
Abstract
D-type cyclins play a pivotal role in cell cycle regulation and their abnormal expression was associated with several human malignancies. To assess Cyclin D2 promoter methylation status and expression levels in prostate tissues, quantitative methylation-specific PCR and quantitative reverse transcription PCR assays were performed in a large series of prostate carcinomas, high-grade prostatic intraepithelial neoplasias (HGPIN), benign prostate hyperplasias (BPH), normal prostate tissue (NPT) samples, and prostate cancer (PCa) cell lines (before and after demethylating treatment). Methylation levels were correlated with mRNA expression levels and key clinicopathologic parameters. Cyclin D2 promoter methylation was found in 117/118 PCa, 38/38 HGPIN, 24/30 BPH, 11/11 NPT, and 4/4 cell lines. Methylation levels were significantly higher in PCa compared with HGPIN, NPT, and BPH (P<0.0001), correlating with tumor stage and Gleason score (r=0.29, P=0.0014; and r=0.32, P=0.0005, respectively). Conversely, Cyclin D2 mRNA levels were significantly lower in PCa (P<0.01) and a significant inverse correlation between Cyclin D2 methylation and expression levels was found in prostatic tissues (r=−0.61, P<0.000001). Demethylating treatment induced a substantial increase in Cyclin D2 mRNA in LNCaP cells whereas decreased levels were observed in DU-145 and PC-3 cells. We concluded that Cyclin D2 promoter methylation downregulates gene transcription and occurs with high frequency at low levels in normal, hyperplastic, and preneoplastic prostate tissues. Conversely, high Cyclin D2 methylation levels characterize invasive prostatic carcinoma, correlating with clinicopathologic features of tumor aggressiveness.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Loss of control of cell growth and proliferation is a distinctive feature of human cancer [1]. Progression through the cell cycle is controlled by cyclin-dependent kinases (CDKs) and their activity is upregulated by cyclins and inhibited by CDK inhibitors [2]. Among cyclins, the D-type cyclins (D1, D2, and D3) are mainly implicated in G1 to S phase transition [3], with corresponding mRNA synthesis attaining a maximum at the former and a minimum at the latter phase [4]. Owing to their pivotal role in cell cycle regulation, their abnormal or untimely expression is likely to disturb cell cycle control and might promote neoplastic transformation [5].
Cyclin D2/CCND2, located at chromosome 12p13, is generally considered a protooncogene because its transcription is activated when Epstein–Barr virus immortalizes primary B lymphocytes [6, 7]. Indeed, overexpression of Cyclin D2 was reported in testicular germ cell tumor cell lines [8], granulosa cell tumors of the ovary [9], colon cancer [10], and gastric cancer, correlating with disease progression and poor prognosis [4, 11]. However, the role of Cyclin D2 as a protooncogene was challenged by several studies that found loss of expression in breast [5, 12–14], lung [14], pancreatic [15], and gastric cancer [16]. It is interesting to note that the mechanism underlying Cyclin D2 silencing in most cases is promoter methylation [5, 13–16], an epigenetic alteration that is commonly associated with partial or complete transcriptional disruption of cancer-related genes [17].
Aberrant promoter methylation of several genes is a common feature of prostate cancer (PCa) [18–20] and a promising tool for early cancer detection [21]. Indeed, Cyclin D2 promoter methylation was reported in 32% of prostate carcinomas, but it was seldom observed in nonmalignant prostate tissues [22]. Moreover, methylation of Cyclin D2 promoter was more frequent in high Gleason score tumors and a high degree of concordance with promoter methylation at other cancer-related genes was observed [22]. Recently, Cyclin D2 hypermethylation was also reported to predict time to progression in a defined subset of prostate carcinomas [23].
Owing to the controversial role of Cyclin D2 deregulation in carcinogenesis and the lack of expression studies in PCa, we aimed to quantitatively assess Cyclin D2 promoter methylation and gene expression in neoplastic, preneoplastic, nonmalignant prostate tissues, and in PCa cell lines. For that purpose, real-time quantitative methylation-specific PCR (QMSP) and real-time quantitative reverse-transcription PCR (QRT-PCR) were performed in a large series of prostate tissue samples and the results were correlated with clinicopathological parameters. Finally, the effect of a demethylating agent [5-aza-2′deoxycytidine (Aza-C)] on Cyclin D2 expression in PCa cell lines was tested.
Materials and methods
Patients, sample collection, and DNA extraction
Primary tumors from 118 patients with clinically localized prostate adenocarcinoma (stage II T1c N0 M0 or T2 N0 M0), according to the TNM staging system [24]), consecutively diagnosed and primarily treated with radical prostatectomy at the Portuguese Oncology Institute, Porto, Portugal, were prospectively collected. In 38 radical prostatectomy specimens with PCa a HGPIN lesion was identified and collected for further analysis. For control purposes, nonneoplastic prostate tissue samples were obtained from 30 randomly selected patients with BPH that underwent transurethral resection of the prostate, and also from the peripheral zone of 11 prostates that did not harbor PCa collected from cystoprostatectomy specimens of bladder cancer patients [normal prostate tissue (NPT)]. All tissue specimens were promptly frozen immediately after surgery and stored at −80°C for further analysis. Five-micron thick sections were cut and stained for the identification of the areas of PCa (i.e., the index or dominant tumor), HGPIN, BPH, and normal tissue. Then, the tissue block was trimmed to maximize the yield of target cells (>70% of target cells). Subsequently, an average of 50 12-μm thick sections were cut and every fifth section was stained to ensure an uniform percentage of target cells and to exclude contamination from neoplastic cells in normal and BPH tissue samples. DNA was extracted from all samples with phenol/chloroform and precipitated with ethanol [25].
Histological slides from formalin-fixed, paraffin embedded tissue fragments were obtained from the same surgical specimens and assessed for Gleason grade [26] and TNM stage [24]. Relevant clinical data were abstracted from the clinical records. The institutional review board (IRB) of Portuguese Oncology Institute, Porto, approved these studies and permission to test these samples without identifiers was also granted by exemption from the IRB of Johns Hopkins University.
Cell culture conditions and demethylating treatment
Human PCa cell lines LNCaP, DU-145, PC-3, and 22RV1 were obtained from American Type Culture Collection (Rockville, MD) and cultured in RPMI 1640 (InVitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum for isolation of DNA and RNA. Demethylating treatment was performed as previously described [27]. In brief, the cells were split to low density (5×105 per T-25 flask) 12–24 h before treatment. Then, cells were treated at day 1 and day 4 with 5 μM Aza-C (Sigma, St Louis, MO, USA) from 100 mM 50% acetic acid dissolved stock. Cells were harvested at day 5 and processed for DNA and RNA extraction.
Bisulfite treatment and QMSP
Sodium bisulfite conversion of unmethylated (but not methylated) cytosine residues to uracil of genomic DNA obtained from patient tissue samples and cell lines was performed as described previously [28]. Four micrograms of DNA were used for the chemical treatment. DNA samples were then purified using the Wizard purification resin kit (Promega, Madison, WI, USA), treated again with sodium hydroxide, precipitated with ethanol, and resuspended in 240 μl of water and stored at −80°C.
The Cyclin D2 (GenBank accession no. AF518005) promoter includes a CpG-rich region, 1,000–1,600 bp 5′ to the translational start site [5]. Thus, we designed a set of primers and probe which encompasses CpG sites found to be methylated in lung and breast cancer cells exhibiting Cyclin D2 underexpression [5, 14]. Specifically, they are: sense, 5′-TTT GAT TTA AGG ATG CGT TAG AGT ACG-3′; antisense, 5′-ACT TTC TCC CTA AAA ACC GAC TAC G-3′; and probe, 6FAM-5′-AAT CCG CCA ACA CGA TCG ACC CTA-3′-TAMRA. The primers and probe used for the internal reference gene (beta actin, ACTB) were previously published [18]. To determine the relative levels of methylated promoter DNA in each sample, the values of the target gene were compared with the values of the internal reference gene to obtain a ratio that was then multiplied by 1,000 for easier tabulation (Cyclin D2/ACTB ×1,000).
Fluorogenic QMSP assays were carried out in a reaction volume of 20 μl in 384-well plates in an Applied Biosystems 7900 Sequence Detection System (Perkin-Elmer, Foster City, CA, USA). PCR was performed in separate wells for each primer/probe set and each sample was run in triplicate. The final reaction mixture consisted of 600 nM of each primer (InVitrogen); 200 nM probe (Applied Biosystems, Foster City, CA, USA); 0.75 U of platinum Taq polymerase (InVitrogen); 200 μM each of dATP, dCTP, dGTP, and dTTP; 16.6 mM ammonium sulfate; 67 mM Trizma; 6.7 mM magnesium chloride; 10 mM mercaptoethanol; 0.1% DMSO; and 3 μl bisulfite-converted genomic DNA. PCR was performed using the following conditions: 95°C for 2 min, followed by 50 cycles at 95°C for 15 s, and 60°C for 1 min. Each plate included multiple nontemplate controls, a negative control, and serial dilutions of a positive control for constructing the standard curve on each plate. Leukocyte DNA collected from healthy individuals was utilized as negative control. The same leukocyte DNA was methylated in vitro with SssI bacterial methyltransferase (New England Biolabs, Beverly, MA, USA) and used as positive control.
A given sample was considered positive for Cyclin D2 hypermethylation when amplification was detected in at least two of the triplicates of the respective QMSP analysis. The QMSP threshold was determined for each run adjusting the best fit of the slope and R 2, using the standard curve.
RNA extraction and QRT-PCR
Total RNA was extracted from all PCa cell lines and also from 14 BPH, 10 NPT, 20 HGPIN, and 34 PCa samples (from which fresh frozen tissue was still available) using FastRNA Kit Green for 45 s with a speed rating of 6.0 in a FastPrep FP120 Instrument (QBiogene, Carlsbad, CA, USA) and it was reverse-transcribed using the SuperScript III First-Strand Synthesis System for RT-PCR (InVitrogen). Primers and probes for Cyclin D2 and for beta-glucuronidase (β-GUS), the internal reference gene, were purchased from Applied Biosystems. To determine the relative expression levels of Cyclin D2 mRNA in each sample, the values of the target gene were normalized with the values of the internal reference gene to obtain a ratio that was then multiplied by 100 for easier tabulation (Cyclin D2/β-GUS ×100).
PCR was performed according to manufacturer’s specifications (Applied Biosystems) in separate wells for each primer/probe set and each sample was run in triplicate. The final reaction mixture consisted of 10 μl of TaqMan® Universal PCR Master Mix, AmpErase® UNG (2×), 1 μl of 20× Assays-on-Demand™ Gene Expression Assay Mix (Cyclin D2 or β-GUS) and 9 μl of cDNA. PCR was performed using the following conditions: 50°C for 2 min, 95°C for 10 min, followed by 50 cycles at 95°C for 15 s, and 60°C for 1 min. Each plate included multiple nontemplate controls and serial dilutions of a positive control for constructing the standard curve on each plate.
Statistical analysis
The Shapiro–Wilk’s W test allowed for the examination of the appropriateness of a normal distribution assumption for each of the parameters (data not shown). Then, the median and interquartile range of the methylation levels and of the mRNA expression levels for each group of samples was determined and analyzed using nonparametric tests, i.e., the Kruskal–Wallis one-way analysis of variance (ANOVA), followed by Mann–Whitney U test when appropriate. The Kruskal–Wallis one-way ANOVA, followed by the Mann–Whitney U test was also used to compare the age distribution among the three patient populations (PCa, BPH, and NPT). The Mann–Whitney U test was performed to compare age and PSA levels between patients with BPH or prostate adenocarcinoma. For multiple comparisons the Bonferroni method was used to adjust the P values. The correlations between the tumor methylation level on the one hand, and age, PSA level, Gleason score, and pathological stage, on the other, were determined by calculating a Spearman’s correlation coefficient. The same statistical test was used to assess the association between Cyclin D2 methylation levels and mRNA expression levels. All statistical tests were two-sided. Statistical analyses were performed using a computer-assisted program (Statistica for Windows, version 6.0, StatSoft, Tulsa, OK, USA).
Results
Clinical and pathological data
Tissue samples from 118 prostate adenocarcinomas, 38 HGPIN lesions, 30 BPH, and 11 normal tissue samples were tested. The clinical and pathological characteristics of the patients are depicted in Table 1. Although PSA levels were higher in patients with PCa (P=0.0018), there was considerable overlap with BPH cases. Statistically significant differences in patient’s age were detected among the three groups of patients (P=0.0088). The Mann–Whitney U test disclosed a significant difference only between the median age of BPH and PCa patients (P=0.0019).
Cyclin D2 promoter methylation in prostatic tissues and clinicopathologic correlations
The frequency and levels of Cyclin D2 promoter methylation in NPT and prostate lesions is displayed in Table 2. Except for one case of PCa and six cases of BPH, all prostate tissue samples harbored some degree of promoter methylation at the Cyclin D2 locus. However, QMSP detected significant differences in methylation levels among the four groups of tissue samples (Kruskal–Wallis test, P<0.0001) (Fig. 1a). Pairwise comparison using Mann–Whitney U test showed that methylation levels were significantly higher in PCa compared with HGPIN, BPH, and NPT (P<0.0001 for all comparisons). Moreover, HGPIN and NPT methylation levels were higher than those of BPH (P<0.0001 for both), but did not significantly differ between them (P=0.08).

Distribution of Cyclin D2 a promoter methylation levels and b mRNA expression levels in prostate tissues: benign prostatic hyperplasia (BPH), normal prostate tissue (NPT), high-grade prostatic intraepithelial neoplasia (HGPIN), and prostate cancer (PCa)
Methylation levels were found to significantly differ among Gleason score categories (P=0.0027) and nonorgan-confined PCa displayed higher Cyclin D2 methylation levels than organ-confined tumors (P=0.00056) (Fig. 2). Moreover, Cyclin D2 methylation levels correlated positively with the Gleason score (r=0.32, P=0.0005) and pathologic tumor stage (r=0.29, P=0.0014), although no correlation was found with age or serum PSA.

Distribution of Cyclin D2 promoter methylation levels according to a Gleason score (GS) and b pathological stage (pT) categories in PCa cases
Cyclin D2 mRNA relative expression levels and promoter methylation in prostate tissues
The distribution of Cyclin D2 mRNA relative expression levels in prostate tissues is depicted in Table 3 and graphically displayed in Fig. 1b. BPH disclosed the highest Cyclin D2 mRNA levels and these significantly differed from those of the remaining tissue samples (P<0.01 for all). Cyclin D2 mRNA levels in HGPIN and NPT did not significantly differ (P=0.17), but were higher than those of PCa (P<0.01 for both). A significant inverse correlation between Cyclin D2 promoter methylation levels and mRNA expression levels in prostate tissues (r=−0.61, P<0.000001) was found.
Cyclin D2 promoter methylation and mRNA relative expression levels in PCa cell lines
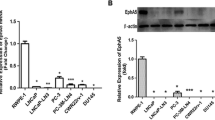
All PCa cell lines (LNCaP, DU-145, PC-3, and 22RV1) were found to be methylated at the Cyclin D2 locus, although the methylation levels were rather variable (Table 4). The highest methylation levels were observed in LNCaP cell line, which concurrently displayed low Cyclin D2 mRNA relative expression level (Table 4). After treatment with Aza-C, an 146.9-fold increase in Cyclin D2 mRNA levels was observed in LNCaP cells. It is intriguing to note that a decrease in Cyclin D2 mRNA levels after demethylation was apparent in DU-145 and PC-3 cancer cell lines, which originally depicted modest Cyclin D2 methylation levels (Table 4).
Discussion
In this study, we found that Cyclin D2 methylation levels were significantly higher in PCa compared to HGPIN, NPT, or BPH, whereas mRNA expression levels followed the opposite trend, resulting in a significant inverse correlation. Moreover, we were able to demonstrate that higher Cyclin D2 methylation levels correlate with clinicopathological parameters of tumor aggressiveness.
The almost ubiquitous presence of Cyclin D2 promoter methylation in prostate tissues observed in the present study contrasts with the lower frequencies reported in the only available study on Cyclin D2 promoter methylation prevalence in a large set of primary prostate carcinomas [22]. In addition to differences in patient population background and primer sets used, it should be emphasized that we used a highly sensitive and specific QMSP assay [29], which was shown to reliably detect promoter methylation in the presence of 10,000-fold excess of unmethylated alleles, i.e., a tenfold increase in sensitivity over the conventional MSP used in the aforementioned report [22]. This methodological difference is likely to account for the discrepancy because similar results concerning the methylation frequencies at other gene promoters (including GSTP1, APC, and RARB2) in prostate carcinoma are apparent in previous reports [18, 19, 30]. Nonetheless, despite the similar frequencies of Cyclin D2 promoter methylation among cancerous and noncancerous prostate tissues, significant differences in methylation levels were detected, paralleling our previous observations on the epigenetic alteration of other cancer-related genes in PCa [18, 19, 31]. Altogether, these results demonstrate that detection of methylation alone is not sufficient evidence of malignancy and emphasize the usefulness of quantitative assays.
The importance of quantitative assays was also underlined in the present study by the significant inverse correlation found between Cyclin D2 promoter methylation levels and the respective mRNA relative expression levels. Indeed, our results suggest that promoter methylation modulates Cyclin D2 expression, resulting in a variable degree of loss of mRNA levels depending on the parallel increase in methylation levels. This finding might also explain the coexistence of Cyclin D2 promoter methylation and expression in some breast cancers [5]. Moreover, the inverse correlation between Cyclin D2 promoter methylation and mRNA expression levels provides a biological rationale for the association between promoter methylation levels and clinicopathological features of tumor aggressiveness observed in our study. It is interesting to note that in a defined subset of prostate carcinomas (i.e., tumors with Gleason score of 3+4=7) Cyclin D2 hypermethylation in conjunction with APC hypermethylation predicted time to progression in multivariate analysis [23]. Thus, quantitative Cyclin D2 promoter methylation might be a useful PCa biomarker, eventually in combination with other genetic or epigenetic alterations, for the identification of the more aggressive prostate carcinomas that might benefit from different therapeutic modalities.
It is noteworthy that significant differences in Cyclin D2 promoter methylation and mRNA expression levels between normal prostate tissue from the peripheral zone and HGPIN, a putative precursor of prostate adenocarcinoma, were not found. This result suggests that this epigenetic alteration is acquired when morphological evidence of neoplastic transformation is still not apparent and heterogeneous nonclonal levels are maintained during the development of a preneoplastic lesion. Thus, Cyclin D2 promoter methylation might not confer a significant growth advantage at this stage of prostatic carcinogenesis. However, the progressive downregulation of Cyclin D2 expression mediated by promoter methylation might become advantageous for cells with an established malignant phenotype, resulting in clonal outgrowth of the highly methylated cells. This hypothesis is further supported by the correlation between Cyclin D2 hypermethylation levels and increased Gleason score and pathological stage, suggesting that this alteration might contribute to enhanced proliferation and loss of differentiation often found in more aggressive prostate carcinomas.
Due to its anticipated role as a protooncogene [6, 7], the transcriptional silencing of Cyclin D2 might seem more deleterious than beneficial for PCa cells. In fact, overexpression of Cyclin D2 was reported in ovarian granulosa cell tumors [9], colon cancer [10], and gastric cancer [4, 11], a finding which is consistent with a protooncogene function. However, results similar to ours were reported for other solid tumors, including breast, lung, pancreatic, and gastric cancer [5, 14–16] and, thus, a tumor-suppressor role for Cyclin D2 late in cancer progression seem plausible. Indeed, Cyclin D2 overexpression was linked with increased cell proliferation [7], but it might also negatively influence cell growth. Previous studies demonstrated that the early G0 and G1 phases of the fibroblast cell cycle are efficiently blocked by Cyclin D2 overexpression, leading to a nonproliferative state [32]. If the same principle holds true for epithelial prostate cells, then Cyclin D2 promoter methylation and consequent decrease in gene expression would release cells from cell cycle blockade and foster cell growth and proliferation. It is acknowledged that this is a rather speculative hypothesis concerning the purported dual role of the Cyclin D2 gene in tumorigenesis. However, to the best of our knowledge, no alternative explanation was provided for the increased levels of Cyclin D2 in some neoplasms and its downregulation in others.
This hypothesis might be reflected in the results we obtained in PCa cell lines. LNCaP cells, which are androgen-dependent, showed concurrently the highest Cyclin D2 promoter methylation levels and very low mRNA levels. Remarkably, demethylating treatment triggered a massive increase in Cyclin D2 mRNA. These results suggest that in this cancer cell line and in a subset of prostate carcinomas (i.e., those prostate carcinomas with high Cyclin D2 methylation levels and which are more likely to be clinically aggressive), Cyclin D2 might play a tumor suppressor function and its inactivation might be accomplished through promoter methylation.
In conclusion, we showed that Cyclin D2 aberrant promoter methylation and decreased gene expression is a common event in the prostate gland, quantitatively differing between malignant and nonmalignant tissues. Moreover, this epigenetic alteration is associated with clinicopathologic features of tumor aggressiveness. Finally, the consistent increase in methylation levels and the concurrent loss of expression depicted along the progression from normal prostate tissue and HGPIN to invasive cancer suggests that Cyclin D2 silencing might play an important functional role in prostate carcinogenesis.
References
Hanahan D, Weinberg R (2000) The hallmarks of cancer. Cell 100:57–70
Tsihlias J, Kapusta L, Slingerland J (1999) The prognostic significance of altered cyclin-dependent kinase inhibitors in human cancer. Annu Rev Med 50:401–423
Zhang P (1999) The cell cycle and development: redundant roles of cell cycle regulators. Curr Opin Cell Biol 11:655–662
Takano Y, Kato Y, Masuda M, Ohshima Y, Okayasu I (1999) Cyclin D2, but not cyclin D1, overexpression closely correlates with gastric cancer progression and prognosis. J Pathol 189:194–200
Evron E, Umbricht CB, Korz D, Raman V, Loeb DM, Niranjan B, Buluwela L, Weitzman SA, Marks J, Sukumar S (2001) Loss of cyclin D2 expression in the majority of breast cancers is associated with promoter hypermethylation. Cancer Res 61:2782–2787
Sinclair AJ, Palmero I, Peters G, Farrell PJ (1994) EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein–Barr virus. EMBO J 13:3321–3328
Sinclair AJ, Palmero I, Holder A, Peters G, Farrell PJ (1995) Expression of cyclin D2 in Epstein–Barr virus-positive Burkitt’s lymphoma cell lines is related to methylation status of the gene. J Virol 69:1292–1295
Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD, Eppig JJ, Bronson RT, Elledge SJ, Weinberg RA (1996) Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature 384:470–574
Dhillon VS, Shahid M, Husain SA (2004) CpG methylation of the FHIT, FANCF, cyclin-D2, BRCA2 and RUNX3 genes in granulosa cell tumors (GCTs) of ovarian origin. Mol Cancer 3:33
Mermelshtein A, Gerson A, Walfisch S, Delgado B, Shechter-Maor G, Delgado J, Fich A, Gheber L (2005) Expression of D-type cyclins in colon cancer and in cell lines from colon carcinomas. Br J Cancer 93:338–345
Takano Y, Kato Y, van Diest PJ, Masuda M, Mitomi H, Okayasu I (2000) Cyclin D2 overexpression and lack of p27 correlate positively and cyclin E inversely with a poor prognosis in gastric cancer cases. Am J Pathol 156:585–594
Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, deFazio A, Watts CK, Musgrove EA, Sutherland RL (1993) Expression and amplification of cyclin genes in human breast cancer. Oncogene 8:2127–2133
Fackler MJ, McVeigh M, Evron E, Garrett E, Mehrotra J, Polyak K, Sukumar S, Argani P (2003) DNA methylation of RASSF1A, HIN-1, RAR-beta, Cyclin D2 and twist in in situ and invasive lobular breast carcinoma. Int J Cancer 107:970–975
Virmani A, Rathi A, Heda S, Sugio K, Lewis C, Tonk V, Takahashi T, Roth JA, Minna JD, Euhus DM, Gazdar AF (2003) Aberrant methylation of the cyclin D2 promoter in primary small cell, nonsmall cell lung and breast cancers. Int J Cancer 107:341–345
Matsubayashi H, Sato N, Fukushima N, Yeo CJ, Walter KM, Brune K, Sahin F, Hruban RH, Goggins M (2003) Methylation of cyclin D2 is observed frequently in pancreatic cancer but is also an age-related phenomenon in gastrointestinal tissues. Clin Cancer Res 9:1446–1452
Yu J, Leung WK, Ebert MP, Leong RW, Tse PC, Chan MW, Bai AH, To KF, Malfertheiner P, Sung JJ (2003) Absence of cyclin D2 expression is associated with promoter hypermethylation in gastric cancer. Br J Cancer 88:1560–1565
Herman JG, Baylin SB (2003) Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 349:2042–2054
Jerónimo C, Henrique R, Hoque MO, Ribeiro FR, Oliveira J, Fonseca D, Teixeira MR, Lopes C, Sidransky D (2004) Quantitative RARβ2 hypermethylation: a promising prostate cancer marker. Clin Cancer Res 10:4010–4014
Jerónimo C, Henrique R, Hoque MO, Mambo E, Ribeiro FR, Varzim G, Oliveira J, Teixeira MR, Lopes C, Sidransky D (2004) A quantitative promoter methylation profile of prostate cancer. Clin Cancer Res 10:8472–8478
Li LC, Carroll PR, Dahiya R (2005) Epigenetic changes in prostate cancer: implication for diagnosis and treatment. J Natl Cancer Inst 97:103–115
Henrique R, Jerónimo C (2004) Molecular detection of prostate cancer: a role for GSTP1 hypermethylation. Eur Urol 46:660–669
Padar A, Sathyanarayana UG, Suzuki M, Maruyama R, Hsieh JT, Frenkel EP, Minna JD, Gazdar AF (2003) Inactivation of cyclin D2 gene in prostate cancers by aberrant promoter methylation. Clin Cancer Res 9:4730–4734
Rosenbaum E, Hoque MO, Cohen Y, Zahurak M, Eisenberger MA, Epstein JI, Partin AW, Sidransky D (2005) Promoter hypermethylation as an independent prognostic factor for relapse in patients with prostate cancer following radical prostatectomy. Clin Cancer Res 11:8321–8325
Hermanek P, Hutter RVP, Sobin LH, Wagner G, Wittekind C (1997) Prostate. In: Hermanek P, Hutter RVP, Sobin LH, Wagner G, Wittekind C (eds) Illustrated guide to the TNM/pTNM classification of malignant tumors. Springer, Berlin Heidelberg New York, pp 278–280
Ahrendt SA, Chow JT, Xu L-H, Yang SC, Eisenberger CF, Esteller M, Herman JG, Wi L, Decker PA, Jen J, Sidransky D (1999) Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J Natl Cancer Inst 91:332–339
Gleason DF, Mellinger GT, Veterans Administration Cooperative Urological Research group (1974) Prediction of prognosis for prostatic adenocarcinoma by combined histologic grading and clinical staging. J Urol 111:58–64
Henrique R, Jerónimo C, Hoque MO, Carvalho AL, Costa VL, Oliveira J, Teixeira MR, Lopes C, Sidransky D (2005) MT1G hypermethylation is associated with higher tumor stage in prostate cancer. Cancer Epidemiol Biomarkers Prev 14:1274–1278
Olek A, Oswald J, Walter JA (1996) A modified and improved method of bisulfite based cytosine methylation analysis. Nucleic Acids Res 24:5064–5066
Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW (2000) MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res 28:E32
Maruyama, R, Toyooka S, Toyooka KO, Virmani AK, Zochbauer-Muller S, Farinas AJ, Minna JD, McConnell J, Frenkel EP, Gazdar AF (2002) Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res 8:514–519
Jerónimo C, Usadel H, Henrique R, Oliveira J, Lopes C, Nelson WG, Sidransky D (2001) Quantitation of GSTP1 hypermethylation in non-neoplastic prostatic tissue and organ confined prostate adenocarcinoma. J Natl Cancer Inst 93:1747–1752
Meyyappan M, Wong H, Hull C, Riabowol KT (1998) Increased expression of cyclin D2 during multiple states of growth arrest in primary and established cells. Mol Cell Biol 18:3163–3172
Acknowledgements
The expert statistical advice of Dr. Maria José Bento is gratefully acknowledged. R. H. and A. L. C. are the recipients of grants from Liga Portuguesa Contra o Cancro—Núcleo Regional do Norte, Portugal, Portugal, and CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) (BEX 21303-7), Brazil, respectively. V. L. C., M. R. T., and C. J. are supported by grants from Fundação para a Ciência e a Tecnologia [SFRH/BD/23374/2005 and Projecto de Investigação Plurianual do Centro de Investigação do IPO-Porto (03-05)]. This study was also supported by the “Comissão de Fomento da Investigação em Cuidados de Saúde—Ministério da Saúde,” Portugal. This study was supported by the NIH Grant U01CA84986-04 entitled, “Integrated Development of Novel Molecular Markers—The Early Detection Research Network: Biomarkers Developmental Laboratories” (EDRN Grant).
Disclaimer
Under a licensing agreement between Oncomethylome Sciences SA and the Johns Hopkins University, Dr. Sidransky is entitled to a share of royalty received by the University on sales of products described in this article. Dr. Sidransky owns Oncomethylome Sciences, SA stock, which is subject to certain restrictions under University policy. Dr. Sidransky is a paid consultant to Oncomethylome Sciences SA and is a paid member of the company’s Scientific Advisory Board. The term of this arrangement is being managed by the Johns Hopkins University in accordance with its conflict of interest policies. The remaining authors do not declare any conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Henrique, R., Costa, V., Cerveira, N. et al. Hypermethylation of Cyclin D2 is associated with loss of mRNA expression and tumor development in prostate cancer. J Mol Med 84, 911–918 (2006). https://doi.org/10.1007/s00109-006-0099-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-006-0099-4