cells seeded in 96-well plate were treated with 50 μM of dye and incubated at 37°C for 1 h and the fluorescence was measured using 96-well plate Wallac reader (Wallac, Turku, Finland).
Animals
Wild-type C57BL/6 (Charles River Breeding Laboratories; Schöneiche, Germany) and nude mice (genetic background athymic Nude-Foxn1, Harlan, Holland) were housed with a 12-h light–dark cycle and received food ad libitum. All governmental and institutional regulations regarding animal ethics were followed.
In vivo studies
Tumor xenografts were obtained by subcutaneous injection of one million cells of U373wt, U373EtsDn, and TfR/U373 EtsDn into the right and left flanks of female nude mice and two more groups which were injected with U373wt were given deferoxamine (DFO; 200 μM) and N-acetylcysteine (NAC; 200 μM) in drinking water.
The tumors were measured in two diameters and the tumor volume (TV) was calculated by the formula TV: length (mm) × width2 (mm) / 2 = mm3 [26] over the time course of 2 months.
Results
Transferrin receptor expression in glioma is regulated by an ets transcription factor binding site in the TfR promoter
We confirmed by Western blotting that TfR is overexpressed in human U373 glioma cells, compared to low-malignancy 1321N1 astrocytomas and to primary astrocytes (Fig. 1a, left; please note that raw data of all blot and gels are available as supplementary data on the JMM website). In the same blot, we show that blockade of ets transcription factors, by overexpression of an ets1 dominant-negative protein etsDN [27] substantially downregulated TfR in U373 cells; the TfR expression in ets dominant-negative cells could be rescued by forced expression of human TfR (Fig. 1a, left). Next, we investigated if other transcription factors could also control TfR expression in U373 cells. Therefore, we used a synthetic luciferase-coupled TfR promoter construct containing an overlapping consensus sequence for the binding of AP1, CREB, and ATF in addition to an ets binding site [16]. We determined the relative contribution of the respective transcription factors to TfR expression by mutating their binding sites in the TfR promoter as described [15]. Mutation of the ets binding site (m-EBS) strongly attenuated TfR expression, whereas mutation of the AP1 (m-AP1) and CRE/ATF (m-Crem) site remained largely without effect. However, a triple mutant (containing m-EBS, m-AP1, and m-Crem) abrogated the promoter activity; altogether, this suggests that ets transcription factors drive the expression of TfR in gliomas (Fig. 1a, right).

Overexpression of TfR in gliomas is controlled by Ets factors and reactive oxygen but not by iron or Hif1. a Western blot for transferrin receptors (TfR) in wild-type (WT) U373 glioma vs. primary astrocytes and a low-malignancy astrocytoma line (1321N1). TfR levels were also compared between WT and Ets dominant-negative U373 (EtsDN) and in EtsDN cells with forced TfR expression (EtsDN + TfR); actin control indicates equal loading (left). TfR promoter luciferase-reporter assay showing high activity in a promoter containing an Ets, Ap1, and ATF/Cre binding sites (nonmutant) compared to basic vector. Reporter activity is reduced after mutation of the Ets binding site (m-EBS) but not after mutation of Ap1 and ATF/Cre sites (m-Ap1 and m-Crem; right). b WT, etsDN, or etsDN + TfR cells were taken for chromatin immunoprecipitation assays, either using an anti-Ets1 (C-20) antibody or an unspecific IgG (Neg IgG) as a control. Total cell lysates were used as a positive control (input). The specific PCR product is a 110-bp sequence of the promoter region of the TfR gene. Positive chIP is indicated by an arrow (left panel). A control immunoprecipitation and PCR reaction with GAPDH primers was carried out in parallel on WT cells (right panel). c Western blotting for phosphorylated ets1 on nuclear and cytosolic fractions of WT, etsDN, and etsDN-TfR cells; cross-reactivity of an ets1 antibody with annexin-V was used as a loading control. d Ds-Red expressing U373 cells strongly immunolabeled for TfR (arrowhead) while GFP-expressing EtsDn cells show low TfR levels in the same culture; note TfR localization in a magnified image (rectangle). e Immunoblot for TfR from U373 WT cells subjected to normoxia or hypoxia for 24 h, with and without the HIF-1 blocker YC1; note that TfR expression is independent of hypoxia or Hif1 (left) and showing nuclear localization and activity of HIF-1 in U373 WT cells under normoxia or hypoxia for 24 h, with and without HIF-1; note that hypoxia enables Hif1 function that can be blocked by YC1 (middle and right). f Immunoblot reveals no change of TfR expression in U373 WT cells exposed to the iron chelator deferoxamine (DFO) or the iron donator ferric ammonium citrate (FAC; left), Western blot showing altered TfR expression upon treatment of U373 WT cells with the oxidant scavenger N-acetyl cysteine (NAC) and the oxidant hydrogen peroxide (H202; right). Scale bars in d are 30 μm
A chIP revealed that ets1 directly binds to the promoter region of the TfR gene (Fig. 1b, left). Ets1 binding is seen only in WT cells (see arrow in Fig. 1b, left) and is prevented in etsDN-expressing cells. These data show that etsDN overexpression directly blocks ets1 function. Western blotting for a phosphorylated (and therefore hyperactivated) form of ets1 in nuclear and cytosolic fractions of U373 cells (Fig. 1c) indicated phosphorylation of ets1 in WT cells, which was attenuated by etsDN expression. However, forced expression of TfR rescued the phosphorylation of ets1 in etsDN-expressing cells, suggesting a positive feedback signal from TfR for ets1 activation.
Under hypoxic conditions or during iron deficiency, TfR expression may be induced by the transcription factor Hif1α, which can also bind to the DNA sequence used in our reporter gene assay (see above). However, TfR expression did not change under hypoxic conditions (5% CO2, pO2 < 0.2 h Pa, for 24 h) and after pharmacological inhibition of Hif1α with YC1 [28] in hypoxic glioma (Fig. 1e, left panel). The corresponding controls show that hypoxia induced and YC1 efficiently blocked accumulation of nuclear Hif1α (Fig. 1e, center). Hypoxic U373 cells also had higher Hif1 activity as measured with a luciferase-based reporter construct containing Hif1 binding sites (Fig. 1e, right).
In an additional experimental approach, we cocultured etsDN-expressing U373 cells together with wild-type cells ensuring that both cell types grow under identical conditions, especially with respect to the iron content in the culture. As shown by immunocytochemistry (Fig. 1d), in these cocultures, the vast majority of wild-type cells (74%, ±4%) overexpressed TfR, whereas only very few U373-etsDN cells (5%, ±2%) overexpressed TfR.
TfR expression in glioma is increased by oxidants, but largely independent of the cellular iron concentration
Western blotting for TfR from U373 cells cultivated under control conditions or together with the iron chelator DFO (200 μM, for 24 h) or the iron donor ferric ammonium citrate (50 μg/ml, for 24 h) revealed unchanged levels of TfR expression (Fig. 1f, left). Whereas transcription factors besides ets factors and also the cellular iron level had no major effect on TfR expression, the cellular oxidant content induced TfR expression. Application of the oxidant H2O2 (50 μM, for 24 h) further increased TfR expression in U373 WT cells, whereas the antioxidant NAC (200 μM, for 24 h) reduced TfR protein expression (Fig. 1f, right).
Transferrin receptors control redox signaling and proliferation
Next, we addressed the question whether the ets transcription-factor-mediated increase in TfR expression had physiological consequences like increased intracellular iron accumulation and increased oxidant production by the labile iron pool [12]. We clonally expanded U373 glioma cells that were derived from wild-type (U373-WT) cells, from cells containing a tetracycline-inducible etsDN construct (U373-estDN) and from cells expressing etsDN plus the human TfR (U373-etsDN + TfR). For iron and ROS measurements and BrdU assays with these cells, we averaged data obtained from three individual clonal lines.
Tetracycline-induced expression of etsDN in U373 cells strongly attenuated intracellular iron accumulation, when compared to U373-WT and U373-etsDN cells without tetracycline, as measured by iron imaging using PhenGreen-SK as an indicator [25]. In U373-etsDN+TfR cells, intracellular iron accumulation was fully rescued, i.e., the intracellular iron content was comparable to U373-WT cells and to U373-etsDN without tetracycline (Fig. 2a).

Induction of transferrin-receptor-mediated redox signaling is a central proto-oncogenic function of Ets factors, leading to accelerated glioma growth. Intracellular iron content and ROS production and proliferation were measured in U373 (WT) cells, tetracycline-inducible EtsDN-expressing U373 cells (EtsDN), TfR-overexpressing EtsDN U373 (EtsDN + TfR) cells, and a low-malignancy astrocytoma with and without forced TfR expression (1321N1-TfR, respectively, WT). a High intracellular iron levels are observed in U373 cells having intact Ets signaling or in U373 EtsDN cells overexpressing TfR but not in U373 cells expressing EtsDN alone. Higher intracellular iron accumulation was also measured in 1321N1 cells overexpressing TfR compared to WT 1321N1. b Similarly, high oxidant levels were measured in U373 cells with intact Ets signaling or in TfR-overexpressing EtsDN cells but not in U373 cells expressing EtsDN alone; additionally, high oxidant generation was observed in TfR-overexpressing 1321 cells (left). Flow cytometry revealed that increased oxidant levels in U373 cells with high TfR expression were homogenous throughout the cell populations (right). c BrdU incorporation assay showed high proliferation levels in U373 cell having intact Ets signaling or in U373 EtsDN cells overexpressing TfR but not in U373 cells expressing EtsDN alone; proliferation was also drastically increased after TfR overexpression in 1321N1 cells. d Levels of BrdU incorporation in U373 WT cells are strongly decreased after iron (DFO) or oxidant (NAC) chelation. Statistical significance is indicated: **p < 0.005; ***p < 0.001
Induced expression of etsDN substantially reduced ROS levels in U373 gliomas, as compared to U373-WT and U373-etsDN without tetracycline, which was evaluated by ROS imaging using DCFH-DA as an indicator [29]. Re-expression of TfR in etsDN cells fully restored iron accumulation in gliomas (Fig. 2b, left). Fluorescence-activated cell sorting (FACS) analysis of DCFH-DA fluorescence in U373-WT, tetracycline-treated U373-estDN, and U373-etsDN + TfR revealed that ROS levels specifically in the U373-estDN cells were homogenously diminished throughout the whole cell population (Fig. 2b, right). This figure indicates that the entire population of U373-etsDN cells produces a very low fluorescence signal for ROS signaling (blue line, which is on the level of the background signal—the background signal is not shown), whereas the populations of WT (red line) and etsDN + TfR (black line) cells homogenously have a much higher signals for ROS production.
Measuring BrdU incorporation of our glioma lines showed that in tetracycline-treated U373-etsDN cells proliferation levels were profoundly decreased, as compared to U373-WT, U373-etsDN without tetracycline, and U373-etsDN + TfR cells (Fig. 2c). The proliferation of U373 cells directly depended on iron and ROS levels since DFO and the NAC largely attenuated glioma proliferation (Fig. 2d).
Further, we compared the results in the U373 glioma line with those of the low-grade astrocytoma line 1321N1. These cells constitutively had a significantly lower iron content, ROS levels, and proliferation rate as compared to U373 cells. Overexpression of TfR in the low-malignancy 1321N1cells increased all three pathological parameters (iron accumulation, oxidant production, and excessive growth) as shown in Fig. 2. The role of oxidant production and ets factors for cancer cell proliferation is also highlighted by another observation: U373 cells constitutively expressing etsDN do not grow at clonal level, i.e., clonal U373-etsDN cells could only be generated when etsDN expression was induced (by addition of tetracycline to the culture medium) after the cells had reached 30% confluency. However, when we overexpressed TfR in bulk etsDN cultures we could clonally expand these cells (U373-etsDN + TfR cells; not shown).
LMW-PTP is a target of TfR-mediated redox signaling
After establishing that high proliferation levels in gliomas are maintained by ets-driven TfR overexpression and subsequent iron accumulation and ROS generation, we investigated which molecules downstream of ROS mediate signals for the accelerated cell division rate. Hence, we searched for proteins, which are known to transduce growth signals and which are sensitive to ROS. Therefore, we electrophoretically separated whole protein contents of U373-WT, U373-estDN, and U373-etsDN + TfR and in parallel performed an immunoblotting procedure for the detection of oxidized proteins (oxyblot). From the blots, we identified bands containing high amounts of differentially oxidized proteins among our cell lines. The corresponding bands were excised from gels separating the total protein contents of glioma cells and were analyzed by mass spectrometry. With this procedure, we identified low-molecular-weight protein tyrosine phosphatase (LMW-PTP) [30, 31] as a potential target for ROS-mediated growth signaling (Fig. 3a).

LMW-PTP inactivation is downstream of Ets/TfR-mediated redox signaling and permits accelerated glioma growth. a shows the differential expression of oxidized proteins (Oxyblot) in U373 (WT), U373 EtsDn (EtsDN), and EtsDN + TfR U373 (EtsDN + TfR) cells and a corresponding (18 kDa) band, which was subsequently excised from a SDS-PAGE gel (total protein) and taken for proteomics analysis. One of the proteins in this band was identified by Maldi-MS as low-molecular-weight protein tyrosine phosphatase (LMW-PTP). b shows that the total expression of LMW-PTP is higher in U373WT and EtsDN + TfR cells compared to U373 EtsDN cells (left). For immunoprecipitation, the amount of LMW-PTP was brought to equal level in U373WT, EtsDn, and EtsDN + TfR cells (input); then, oxidized proteins were immunoprecipitated (IP: oxidized proteins) and immunoblotted for LMW-PTP (blot: LMW-PTP); note that the amount of oxidized LMW-PTP is much higher in U373WT and EtsDn + TfR cells as compared to U373 cells expressing EtsDN alone (right). c BrdU incorporation assay showing the proliferation of U373 EtsDN cells compared to U373 EtsDN cells overexpressing a mutated (catalytically inactive) LMW-PTP and compared to (WT) U373 cells; note that the mutant LMW-PTP rescues proliferation in U373 EtsDn cells. d Catalytically active LMW-PTP suppresses PI3K and Ras pathways, whereas oxidized LMW-PTP facilitates Ras activation and MAPK phosphorylation and permits PI3K signaling and Akt activation (details, see text). Statistical significance is indicated: **p < 0.005; ***p < 0.001
The total amount of LMW-PTP expression was increased in U373-WT and U373-etsDN + TfR cells compared to U373-estDN (Fig. 3b, left). However, LMW-PTP was also differentially oxidized (and hence inactivated) in these three lines. In cells with high TfR and ROS content (U373-WT and U373-etsDN + TfR), we detected much higher amounts of oxidized LMW-PTP than in U373-estDN (containing low TfR and ROS levels; Fig. 3b, right).
Importantly, we observed that oxidation of LMW-PTP can account for the increased proliferation in cells with high ROS content. We found that overexpression of a point mutated form of LMW-PTP—with an inactivated catalytic site mimicking the oxidized form of LMW-PTP [32]—in U373etsdDN cells fully rescued glioma proliferation (Fig. 3c). This suggests that the high redox levels in glioma may facilitate growth factor signaling in the brain tumors, partly through oxidizing LMW-PTP.
Catalytically active LMW-PTP antagonizes Akt and MAPK signaling, but an elevated redox level inactivates LMW-PTP and augments Akt and MAPK activity ([32] and Fig. 3d). In gliomas, Akt and MAPK signaling are the main pro-proliferative pathways [33].
TfR expression triggers Akt and MAPK signaling and inhibits p21 and pRB
Next, we sought to establish if U373-WT and U373-etsDN + TfR have altered MAPK and Akt signaling compared to U373-etsDN cells. Indeed, cells with high TfR levels like U373-WT, U373-etsDN + TfR, and U373-etsDN cells without tetracycline exhibited high levels of phosphorylated Akt indicating the activation of the Akt pathway. Conversely, cells with low iron and ROS levels like tetracycline-stimulated U373-etsDN and DFO or NAC-treated U373-WT cells had lower levels of phosphorylated Akt (Fig. 4a, left). However, the total Akt levels in U373-etsDN were even increased compared to all other cells, suggesting some compensatory mechanism in these glioma cells, e.g., to cope with low levels of Akt phosphorylation under low ROS conditions. Akt signaling in U373 cells was phosphatidylinositol 3′-kinase (PI3K) dependent as indicated by the inhibition of phosphorylated Akt levels by wortmannin (Fig. 4a, right).

Pro-proliferative AKT and MAPK signaling and facilitated S-phase entry is restricted to TfR-overexpressing glioma cells. We compared the activity of the Akt and MAPK signaling by Western blotting in U373 (WT), U373 EtsDn (EtsDN), and EtsDN + TfR U373 (EtsDN + TfR) cells and investigated the effect of altered redox signaling on G1 checkpoint control and cell cycle progression. a Levels of active Akt (phopho-AKT; P-Akt) compared to total AKT expression (Akt) in the various U373 cell lines; note that P-Akt is reduced after tetracycline-induced EtsDN expression and after iron (DFO) and ROS (NAC) chelation but remains high in all other cells (left). P-AKT levels are reduced after application of the PI3 kinase blocker wortmannin (right). b MAPK (p38, Erk, and JNK) are inactive (nonphosphorylated; P-p38, p-Erk, P-JNK) after tetracycline-induced EtsDN expression in U373 cells, but expression of total MAPK is not affected; note that MAPK signaling is rescued in EtsDN cells after TfR overexpression (EtsDN + TfR). Phosphorylation of the RB protein (P-RB) is absent after tetracycline-induced EtsDN expression, whereas pRB is strongly phosphorylated (inactivated) in U373-WT and U373-EtsDN + TfR cells. Likewise, p21 is active (nuclear localization) after tetracycline-induced EtsDN expression but inactive (cytoplasmic localization) in U373-WT and U373-EtsDN + TfR cells (loading controls are for cytoplasm: actin; for nuclear fraction: TATA-binding protein; TBP). c Immunostaining also identifies cytoplasmic localization (nuclei are blue) of p21 (red) in U373-WT and U373-EtsDN + TfR cells but nuclear p21 in tetracycline-induced EtsDN cells. d Cell cycle analysis for WT, EtsDN, and EtsDN + TfR U373 cells by flow cytometry using propidium iodide staining; note that in WT and EtsDN + TfR the number of cells entering S-phase is twice as high as after tetracycline-induced EtsDN expression
Similar to the findings obtained for Akt phosphorylation, we observed that all three major MAPK pathways are phosphorylated in cells with high iron and ROS content (U373-WT, U373-etsDN + TfR) and that tetracycline-induced expression of etsDN and subsequent attenuation of ROS generation abrogated MAPK signaling (Fig. 4b). Importantly, TfR-overexpressing cells showed increased activation (phosphorylation) of p38, ERK, and JNK MAPKs, but the expression of total (nonphosphorylated) MAPKs was at equal level in all three cell lines or even increased in the etsDN-expressing glioma. This indicates that etsDN only interferes with MAPK signaling but does not attenuate MAPK expression.
Altered Akt and MAPK signaling can have direct impact on cell proliferation through phosphorylation of pRB by p38 MAPK [34] and through phosphorylation and translocation of p21(cdkn1a/waf1/cip1) from the nucleus to the cytosol, which inactivates p21 [35]. Indeed, we observed that in TfR-overexpressing gliomas pRB was overphosphorylated and therefore inactivated (Fig. 4c). Likewise, p21 had a predominantly cytosolic localization in glioma cells with high TfR expression, active iron accumulation, and abundant ROS generation. By quantification of p21 localization in the respective cell lines, we observed that 95% of all U373-WT cells exhibited cytosolic p21; only 19% of all tetracycline-treated U373-etsDN had cytosolic p21 localization (consequently, 81% of p21 was nuclear) whereas U373-etsDN + TfR had again cytosolic p21 in 89% of all cells.
The tumor suppressors p21 and pRB control cell cycle entry in G1 and prevent cells from entering S-phase. To investigate if p21 and pRB activation in the TfR-overexpressing gliomas can account for the increased proliferation of these cell lines, we quantified cell cycle phases in our glioma cell lines by FACS analysis, measuring ploidity of the cellular DNA with propidium iodide. We observed that in U373-WT and U373-etsDN + TfR the amount of cells, which had entered S-phase, was approximately 50% higher than in tetracycline-stimulated U373-etsDN glioma cells. These data indicate that TfR overexpression can facilitate S-phase entry and proliferation of gliomas (Fig. 4d).
TfR-mediated redox signaling accelerates glioma growth in vivo
Experimental gliomas were induced by injection of a glioma cell line (GL261 cells overexpressing EGFP) into syngeneic mice [36]. The majority of the glioma cells showed high expression of TfR when compared to normal parenchyma (Fig. 5a, b). To investigate if TfR expression correlated with proliferation, we systemically applied a single dose of BrdU to mice bearing a glioma in the caudate putamen, thereby labeling all cells in S-Phase. Two hours after injection, the animals were sacrificed and subsequently the brains were inspected for TfR and BrdU immunolabeling. We found that all BrdU + cells had high TfR expression but not all TfR + cells were BrdU-labeled (Fig. 5c).To examine if the TfR-mediated increase in iron accumulation, redox signaling, and cell cycle acceleration had direct consequences for the progression of gliomas in vivo, we subcutaneously implanted the U373 wild-type cells, U373 cells overexpressing etsDN, and the etsDN + TfR-expressing glioma line in immune-compromised mice and monitored tumor growth over a time course of 8 weeks. Additionally, we examined if the suppression of iron and ROS accumulation in gliomas could be used as a therapeutic approach for glioma treatment by supplying mice with DFO or NAC in the drinking water. The tumor sizes were determined weekly. We observed that gliomas with high TfR activity had the strongest growth over the whole time course examined, whereas iron chelation by DFO greatly reduced tumor growth. In U373 cells overexpressing etsDN, we detected only little tumor growth during the 8 weeks of the experiment. Most striking therapeutic results were obtained with the ROS chelator NAC, which reduced in vivo glioma growth almost to the level of etsDN-expressing cells (Fig. 5d, left).

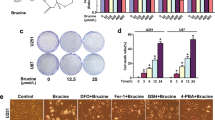
TfR-mediated redox signaling accelerates glioma growth in vivo. a Immunostaining for TfR (blue) in an experimental glioma induced by injection of the mouse glioma cell line, Gl261(expressing Ds-Red) into syngeneic BL/6 mice; note that TfR is strongly overexpressed only in the glioma. An area marked by the rectangle was magnified in b. TfR overexpression colocalizes with the glioma cells in the tumor area and on the single-cell level; a single cell (highlighted by arrow) was 3D reconstructed cell (insert in b). c shows strong labeling for TfR (red) in GL261 glioma cells (expressing GFP), which have incorporated BrdU (labeling in blue), after systemic BrdU administration. d Tumor growth of subcutaneously implanted U373 WT, EtsDn, and TfR + EtsDn glioma in nude mice (n = 6 per group) was followed over a time course of 8 weeks. Additionally, DFO and NAC were given with the drinking water to nude mice implanted with WT cells. Glioma grew rapidly in WT and EtsDN + TfR-implanted mice but progressed much slower in EtsDN-inoculated animals or in mice receiving DFO or NAC (left; error bars are smaller than symbols). Finally, the tumors were resected from the mice and glioma masses were measured by weighing the resections; note that tumor masses are much higher in WT and EtsDN + TfR-implanted mice but significantly lower (p < 0.001) in EtsDN-inoculated animals or in mice receiving DFO or NAC with the drinking water (right)
Next, we compared the tumor sizes we had obtained by volumetric measurement over time with the actual tumor mass at the end point of our experiment. Animals were sacrificed 8 weeks after glioma injection and the tumor was isolated. Again, the TfR-overexpressing cells and gliomas with active iron accumulation and redox signaling (U373-WT and U373-etsDN + TfR) had drastically increased tumor weights compared to U373 expressing etsDN and DFO- or NAC-treated gliomas (Fig. 5d, right).
TfR expression in glioma cells induces glutamate secretion and NMDA-receptor-mediated reduction of neuron mass
We had initially found that TfR expression is not modulated by cellular iron levels and therefore appears to be independent of iron regulatory proteins (IRPs, see also Fig. 1f). However, IRPs have a dual role and acquire aconitase activity when iron concentration is high [37]. Under high iron conditions, this enzyme has a key role in the biosynthesis of glutamate [38]. Hence, we investigated whether the expression level of TfR is linked to glutamate release. Indeed, we found glioma cells with a high expression level of TfR, namely U373-WT and U373-etsDN + TfR, released large amounts of glutamate into the supernatant. Glutamate levels reached 400 to 500 μM within 8 h and a plateau value of approximately 700 μM within 72 h. In contrast, glioma cells with low level of TfR expression, the U373-etsDN, released only 15 μM glutamate within 8 h and approximately 50 μM within 72 h (Fig. 6d).

Only TfR-overexpressing U373 glioma induces excitotoxic neuronal death. a MAP2 staining of hippocampal neurons cultured for 24 h under control conditions or cocultivated with U373 cells. Control neurons without U373 cells have numerous and elongated dendrites without dendrite fragmentation; note that neurons intensely label for MAP2 and display nuclear integrity (insert in a). b In the presence of U373 cells, hippocampal neurons display a strongly altered dendrite morphology. The majority of neurons have short (if any) dendrites, often characterized by dendrite fragmentation, not that these neurons also lose their nuclear integrity (as indicated by DAPI staining in the insert in b). c Quantification of the fraction of neurons with abnormal dendrite morphology cultured under different experimental conditions (left panel); note that reduction of neuron mass occurs only in coculture with TfR-overexpressing glioma and that this process is prevented by the NMDA receptor antagonist MK801 (left). Likewise, the total number of MAP2-positive neurons declines specifically in coculture with TfR-overexpressing glioma; this neuronal loss is prevented by MK801 (right panel). d Time-course experiment (n = 3) quantifying glutamate release from U373 glioma; note that excessive glutamate release is restricted to TfR-overexpressing glioma (error bars are smaller than symbols). e Schematic drawing summarizing the findings: Ets factors induce TfR expression; TfR activity increases the labile iron pool and catalyzes reactive oxygen formation (ROS); ROS facilitates proto-oncogenic pathways (MAPK, Akt) and blunts tumor suppressor pathways (p21, pRB) leading to accelerated growth; simultaneously, ROS enables glutamate synthesis and release which leads to neuronal loss, i.e., creates space for tumor expansion (details, see text). Significance levels (Student’s t test and ANOVA) are indicated as p < 0.05 (*) and p < 0.001 (***). Scale bar 100 μm
To study the pathological impact of the glioma-derived glutamate, we cocultivated the tumors with hippocampal neurons (Fig. 6a–c). In the presence of U373-WT and U373-etsDN + TfR, we observed a massive reduction of neuron mass, i.e., the majority of hippocampal neurons (more than 80%) had drastically shortened or even fragmented dendrites (Fig. 6b, c, left). This effect was almost fully reversed (to about 15% of neurons with morphological changes) in cultures treated with an NMDA receptor blocker (1 μM MK801) or in glioma with little TfR signaling and low iron content (U373-etsDN). Likewise, the total number of neurons was reduced (Fig. 6c, right) in cocultures with U373-WT and U373-etsDN + TfR gliomas (by about 45%) compared to U373-etsDN, U373 + MK801, and neurons alone. Finally, the remaining neurons exhibited a faint DAPI signal that appeared unusually homogenous, as if the nuclear compartment was dissolving (see inset in Fig. 6b); this effect has previously been described as karyolysis and occurs during necrosis [39].
Discussion
In our present study, we showed that ets factors promote brain tumor progression by increasing cellular redox levels. Increased oxidant production in gliomas occurred downstream of TfR overexpression and excess iron accumulation. ROS production led to LMW-PTP oxidation (inactivation) and MAPK and Akt activation and resulted in loss of G1 checkpoint control and enhanced glioma proliferation. Concomitantly, TfR mediated high glutamate release, making iron-accumulating glioma cells excitotoxic for neurons. Overall, our data indicate that in glioma TfR coordinately promotes tumor growth and boosts excitotoxicity to provide space for the advancing tumor.
Under physiological conditions, the expression of TfR is regulated in a complex fashion on the transcriptional [14, 15] and posttranscriptional [16] level. However, in the U373 glioma cells, TfR expression remained largely unchanged after iron chelation or iron administration. This suggests that the diverse cellular iron regulatory mechanisms, which cocontrol TfR mRNA stability according to the intracellular iron level under physiological conditions [12], have a minor role for TfR expression in glioma. Using a reporter gene assay, we screened for factors exerting transcriptional control over TfR expression and observed that mainly ets factors are responsible for the elevated TfR levels in glioma. Furthermore, interfering with ets signaling strongly attenuated TfR expression, which could be rescued to control levels by overexpressing TfR from a vector. Hif1 and hypoxia did not alter TfR expression in glioma.
Downregulation of TfR expression by attenuating ets activity decreased intracellular iron accumulation and decreased the redox level in glioma. The generation of reactive oxygen species is catalyzed by excess intracellular iron and contributes to brain pathology [12] and high oxidant levels can promote cancer cell proliferation [3]. Proliferation of U373-etsDN cells was largely diminished compared to wild-type cells but could be fully rescued by forced TfR expression. Furthermore, the overexpression of TfR was sufficient to rescue clonal growth of etsDN cells. These data show for the first time that modulation of redox signaling is an important protumorigenic effect of ets transcription factors. Next, we identified LMW-PTP as a target of ROS in brain tumor cells. We showed that oxidized LMW-PTP was much more abundant in cells expressing TfR at high level than in U373etsDN cells containing TfR only at low level. Forced expression of a point mutated (inactive) form of LMW-PTP rescued U373etsDN cell growth, showing the importance of the oxidization (inactivation) of this molecule for glioma proliferation.
LMW-PTP counteracts activation of the Ras/MAPK and PI3Kinase/Akt signaling pathways [32, 40, 41]. In the present study, we provided evidence that the MAPK and Akt pathways are only fully active in gliomas with high TfR expression. Elevated Akt and MAPK signaling is found in most glioma and may be one of the primary pathways driving brain tumor initiation and progression [7]. Our study suggests that TfR-mediated redox signaling—in addition to mutations of growth factor receptors and of PTEN—is important for the activation of Akt and MAPK.
Akt and MAPKs exert growth control by inactivation of G1 checkpoint proteins p21 and pRB [35, 42, 43]. In glioma cells with high redox signaling (U373WT and U373etsDN+TfR cells), we observed hyperphosphorylated (inactivated) pRB, whereas U373etsDN cells contained hypophosporylated pRB, which actively blocks cell cycle entry in G1. Likewise, cells with high TfR expression and activated Akt showed inactivation of the G1 cell cycle arrest protein p21. The tumor suppressor p21 is phosphorylated by Akt, which induces shuffling of p21 out of the nucleus and loss of tumor suppressor function [35]. We observed high nuclear p21 in U373etsDN cells and high cytoplasmic p21 in U373WT and U373etsDN + TfR cells. On the contrary, the TfR-low cells (U373-etsDN) were arrested in G1 and in G2.
The increased cell cycle arrest in TfR-low cells explains the results from our BrdU assay, which showed that U373etsDN cells proliferate less than U373WT and U373etsDN + TfR cells. Furthermore, in an in vivo model, tumors were growing at much higher pace in glioma expressing TfR at high level. Systemic abrogation of high iron and ROS levels with an iron chelator or an antioxidant yielded reduced tumor growth, further indicating the relevance of redox signaling for glioma progression.
Accelerated proliferation rates in glioma could not lead to continuous glioma progression without coordinated provision of space for the expanding tumor mass. On this point, gliomas have requirements different from other solid tumors since the skull prevents the brain tissue to be invariably be pushed aside. Gliomas circumvent this problem by actively creating space for their own growth, i.e., by killing surrounding neurons [8, 9]. We show that TfR promotes both excessive proliferation and massive reduction of neuron mass, which most likely reflects NMDA-receptor-mediated excitotoxicity [23]. Hence, TfR represents an important target for glioma therapy, by which neural tumor cell cycles [44, 45], glioma expansion [46], and neurological problems—that are frequently occurring with these tumor—may be suppressed.
References
Terada LS (2006) Specificity in reactive oxidant signaling: think globally, act locally. J Cell Biol 174(5):615–623
Veal EA, Day AM, Morgan BA (2007) Hydrogen peroxide sensing and signaling. Mol Cell 26(1):1–14
Hussain SP, Hofseth LJ, Harris CC (2003) Radical causes of cancer. Nat Rev Cancer 3(4):276–285
Martin V, Herrera F, Garcia-Santos G, Antolin I, Rodriguez-Blanco J, Rodriguez C (2007) Signaling pathways involved in antioxidant control of glioma cell proliferation. Free Radic Biol Med 42(11):1715–1722
Kleihues P, Burger PC, Scheithauer BW (1996) Histological typing of the tumours of the central nervous system, 2nd edn. Springer, Stuttgart
Holland EC (2000) Glioblastoma multiforme: the terminator. Proc Natl Acad Sci USA 97(12):6242–6244
Holland EC (2001) Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet 2(2):120–129
Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M (2001) Glutamate release promotes growth of malignant gliomas. Nat Med 7(9):1010–1015
Sontheimer H (2003) Malignant gliomas: perverting glutamate and ion homeostasis for selective advantage. Trends Neurosci 26(10):543–549
Recht L, Torres CO, Smith TW, Raso V, Griffin TW (1990) Transferrin receptor in normal and neoplastic brain tissue: implications for brain-tumor immunotherapy. J Neurosurg 72(6):941–945
Richardson DR (2002) Iron chelators as therapeutic agents for the treatment of cancer. Crit Rev Oncol Hematol 42(3):267–281
Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR (2004) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5(11):863–873
Cavanaugh PG, Jia L, Zou Y, Nicolson GL (1999) Transferrin receptor overexpression enhances transferrin responsiveness and the metastatic growth of a rat mammary adenocarcinoma cell line. Breast Cancer Res Treat 56(3):203–217
Sieweke MH, Tekotte H, Frampton J, Graf T (1996) MafB is an interaction partner and repressor of Ets-1 that inhibits erythroid differentiation. Cell 85(1):49–60
Marziali G, Perrotti E, Ilari R, Lulli V, Coccia EM, Moret R, Kuhn LC, Testa U, Battistini A (2002) Role of Ets-1 in transcriptional regulation of transferrin receptor and erythroid differentiation. Oncogene 21(52):7933–7944
Owen D, Kuhn LC (1987) Noncoding 3′ sequences of the transferrin receptor gene are required for mRNA regulation by iron. Embo J 6(5):1287–1293
Sharrocks AD (2001) The ETS-domain transcription factor family. Nat Rev Mol Cell Biol 2(11):827–837
Kitange G, Kishikawa M, Nakayama T, Naito S, Iseki M, Shibata S (1999) Expression of the Ets-1 proto-oncogene correlates with malignant potential in human astrocytic tumors. Mod Pathol 12(6):618–626
Dittmer J (2003) The biology of the Ets1 proto-oncogene. Mol Cancer 2:29
Kita D, Takino T, Nakada M, Takahashi T, Yamashita J, Sato H (2001) Expression of dominant-negative form of Ets-1 suppresses fibronectin-stimulated cell adhesion and migration through down-regulation of integrin alpha5 expression in U251 glioma cell line. Cancer Res 61(21):7985–7991
Meier J, Grantyn R (2004) A gephyrin-related mechanism restraining glycine receptor anchoring at GABAergic synapses. J Neurosci 246:1398–1405
Ferrer I (1999) Neurons and their dendrites in frontotemporal dementia. Dement Geriatr Cogn Disord 10(Suppl 1):55–60
Eichler SA, Kirischuk S, Juttner R, Legendre P, Lehmann TN, Gloveli T, Grantyn R, Meier JC (2008) Glycinergic tonic inhibition of hippocampal neurons with depolarising GABAergic transmission elicits histopathological signs of temporal lobe epilepsy. J Cell Mol Med doi:10.1111/j.1582-4934.2008.00357.x
Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA (2002) Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med 33(4):562–571
Petrat F, de Groot H, Rauen U (2001) Subcellular distribution of chelatable iron: a laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem J 356(Pt 1):61–69
Geran RI, Greenberg NH, Macdonald MM, Abbott BJ (1977) Modified protocol for the testing of new synthetics in the L1210 lymphoid leukemia murine model in the DR&D program, DCT, NCI. Natl Cancer Inst Monogr 45:151–153
Nakada M, Yamashita J, Okada Y, Sato H (1999) Ets-1 positively regulates expression of urokinase-type plasminogen activator (uPA) and invasiveness of astrocytic tumors. J Neuropathol Exp Neurol 58(4):329–334
Sun HL, Liu YN, Huang YT, Pan SL, Huang DY, Guh JH, Lee FY, Kuo SC, Teng CM (2007) YC-1 inhibits HIF-1 expression in prostate cancer cells: contribution of Akt/NF-kappaB signaling to HIF-1alpha accumulation during hypoxia. Oncogene 26:3941–3951
Mattia CJ, Adams JD Jr, Bondy SC (1993) Free radical induction in the brain and liver by products of toluene catabolism. Biochem Pharmacol 46(1):103–110
Chiarugi P, Taddei ML, Schiavone N, Papucci L, Giannoni E, Fiaschi T, Capaccioli S, Raugei G, Ramponi G (2004) LMW-PTP is a positive regulator of tumor onset and growth. Oncogene 23(22):3905–3914
Ostman A, Hellberg C, Bohmer FD (2006) Protein-tyrosine phosphatases and cancer. Nat Rev Cancer 6(4):307–320
Giannoni E, Raugei G, Chiarugi P, Ramponi G (2006) A novel redox-based switch: LMW-PTP oxidation enhances Grb2 binding and leads to ERK activation. Biochem Biophys Res Commun 348(2):367–373
Martin V, Herrera F, Carrera-Gonzalez P, Garcia-Santos G, Antolin I, Rodriguez-Blanco J, Rodriguez C (2006) Intracellular signaling pathways involved in the cell growth inhibition of glioma cells by melatonin. Cancer Res 66(2):1081–1088
Nath N, Wang S, Betts V, Knudsen E, Chellappan S (2003) Apoptotic and mitogenic stimuli inactivate Rb by differential utilization of p38 and cyclin-dependent kinases. Oncogene 22(38):5986–5994
Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC (2001) Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 3(3):245–252
Szatmari T, Lumniczky K, Desaknai S, Trajcevski S, Hidvegi EJ, Hamada H, Safrany G (2006) Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci 97(6):546–553
Hentze MW, Kuhn LC (1996) Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci USA 93(16):8175–8182
McGahan MC, Harned J, Mukunnemkeril M, Goralska M, Fleisher L, Ferrell JB (2005) Iron alters glutamate secretion by regulating cytosolic aconitase activity. Am J Physiol Cell Physiol 288(5):C1117–C1124
Segerer S, Eitner F, Cui Y, Hudkins KL, Alpers CE (2002) Cellular injury associated with renal thrombotic microangiopathy in human immunodeficiency virus-infected macaques. J Am Soc Nephrol 13(2):370–378
Xing K, Raza A, Lofgren S, Fernando MR, Ho YS, Lou MF (2007) Low molecular weight protein tyrosine phosphatase (LMW-PTP) and its possible physiological functions of redox signaling in the eye lens. Biochim Biophys Acta 1774(5):545–555
Pandey SK, Yu XX, Watts LM, Michael MD, Sloop KW, Rivard AR, Leedom TA, Manchem VP, Samadzadeh L, McKay RA, Monia BP, Bhanot S (2007) Reduction of low molecular weight protein-tyrosine phosphatase expression improves hyperglycemia and insulin sensitivity in obese mice. J Biol Chem 282(19):14291–14299
Suyama H, Igishi T, Sano H, Matsumoto S, Shigeoka Y, Nakanishi H, Endo M, Burioka N, Hitsuda Y, Shimizu E (2004) ERK activation and subsequent RB phosphorylation are important determinants of the sensitivity to paclitaxel in lung adenocarcinoma cells. Int J Oncol 24(6):1499–1504
Fernandes DJ, Ravenhall CE, Harris T, Tran T, Vlahos R, Stewart AG (2004) Contribution of the p38MAPK signalling pathway to proliferation in human cultured airway smooth muscle cells is mitogen-specific. Br J Pharmacol 142(7):1182–1190
Renton FJ, Jeitner TM (1996) Cell cycle-dependent inhibition of the proliferation of human neural tumor cell lines by iron chelators. Biochem Pharmacol 51(11):1553–1561
Jeitner TM, Renton FJ (1996) Inhibition of the proliferation of human neural neoplastic cell lines by cysteamine. Cancer Lett 103(1):85–90
Basset P, Zwiller J, Revel MO, Vincendon G (1985) Growth promotion of transformed cells by iron in serum-free culture. Carcinogenesis 6(3):355–359
Acknowledgements
We would like to thank Dr. L. Kühn (ISREC, Epalinges, Switzerland), Dr. M Gossen (Max Delbrück Center, Berlin, Germany), and Dr. H. Sato (Kanazawa University; Kanzawa, Japan) for providing us with expression plasmids.
The research was supported by Bundesministerium für Forschung und Technologie (H. K.); J.C.M and S.A.E. were financed by grants from the DFG (ME2075/3-1 and Sonderforschungsbereich Grant TR3/B5 to JCM) and Helmholtz Association (VH-NG-246 to JCM).
Author information
Authors and Affiliations
Corresponding author
Additional information
H. Kettenmann and R. Glass contributed equally to this work.
Rights and permissions
About this article
Cite this article
Chirasani, S.R., Markovic, D.S., Synowitz, M. et al. Transferrin-receptor-mediated iron accumulation controls proliferation and glutamate release in glioma cells. J Mol Med 87, 153–167 (2009). https://doi.org/10.1007/s00109-008-0414-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-008-0414-3