
Abstract
Genes involved in carbohydrate and lipid metabolism are nutritionally regulated at the transcriptional level in a coordinated fashion. SREBP-1c is a bHLH transcription factor that controls lipogenesis and is induced during overnutrition to facilitate the conversion of glucose to fatty acids and triglycerides for the storage of the excess energy. Uncontrolled activation of nuclear SREBP-1c in the liver can cause hepatosteatosis, hypertriglyceridemia, and hepatic insulin resistance due to direct suppression of insulin signaling pathways, precipitating development of metabolic syndrome. Conversely, TFE3 is a novel bHLH transcription factor that strongly activates various insulin signaling molecules, protecting against the development of insulin resistance and the metabolic syndrome. Regulation of IRS-2 is the primary site where TFE3 in synergy with Foxo1, and SREBP-1c converge. Taken together, TFE3/Foxo1 and SREBP-1c reciprocally regulate IRS-2 expression and insulin sensitivity in the liver. This scenario provides a mechanistic explanation for the physiological link between glucose and lipid metabolism such as physiological switching of glycogen synthesis to lipogenesis. In addition, these two transcription factors may ultimately contribute to pathophysiological effects of overnutrition leading to the development of the metabolic syndrome and diabetes. In this review, I will discuss roles of SREBP-1c and TFE3 in homeostasis of energy metabolism and in metabolic disturbances, focusing on hepatic insulin sensitivity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Nutritional regulation of hepatic lipogenesis by SREBP-1c
Ingestion of excess energy induces fatty acid synthesis in the liver. Dissection of its molecular mechanism is a pathway to understanding of transcriptional regulation of energy metabolism involving physiology of energy storage and pathophysiology linking to hepatosteatosis, dyslipidemia, metabolic syndrome, and diabetes. The sterol regulatory element-binding protein (SREBP) family is a group of transcription factors that control biosynthesis of lipids and play a pivotal role in the homeostasis of cellular sterol regulation [1]. SREBP-1c is the isoform that controls biosynthesis of fatty acids and triglycerides in the liver [2, 3]. Nuclear SREBP-1c up-regulates gene expression of a group of target lipogenic enzymes such as acetyl CoA carboxylase, fatty acid synthase, long chain fatty acid elongase (Evolv-6), and stearoyl-CoA desaturase 1 which coordinately are responsible for the conversion of glucose to oleate, the final end-product of de novo fatty acid synthesis in the liver (Fig. 1). The crucial role for SREBP-1c in hepatic lipogenesis was established primarily through animal studies. Overproduction of hepatic nuclear SREBP-1c in transgenic mice caused activation of these lipogenic genes leading to fatty liver, whereas absence of SREBP-1 by targeted gene disruption abolished nutritional regulation of lipogenic enzymes [4–6]. Of physiological relevance, hepatic SREBP-1c is highly regulated by nutritional conditions. Fasting suppresses and refeeding induces SREBP-1c expression [7, 8]. In addition, diets rich in carbohydrates, sugars, or saturated fatty acids are strong inducers, whereas polyunsaturated fatty acids (PUFA) are inhibitors of SREBP-1c (discussed later).

Nutritional regulation of hepatic lipogenesis controlled by SREBP-1c
SREBPs are synthesized as membrane-bound proteins and reside on rough endoplasmic reticulum. Like SREBP-1a and SREBP-2, SREBP-1c requires a cleavage process to release the amino-terminal portion of the protein into the nucleus for transactivation of its target genes [9, 10]. Whereas this SREBP-cleavage process is the key regulatory step for cellular sterol regulation mediated by SREBP-2, it is not crucial for the activation of hepatic SREBP-1c. The amount of nuclear SREBP-1c protein, the active form, is highly correlated with SREBP-1c mRNA level [7, 11]. Thus, gene expression of SREBP-1c is the key regulatory step for its activity and ultimately determines hepatic lipogenesis.
Upon analysis of the mouse SREBP-1c gene promoter, we identified multiple systems that contribute to nutritional regulation of SREBP-1c and by extension, lipogenesis. The SREBP-1c promoter contains a sterol regulatory element (SRE), which is a canonical binding site for SREBP. This regulatory an auto-loop could explain the phenomenon of overshooting of lipogenic enzyme synthesis when mice are fed high carbohydrate diets after fasting [12]. Upstream of the SRE, there are two LXR response elements or LXREs [13]. LXR plays a crucial role in cholesterol metabolism as an oxysterol receptor and is now also a dominant regulator of SREBP-1c [13, 14]. Control of energy metabolism can be added to the expanding list of LXR functions [15]. It is likely that LXR is involved in the nutritional regulation of SREBP-1c by fasting/refeeding, although more studies are necessary to characterize the relationship between LXR and SREBP-1c in energy metabolism.
Many nutrients and nutrition-related signals, including insulin and glucagon, regulate SREBP-1c expression in a coordinated fashion as summarized in Fig. 2. Nutrients such as carbohydrates (glucose and fructose), as well as saturated fatty acids, are strong inducers of SREBP-1c [7, 8, 11, 16, 17]. Protein kinase A and AMP kinase, both of which are activated during energy depletion, suppress SREBP-1c [18, 19] (Yamamoto T and Shimano H, unpublished data). Leptin also strongly suppresses SREBP-1c [20]. Conversely, insulin and hyperglycemia elevate SREBP-1c expression [11, 16–18, 21, 22]. Some of PKC signals also could be involved in SRBP-1c activation [23] (Yamamoto T and Shimano H, unpublished data). Nevertheless, the precise molecular mechanism by which these nutritional signals control the SREBP-1c promoter is still an enigma. Unlike the dynamic nutritional regulation of hepatic SREBP-1c that occurs in vivo depending upon nutritional states, this level of control is not fully observed in cultured cells including primary hepatocytes, hampering molecular dissection of this system by in vitro experiments.

Nutrients and signals that regulate hepatic SREBP-1c expression. Note LXRs and SREBPs known to directly activate SREBP-1c promoter are not indicated here
Potential involvement of SREBP-1c in metabolic syndrome
Nutritional regulation of SREBP-1c is also involved in the pathogenesis of clinically relevant metabolic disturbances (Fig. 3). Hepatic lipogenesis, which is controlled by SREBP-1c, is linked to production of triglyceride-rich very low-density lipoprotein secreted into circulation as a source of energy for peripheral tissues. Thus, it is conceivable that activation of SREBP-1c can contribute to hyperlipidemia, especially hypertriglyceridemia, which is exacerbated by overnutrition. Experiments in animal provide evidence that overproduction of SREBP-1a and SREBP-1c is associated with hyperlipidemia under conditions in which plasma clearance of apoB-containing lipoproteins is retarded. This has been observed in SREBP-1a transgenic/LDL receptor KO and in ob/ob LDL receptor KO mice [24, 25]. The metabolic syndrome is defined as a cluster of cardiovascular risks such as abdominal obesity, hypertriglyceridemia, low HDL cholesterol level, hypertension, and impaired fasting glucose, all of which enhance atherosclerotic lesion formation. To examine the role of SREBP-1c in the metabolic syndrome, we recently developed a mouse model with SREBP-1c overexpressed in the liver on a background of LDLR deficiency (Takahashi A and Shimano H, unpublished data). These mice exhibited a plasma lipoprotein pattern of increased VLDL triglycerides and decreased HDL cholesterol without changes in LDL cholesterol, a profile similar to that seen in individuals with the metabolic syndrome. These mice develop atherosclerosis, confirming the atherogenicity of the metabolic syndrome-like lipoprotein profiles elicited by overexpression of SREBP-1c. In contrast, SREBP-1 knockout mice have consistently lower triglyceride levels in both plasma and liver than wild-type mice.

Contribution of hepatic SREBP-1c to metabolic syndrome
SREBP-1c causes hepatic insulin resistance
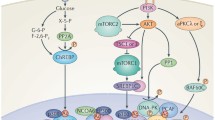
In addition to the regulation of lipid synthesis, SERBP-1c is involved in the regulation of insulin signaling [26] (Fig. 4). Animal models for obesity and insulin resistance such as ob/ob, db/db, KKAy, IRS-2 knockout mice, and aP2-SREBP-1c transgenic mice often exhibit fatty livers with increased SREBP-1c expression [20, 27]. Among insulin signaling molecules, IRS-1 and -2 play the crucial and complementary roles in hepatic insulin signaling. IRS-1 and -2 exhibit differential regulation and roles. IRS-1 is regulated at the protein level and is reported to be more closely linked to glucose homeostasis such as suppression of gluconeogenic genes, whereas IRS-2 controls lipid metabolism [28]. IRS-2 is unique because it is nutritionally regulated at the transcription level. IRS-2 knockout mice exhibited type 2 diabetes, and repression of IRS-2 was observed in the above insulin resistant mice [29, 30]. Thus, IRS-2 is the major hepatic insulin signal molecule in the long-term regulation. Intriguingly, in various nutritional conditions, expression of IRS-2 and SREBP-1c was consistently reciprocal, which led us to speculate that SREBP-1c should repress IRS-2 expression. Through adenoviral overexpression experiments and promoter analyses, it was found that SREBP directly binds to the promoter of IRS-2 and suppresses IRS-2 expression, leading to impaired insulin sensitivity [26]. Adenoviral overexpression of nuclear SREBP-1c decreased IRS-2 protein and autophosphorylation, accompanied by decreased Akt phosphorylation. Glycogen synthesis, a marker of insulin sensitivity, was suppressed by SREBP-1c, whereas fatty acid synthesis was activated. Thus, SREBP-1c could be a molecular mechanism of lipotoxicity in the liver by causing both insulin resistance and fatty liver. Hepatic insulin resistance could lead to hyperglycemia and further activation of hepatic SREBP-1c, forming a vicious circle of metabolic disturbances (Fig. 3). This scenario could also explain lipotoxicity in pancreatic beta cells, as overexpression of SREBP-1c in insulin-promoter transgenic mice exhibits impaired insulin secretion and decreased beta cell mass [31]. Consistently, SREBP-1 knockout mice show higher IRS-2 expression in both liver and beta cells. However, it should also be noticed that these unfavorable lipotoxic effects of SREBP-1c are a dark side of this important lipid transcription factor in the case of chronic activation. In a short term, overexpression of nuclear SREBP-1 consumes glucose for lipogenesis, suppresses PEPCK [32], and could lower blood glucose [33]. Hepatic glucose and lipid metabolism involves many transcription factors and co-factors in a cross-talk network in complex manners and needs to be carefully estimated [3, 34].

SREBP-1c and insulin signaling in hepatocytes
Control of SREBP-1c by fatty acids
It is becoming increasingly clear that a link between pro-inflammatory signals and metabolic disturbances exists. Fatty acids are thought to be inducers of pro-inflammatory signaling cascades, and in the context of cellular stress, this pro-inflammatory state may contribute to the development of insulin resistance. SREBP-1c is highly induced by saturated fatty acids in both hepatocytes and isolated islets. This regulation of SREBP-1c by saturated fatty acids may be linked to their ability to promote inflammation. For example, induction of hepatic SREBP-1c by saturated fatty acids is mediated through PGC-1β [35], and pro-inflammatory signals such as STAT3 and SOCS3 are also involved in SREBP-1c regulation [36]. In contrast to saturated fatty acids, polyunsaturated fatty acids (PUFA) suppress SREBP-1c through multiple mechanisms. PUFAs repress SREBP-1c transcription, enhance degradation of its mRNA, and inhibit SREBP-1c cleavage for nuclear translocation [37–39]. When ob/ob mice were administered with PUFAs such as EPA or fish oil in the diet, the nuclear form of SREBP-1c, and subsequently, hepatic triglyceride content, was significantly decreased. The contribution of hepatic SREBP-1c to fatty liver was also shown by amelioration in ob/ob/SREBP-1 KO double mutant mice [40]. In addition to improvement of fatty liver, PUFA administration decreased both plasma insulin and glucose levels, improving insulin resistance [41]. This effect could be at least partially explained by the suppression of hepatic SREBP-1c. The precise molecular mechanism by which PUFAs inhibit the cleavage of SREBP-1 but not SREBP-2 is yet to be clarified, but will be crucial to understanding the differential regulation of SREBP-1 and -2 activation.
Discovery of TFE3 as a potent enhancer of insulin signaling
As mentioned above, SREBP-1c is likely to play an important role as an upstream regulator of lipogenesis, and when overexpressed, it contributes to metabolic disturbances related to lipotoxicity, including hyperlipidemia and insulin resistance. Based upon this knowledge, we sought to discover new factors that could have insulin signaling enhancing effects by playing a reciprocal role to SREBP-1c. E-boxes are consensus cis-elements for bHLH proteins and are often found to play a role in the nutritional regulation of metabolic genes. Thus, we adopted an expression strategy that utilized a carbohydrate response element including an E-box found in spot 14 gene. By screening an expression library from SREBP-1 KO mice, two clones that activated a reporter luciferase gene fused to this E-box containing cis-element were selected and identified as TFE3 and TFEB [42].
TFE3 is a bHLH protein that has been studied in its relation to immunology and cancer. TFE3 binds to enhancer of the immunoglobulin gene [43] and contributes to TGF-β-mediated PAI-1 gene induction through interaction with Smad proteins [44]. TFE3 is expressed ubiquitously, including energy-organs such as liver and adipose tissue. However, the role of TFE3 in metabolism had not been investigated. To obtain a global blueprint of the metabolic function of TFE3, liver samples from mice infected with adenovirus encoding TFE3 (Ad-TFE3) were subjected to DNA microarray analyses. Intriguingly, genes upregulated by TFE3 include IRS2, Akt1, Insig1, and HKII, all of which are involved in insulin signaling (Table 1). Transactivation of IRS2, HKII, and Insig1 by TFE3 was reconfirmed in primary hepatocytes in which TFE3 was overexpressed. Increased IRS-2 protein was associated with enhancement of phosphorylation of Akt, GSK3, and ERK with concomitant activation of glycogen synthesis, an indication of enhanced insulin signaling.
In vivo action of TFE-3
Consistent with enhanced insulin action in hepatocytes, after infection into normal mice, Ad-TFE3 exhibited a potent glucose-lowering action by similar activation of hepatic insulin-signaling molecules.
Next, we explored potential therapeutic effects of TFE3 on murine models of insulin resistance and diabetes. Ad-TFE3 injection caused marked amelioration of diabetes in KK and db/db mice. Both high glucose and insulin levels were decreased by TFE3 overexpression. In addition to overexpression experiments, knockdown of TFE3 in the liver caused suppression of IRS2 and emergence of insulin resistance, implicating TFE3 in physiological regulation of insulin sensitivity. Both TFE3 and IRS-2 are concomitantly repressed in the livers of insulin resistant ob/ob mice and up-regulated in streptozocin-treated mice. The coordinated regulation of TFE3 and IRS-2 in physiological and pathophysiological livers supported TFE3 regulation of IRS-2.
Activation of IRS-2 promoter by TFE3
Human IRS-2 promoter analysis (reporter and gel shift assays) revealed that TFE3 binds to an E-box in the middle of the SREBP binding site and next to a Foxo binding site. In transfection studies, co-expression of TFE3 and Foxo1 synergistically activated the IRS2 promoter. As expected from the synergistic activation of IRS-2 promoter, Foxo1/TFE3 physically interact as shown by co-immunoprecipitation. Further analysis of Insig-1 and HKII promoters confirmed that HKII and Insig-1 are also direct target genes of TFE3. Physical in vivo binding of these factors to IRS-2 promoter was confirmed by ChIP assays. SREBP binds to the IRS-2 promoter in competition with Foxo1/TFE3 and leads to insulin resistance. As summarized in Fig. 5, TFE3/Foxo1 and SREBP-1c dictate nutritional regulation of IRS-2 expression and insulin sensitivity in the liver. In a fasted state with low plasma insulin levels, TFE3 and Foxo1 bind to the IRS2 promoter and trans-activate IRS2 expression, presumably through recruiting the co-activator, PGC-1α [26]. High expression of IRS2 assures efficient insulin signaling preparing for the next meal with its concomitant rise in insulin. In a re-fed condition or insulin-resistance states, active SREBP-1c accumulates in the nucleus, occupies and suppresses the IRS-2 promoter, and leads to hepatic insulin resistance. Collectively, SREBP-1c, Foxo1, and TFE3 control insulin sensitivity by regulating IRS-2 expression.

Regulation of IRS-2 and insulin sensitivity by TFE3/Foxo1 and SREBP-1c TFE3/Foxo1 and SREBP-1c compete for binding to their overlapped binding site in the IRS-2 promoter. TFE3/Foxo1 activate IRS-2 expression in a fasted or insulin-depleted state. SREBP-1c accumulates in overnutrition and represses IRS-2 expression. When these three factors are co-localized in the nucleus, as observed in an insulin-resistant state, SREBP dominates over TFE3/Foxo1 for binding to the IRS-2 promoter, and insulin resistance persists
Various metabolic impacts of TFE3 overexpression
It is noteworthy that TFE3 markedly improved plasma glucose levels even in STZ-treated diabetic mice, a model of type 1 diabetes characterized by insulin depletion. This effect implicates that in addition to increasing levels of insulin signaling molecules, TFE3 also activates insulin signaling through a novel mechanism other than increases in IRS-2/Akt. TFE3 overexpression experiments in rat primary hepatocytes demonstrated that phosphorylation of IRS-2 and Akt was observed only in the presence of insulin, whereas phosphorylation of GSK3β and ERK was induced by TFE3 even in the absence of insulin. The precise molecular mechanism by which insulin signaling molecules were selectively hyperphosphorylated by TFE3 in the absence of insulin is currently unknown. It is conceivable that in addition to activation of HKII, this unique feature can account for the glucose-lowering effect of TFE3 in STZ-treated mice.
In addition to glucose/insulin metabolism, TFE3 has a profound effect on lipid metabolism. Hepatic triglyceride and cholesterol content as well as plasma triglycerides were diminished in Ad-TFE3-treated mice. This is likely due to the activation of Insig-1 by TFE3. Insig-1 retains SREBP/SCAP at rough ER, prevents their travel to golgi, and thus, functions as an inhibitor of SREBPs. AdTFE3 activated Insig-1 and abolished accumulation of nuclear SREBP-1c protein and lipogenesis.
Furthermore, TFE3 also activates protein synthesis. Ad-TFE3-injected mice demonstrated increased hepatic phosphorylation of p70S6kinase and S6, proteins involved in protein translation. Consistent with the activation of these molecules, serum proteins including albumin were elevated. Ad-TFE3 infection caused enlargement of liver with a hypertrophic change in hepatocytes, which could be a reflection of increased protein synthesis.
Collectively, these data demonstrate that TFE3 has a significant impact on metabolic gene regulation, including increased insulin signaling, increased glycogen synthesis, decreased lipogenesis, and increased protein synthesis (Fig. 6). The unique and remarkable feature of this new metabolic transcription factor is that a single transcription factor has such a deep impact on various metabolic pathways in a coordinated fashion. Enhancement of insulin signaling and hypoglycemic effect, inhibition of lipogenesis, and activation of protein synthesis are all favorable for the prevention of metabolic syndrome and diabetes. The more precise mechanism by which TFE3 controls each of these pathways will require further investigation.

Various actions of TFE3 on hepatic metabolism
Clinical perspective of energy transcription factors as targets of metabolic diseases
Risk factors such as obesity, insulin resistance, hypertriglyceridemia, and hypertension contribute to the metabolic syndrome. Individually and cumulatively, these factors promote the development of diabetes and cardiovascular disease. Efficient control of each of these factors is required for the prevention of life-threatening events. Because insulin resistance is one of the central features of the metabolic syndrome, intervention focused at the underlying pathology for this factor would be more efficient and cause less adverse effects than using multiple drugs for each of the other risks alone. In this respect, modification of energy transcription factors is a reasonable approach. Agonists for PPARs such as fibrates and thiazolidinediones are already clinically used for the improvement of both lipid and glucose metabolism through insulin sensitization. HMGCoA reductase inhibitors (statins) are used most widely for the prevention of atherosclerotic disease. The plasma cholesterol lowering action of statins is mediated through activation of the SREBP-2/LDL receptor pathway. These pieces of well-known clinical evidence demonstrate that energy transcription factors are important targets of metabolic diseases. Meanwhile, cross-talk network of energy transcription factors is complex, and sometimes, drug development that targeted this system might encounter unexpected outcome [3]. For instance, SREBP-2 activation is beneficial in reducing plasma lipid levels because of the up-regulation of LDL receptors; however, activation of endogenous cholesterol synthesis could be harmful to pancreatic beta cells (Ishikawa M and Shimano H, a manuscript in preparation). LXR agonists activate cholesterol efflux in macrophages in atherosclerosis and have been thought to be promising as a future anti-atherosclerosis drug [45]. However, LXR activation in liver directly induces SREBP-1c and phospholipid transfer protein leading to fatty liver and hypertriglyceridemia, implicating double-edged efficacy [46]. To obtain favorable outcomes, it is crucial to modify transcription factors in a targeted manner in specific tissue with the correct intensity as already noticed in the concept of selective estrogen receptor modulators. Based upon our findings, suppression of SREBP-1c and activation of TFE3 in the liver are a favorable strategy to improve insulin resistance for the prevention of diabetes and cardiovascular risks. But their roles in other tissues, especially adipose tissues and skeletal muscle, require further investigation. Roles of these factors in vascular wall are also important issues to be investigated. To seek for the way of TFE3 activation, it is important to clarify physiological mode of regulation of this versatile factor especially at the protein level. These future investigations should also help to understand cross-talk network of energy transcription factors.
References
Brown MS, Goldstein JL (1997) The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89:331–340
Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125–1131
Shimano H (2002) Sterol regulatory element-binding protein family as global regulators of lipid synthetic genes in energy metabolism. Vitam Horm 65:167–194
Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL (1997) Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 99:846–854
Shimano H, Yahagi N, Amemiya-Kudo M et al (1999) Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem 274:35832–35839
Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS (2002) Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem 277:9520–9528
Horton JD, Bashmakov Y, Shimomura I, Shimano H (1998) Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc Natl Acad Sci USA 95:5987–5992
Kim JB, Sarraf P, Wright M et al (1998) Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest 101:1–9
Brown MS, Goldstein JL (1998) Sterol regulatory element binding proteins (SREBPs): controllers of lipid synthesis and cellular uptake. Nutr Rev 56:S1–S3; discussion S54–S75
Brown MS, Goldstein JL (1999) A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci USA 96:11041–11048
Hasty AH, Shimano H, Yahagi N et al (2000) Sterol regulatory element-binding protein-1 is regulated by glucose at the transcriptional level. J Biol Chem 275:31069–31077
Amemiya-Kudo M, Shimano H, Yoshikawa T et al (2000) Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J Biol Chem 275:31078–31085
Yoshikawa T, Shimano H, Amemiya-Kudo M et al (2001) Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol 21:2991–3000
Repa JJ, Liang G, Ou J et al (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14:2819–2830
Laffitte BA, Chao LC, Li J et al (2003) Activation of liver X receptor improves glucose tolerance through coordinate regulation of glucose metabolism in liver and adipose tissue. Proc Natl Acad Sci USA 100:5419–5424
Matsuzaka T, Shimano H, Yahagi N et al (2004) Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes 53:560–569
Nagai Y, Nishio Y, Nakamura T, Maegawa H, Kikkawa R, Kashiwagi A (2002) Amelioration of high fructose-induced metabolic derangements by activation of PPARalpha. Am J Physiol Endocrinol Metab 282:E1180–E1190
Foretz M, Pacot C, Dugail I et al (1999) ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol 19:3760–3768
Zhou G, Myers R, Li Y et al (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Tobe K, Suzuki R, Aoyama M et al (2001) Increased expression of the sterol regulatory element-binding protein-1 gene in insulin receptor substrate-2(−/−) mouse liver. J Biol Chem 276:38337–38340
Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL (1999) Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci USA 96:13656–13661
Ono H, Shimano H, Katagiri H et al (2003) Hepatic Akt activation induces marked hypoglycemia, hepatomegaly, and hypertriglyceridemia with sterol regulatory element binding protein involvement. Diabetes 52:2905–2913
Matsumoto M, Ogawa W, Akimoto K et al (2003) PKClambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. J Clin Invest 112:935–944
Horton JD, Shimano H, Hamilton RL, Brown MS, Goldstein JL (1999) Disruption of LDL receptor gene in transgenic SREBP-1a mice unmasks hyperlipidemia resulting from production of lipid-rich VLDL. J Clin Invest 103:1067–1076
Hasty AH, Shimano H, Osuga J-I et al (2001) Severe hypercholesterolemia, hypertriglyceridemia and atherosclerosis in mice lacking both leptin and the low density lipoprotein receptor. J Biol Chem M01017–M06200
Ide T, Shimano H, Yahagi N et al (2004) SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat Cell Biol 6:351–357
Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL (2000) Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell 6:77–86
Taniguchi CM, Ueki K, Kahn R (2000) Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of metabolism. J Clin Invest 115:718–727
Kido Y, Burks DJ, Withers D et al (2000) Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J Clin Invest 105:199–205
Kubota T, Kubota N, Moroi M et al (2003) Lack of insulin receptor substrate-2 causes progressive neointima formation in response to vessel injury. Circulation 107:3073–3080
Takahashi A, Motomura K, Kato T et al (2005) Transgenic mice overexpressing nuclear SREBP-1c in pancreatic beta-cells. Diabetes 54:492–499
Yamamoto T, Shimano H, Nakagawa Y et al (2004) SREBP-1 interacts with hepatocyte nuclear factor-4 alpha and interferes with PGC-1 recruitment to suppress hepatic gluconeogenic genes. J Biol Chem 279:12027–12035
Barthel A, Schmoll D (2003) Novel concepts in insulin regulation of hepatic gluconeogenesis. Am J Physiol Endocrinol Metab 285:E685–E692
Weickert MO, Pfeiffer AF (2006) Signalling mechanisms linking hepatic glucose and lipid metabolism. Diabetologia 49:1732–1741
Lin J, Yang R, Tarr PT et al (2005) Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 120:261–273
Ueki K, Kondo T, Tseng YH, Kahn CR (2004) Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci USA 101:10422–10427
Kim HJ, Takahashi M, Ezaki O (1999) Fish oil feeding decreases mature sterol regulatory element-binding protein 1 (SREBP-1) by down-regulation of SREBP-1c mRNA in mouse liver. A possible mechanism for down-regulation of lipogenic enzyme mrnas. J Biol Chem 274:25892–25898
Yahagi N, Shimano H, Hasty AH et al (1999) A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J Biol Chem 274:35840–35844
Jump DB, Botolin D, Wang Y, Xu J, Christian B, Demeure O (2005) Fatty acid regulation of hepatic gene transcription. J Nutr 135:2503–2506
Yahagi N, Shimano H, Hasty AH et al (2002) Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem 277:19353–19357
Sekiya M, Yahagi N, Matsuzaka T et al (2003) Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology 38:1529–1539
Nakagawa Y, Shimano H, Yoshikawa T et al (2006) TFE3 transcriptionally activates hepatic IRS-2, participates in insulin signaling and ameliorates diabetes. Nat Med 12:107–113
Beckmann H, Su LK, Kadesch T (1990) TFE3: a helix-loop-helix protein that activates transcription through the immunoglobulin enhancer muE3 motif. Genes Dev 4:167–179
Hua X, Liu X, Ansari DO, Lodish HF (1998) Synergistic cooperation of TFE3 and smad proteins in TGF-beta-induced transcription of the plasminogen activator inhibitor-1 gene. Genes Dev 12:3084–3095
Tontonoz P, Mangelsdorf DJ (2003) Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol 17:985–993
Joseph SB, Tontonoz P (2003) LXRs: new therapeutic targets in atherosclerosis? Curr Opin Pharmacol 3:192–197
Acknowledgment
I am grateful to Drs. Naoya Yahagi, Michiyo Amemiya-Kudo, Tomohiro Yoshikawa, Tomihiro Ide, Akimitsu Takahashi, Motoya Sekiya, Yoshimi Nakagawa for their contribution to our work, Alyssa H Hasty for the critical reading of this manuscript, and to Profs. Nobuhiro Yamada, Toshio Murase for continuous support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shimano, H. SREBP-1c and TFE3, energy transcription factors that regulate hepatic insulin signaling. J Mol Med 85, 437–444 (2007). https://doi.org/10.1007/s00109-007-0158-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-007-0158-5