
Abstract
Nonsyndromic hereditary hearing impairment (NSHHI) is a highly heterogeneous disorder with more than 90 loci mapped, of which nearly one-half of the responsible genes are identified. In dominant NSSHI hearing loss is typically biased towards the high frequencies while low-frequency hearing loss is unusual. Only two NSHHI loci, DFNA1 and DFNA6/14/38, are associated with predominantly low- frequency loss. We mapped the loci harboring the gene responsible for autosomal dominant low-frequency hearing loss in a multigenerational family. The pedigree of a Swiss family with low-frequency hearing loss was established. Using genomic DNA, DFNA1 and DFNA6/14/38 were excluded by linkage analysis or by direct sequencing of the responsible gene. Genome-wide linkage analysis was performed using commercially available microsatellite markers. Two-point linkage analysis demonstrated linkage to chromosome 5q31, the locus for DFNA15, with a lod score of 6.32 at recombination fraction θ=0 for marker D5S436. Critical recombinations were seen at markers D5S1972 and D5S410. Sequencing of the corresponding gene POU4F3 yielded no pathogenic mutation segregating with the affected members. In addition to Wolfram syndrome gene 1 (DFNA6/14/38) and diaphanous (DFNA1) there is evidence for a third gene involved in low-frequency hearing loss located at DFNA15. Because of the differences in auditory phenotype and the absence of pathogenic mutation in the coding region of POU4F3 it is likely that there is a second gene in 5q31, designated DFNA54, associated with NSHHI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nonsyndromic hereditary hearing impairment (NSHHI) is a highly heterogeneous genetic disorder with more than 90 loci mapped and nearly 40 genes identified (G. VanCamp, R.J.H. Smith, “Hereditary hearing loss,” http://www.uia.ac.be/dnalab/hhh). These genes responsible for NSHHI are diverse including members of the gap junction family, cytoskeletal proteins, transcription proteins, ion channels, and genes of unknown function. Hearing loss associated with recessive NSHHI is generally congenital and profound; on the other hand, dominant NSHHI is characterized by delayed-onset, high-frequency hearing loss that progresses to involve all frequencies. Phenotypically hearing loss involving the low frequencies first is uncommon. Not surprisingly, among the nearly 90 NSHHI loci mapped, only two are characterized by low-frequency hearing loss: DFNA1 and DFNA6/14/38 [1, 2, 3]. The latter, DFNA6, DFNA14, and DFNA38 likely represent a single locus as all have hearing loss due to a mutation in Wolfram syndrome gene 1 (WFS1).
The majority of families with low-frequency hearing loss harbor mutations in exon 8 of WFS1 [4]. The protein product of WFS1, called Wolframin, is a membrane glycoprotein which is localized primarily in the endoplasmic reticulum [5]. Its expression in the human cochlea remains unknown. Functional analysis in mutated cell lines suggests that the autosomal-dominant form of low-frequency hearing loss is due to reduced protein dose of Wolframin [6]. Mutations in diaphanous (DIAPH), corresponding gene for the other low-frequency hearing loss locus DFNA1, have been reported in only one family from Costa Rica [2]. DIAPH is present in inner and outer hair cells. Its function may lie in the acteylcholine-activated pathway that regulates outer hair cell contractility [7]. Here we report a third locus, DFNA54, harboring a gene for nonsyndromic autosomal dominant low-frequency hearing loss in a large Swiss family.
Materials and methods
Family
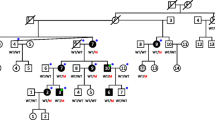
One Swiss family with autosomal-dominant NSHHI spanning four generations and comprising 83 members was identified (Fig. 1). The pedigree was established, and the auditory phenotype was investigated by trained otolaryngologists with pure tone audiometry after otoscopic examination. The onset, severity, presence or absence of progression, and type of hearing loss were noted. Syndromic and environmental causes were ruled out by a questionnaire. Individuals gave informed consent according to the guidelines of the Ethics Committee of the University Hospital of Basel, Switzerland.

Pedigree of the Swiss family. Haplotype analysis is according to the five markers represented in Table 1. Key recombinants are marked by a white square in the haplotype bar
DNA extraction and genotyping
Blood samples were obtained from every member relevant for linkage analysis. Genomic DNA was harvested from whole blood using the Qiagen DNA isolation kit (BloodMaxi Kit, QIAamp, Basel, Switzerland). The ABI Prism Linkage Mapping Set MD-10 (Applied Biosystems, Foster City, Calif., USA) of fluorescent-labeled PCR markers was used to amplify highly polymorphic dinucleotide repeats in the family. PCR amplification reactions were performed following the manufacturer’s recommendation, pooled and separated on an ABI 3700 DNA Analyzer (Applied Biosystems) and sized with ABI GeneMapper software (Applied Biosystems).
Linkage analysis
Positive linkage to DFNA1 was excluded by genotyping four affected and four unaffected family members with the corresponding microsatellite markers already published [2] (data not shown). Two-point linkage analysis and multipoint analysis were performed using the Linkage programs by Ott et al. (“Genetic Linkage Programs,” ftp://linkage.rockefeller.edu/software/). The defect was assumed to be inherited in a dominant manner and fully penetrant. The disease allele frequency was estimated at 10−4; changing the disease allele frequency to 10−3 only slightly modified the lod score value. The allele frequencies of the polymorphic markers were assumed to be equal. The meiotic recombination frequencies for males and females were taken from database provided by the Genetic Epidemiology Research Group.
Sequencing of the genes WFS1 and POU4F3
The DNA from two affected and two unaffected family members were analyzed. Mutations in the coding exons of WFS1 were excluded by sequencing following established protocols (N. Gürtler, 2002, personal communication). Coding exons 1 and 2 of POU4F3 were amplified with primer pairs as follows: for exon 1 5′-AAGCCTGATTCCATGTCACC-3′ and 5′-CATGAAGCTAGTGCCTGTCAA-3′, and for exon 2 (in two fragments) 5′-CATCAAGCTGGGGGTGAC-3′ and 5′-GTGGACAGCCGAATACTTCA-3′and 5′-CATCAAGCTGGGGGTGAC-3′ and 5′-CTTGGAGTTCCCGATAACCA-3′. Exon 1 was amplified using the touchdown program: 94°C for 3 min; 3 cycles of 94°C for 10 s, 65°C for 20 s, 72°C for 40 s; 3 cycles of 94°C for 10 s, 62°C for 20 s, 72°C for 40 s; 3 cycles of 94°C for 10 s, 59°C for 20 s, 72°C for 40 s; 3 cycles of 94°C for 10 s, 56°C for 20 s, 72°C for 40 s; 30 cycles of 94°C for 10 s, 55°C for 20 s, 72°C for 40 s; final extension of 72° for 5 min. Conditions used for PCR for the fragments of exon 2 were 94°C for 3 min, then 35 cycles of 94°C for 10 s, 59° for 15 s, 72° for 20 s and final extension time of 72° for 3 min. After PCR amplification excess primers and deoxyribonucleoside triphosphate were removed by exonuclease I (0.1 µl/10ul sample) and shrimp alkaline phosphatase (0.5 µl/10 µl sample) treatment. Sequencing was performed on a 3700 ABI DNA-analyzer (Applied Biosystems) using the manufacturer’s instructions. Double-pass sequencing was carried out using both the forward and reverse primer. Before loading the amplicons they were purified by running them through a filtration block (Edgebiosystems, Gaithersburg, Md., USA). Each fragment was sequenced twice following independent PCR amplification to assure accurate sequence results. The sequences were analyzed using the Sequencher 4.0 software (Gene Codes, Ann Arbor, Mich., USA).
Assessment of intragenic deletion in POU4F3
To detect large deletions in the POU4F3 region affected and unaffected members were typed for additional markers (in addition to D5S436 and D5S2090 used in linkage analysis) and single-nucleotide polymorphisms (SNPs) in the POU4F3 region: D5S2033, D5S2099, and five SNPs, including a POU4F3 intragenic SNP. Loss of heterozygosity (LOH) with informative markers would be evidence for deletion. To assess the possibility of an intragenic deletion PCR was performed with two separate POU4F3 primers: one within exon 1 and the other for the intragenic SNP. The product was run on agarose gel, stained with ethidium bromide, and the relative amount of product was assessed using public domain NIH ImageJ program on a Apple computer using OSX system software; the program was developed at the United States National Institutes of Health and is available on the internet at http://rsb.info.nih.gov/ij/.
Results
Clinical phenotype
The low-frequency hearing loss was symmetric and of moderate severity (Fig. 2). Hearing loss was sensorineural, as no air-bone gap was observed. In the older affected members hearing loss was noted to have progressed to moderate to high severity. The onset of hearing loss in this family spanned over 20 years, from age 5 to age 40 years. As the hearing loss started gradually, family members found it difficult to define the year it had started. Most audiograms were recorded in adulthood and showed a moderate hearing loss; in the few cases in which an audiogram was performed in childhood hearing loss was mild and progressed to moderate loss within 10 years. Vestibular symptoms, described as vertigo, were reported by only two patients, aged 27 and 31 years, and occurred only once in their life at about age 17 and 21 years; further vestibular assessment was not available.

Pure-tone audiogram of the Swiss family for air conduction showing a symmetrical moderate sensorineural hearing loss in the low frequencies. X, O Left and right ear thresholds, respectively
Linkage analysis
Before genotyping was undertaken, the two previously published loci for low-frequency hearing loss were excluded. For DFNA1, four polymorphic microsatellite markers were used to exclude linkage. For DFNA6 WFS1 was sequenced and no pathogenic mutation was identified in the affected family members. Subsequently a whole-genome scan demonstrated evidence for linkage for marker D5S436 at 5q31, generating a positive lod score for marker D5S436 greater than 3. Maximum lod scores of 6.32 and 6.02 were obtained at recombination fraction θ=0 for markers D5S436 and D5S2090 (Table 1). Key recombinations in the family were identified at markers D5S1972 and D5S410, defining a linked interval of 9.5 cM for the disease-specific haplotype (Fig. 1). This area completely overlaps with DFNA15 with its corresponding gene POU4F3 (Fig. 3).

Linked region of DFNA15 in cM for Israeli family, Chinese family (DFNA42) and Swiss family (DFNA54) with corresponding recombinant markers and type of hearing loss
Sequencing analysis of POU4F3
Sequencing of the two coding exons in two affected and two unaffected family members yielded no pathogenic mutation in the forward or in the reverse sequence. This was confirmed by a second independent run.
Assessment of intragenic deletion in POU4F3
Of the four markers D5S436, D5S2090, and D5S2033 were informative, and there was no LOH; D5S2099 was uninformative. Informative polymorphisms were found in four of the five SNPs tested; however, there was no LOH. The intragenic SNP was not informative. These results are consistent with the absence of a large deletion in the POU4F3 locus. Further, using POU4F3 specific primers there was no statistically significant difference between the quantity of PCR product between affected and unaffected individuals by χ2 analysis, consistent with the absence of a microdeletion in POU4F3.
Discussion
The human cochlea is a spiral shaped sensory organ characterized by tonotopic representation of hearing: low frequencies are located in the apex and high frequency hearing in its base. In contrast to expectation, hearing loss due to a variety of different disorders does not affect all frequencies equally. For reasons that remain elusive, noise-induced hearing loss, genetic deafness, and hearing loss due to tumors, among others, often affect preferentially the high frequencies; with progression the loss can affect the low frequencies as well. Of the nearly 90 NSHHI loci only two are characterized by low-frequency hearing loss. Here we report a third locus, DFNA54, harboring a gene which, when affected, results in low-frequency hearing impairment.
The linked locus, responsible for the low-frequency hearing loss in the current Swiss family, is within the much larger previously mapped region defining DFNA15. In 1998 linkage analysis of a small Israeli Jewish family identified a new locus, DFNA15, for hereditary hearing impairment on chromosome 5q31 [8]. Despite the large linked region of 30 cM sequence analysis of an excellent candidate gene, POU4F3, identified an 8-bp deletion in exon 2, resulting in a truncated protein. Expressed in the mammalian cochlea, targeted deletion of both alleles of pou4f3 caused complete deafness in mice. The linked region for the Swiss family is less than one-half the size of the original locus and contains the corresponding gene for DFNA15, POU4F3. Mutation screening of the two coding exons of POU4F3 did not identify any pathogenic mutation, and there is no evidence for intragenic microdeletion; the possibility of a mutation in the promoter region cannot be excluded. Further, there are important phenotypic differences between the Israeli family and the current Swiss family suggesting that they may in fact have different genotypic etiology. The original family had progressive sensorineural hearing loss affecting all frequencies whereas the current family has progressive low-frequency sensorineural hearing loss. Similarly, DFNA42, linked to 5q31.1–32 in a Chinese family is phenotypically different from current family (Fig. 3) [9].
There are several reasonable candidate genes within the mapped region as 11 are expressed in the inner ear. Unfortunately, their functional role in this organ is either unknown or cannot be compared to published deafness genes with one exception. TCERG1 is a transcription elongation regulator similar to OCP2 (organ of Corti protein). OCP2, a highly specific calcium-binding protein, is expressed in human adult cochlea that peaks at onset of cochlear function at 20 weeks [10]. It colocalizes with connexin 26 along the epithelial gap-junction systems [11]. Function of OCP2 remains unclear but is thought to play a role in maturation and electrochemical maintenance of the cochlear gap-junction system [10]. Transporter proteins constitute a second group of good candidate genes. Five are located within the recombinant region in this family: SLC26A2, SLC6A7, SLC36A1, SLC36A2, and SLC36A3. The mutant SLC26A4 gene causes Pendred syndrome, DFNB4 and large vestibular aqueduct syndrome [12, 13, 14]. The phenotype is different from the Swiss family insofar as in all these forms hearing loss is either congenital, profound or fluctuating, progressive in the high frequencies. Targeted dysruption of the SLC26A4 gene results in completely deaf mice [15]. Further mutations of transporter proteins (SLC12A2, SLC30A4, SLC9A1) are seen in other mouse mutants [16, 17] (MRC Institute of Hearing Research Nottingham: http://www.ihr.mrc.ac.uk/hereditary/MutantsTable.shtml).
Using a positional candidate approach Dixon et al. [17] demonstrated that different mutations in one single gene encoding the basolateral Na+/2Cl−/K+ cotransporter SLC12A2 causes the deafness in sy and nsy mice. In the cochlea K+ is taken up from the intrastrial space across the basolateral membrane of strial marginal cells via the Na+/2Cl−/K+ cotransporter SLC12A2 [18]; nsy is associated with abnormal production of endolymph. A possible role in endolymphatic fluid resorption is also suggested for SLC26A4 based on its expression and putative function as anion transporter [19]. These transporter proteins have variable functions, and their expression in the inner ear is unknown. SLC26A2 acts as a sulfate transporter and is implicated in inherited achondroplasia [20]. SLC6A7 is involved in proline uptake in the central nervous system [21]. SLC36A1, with highest expression levels in the brain, is a lysosomal transporter for neutral amino acids [22]. These transcription factors and transporter proteins are excellent candidate genes for DFNA54.
In this study we present evidence for a third locus for hereditary low-frequency hearing impairment located at 5q31. DFNA15 has been previously mapped to this chromosomal region. Significant differences in the auditory phenotype and absence of mutation in POU4F3 are strongly suggestive of a second distinct locus within 5q31, DFNA54.
Abbreviations
- LOH :
-
Loss of heterozygosity
- NSHHI :
-
Nonsyndromic hereditary hearing impairment
- SNP :
-
Single-nucleotide polymorphism
References
Lesperance MM, Hall JW 3rd, Bess FH, Fukushima K, Jain PK, Ploplis B, San Agustin TB, Skarka H, Smith RJ, Wills M et al (1995) A gene for autosomal dominant nonsyndromic hereditary hearing impairment maps to 4p16.3. Hum Mol Genet 4:1967–1972
Lynch ED, Lee MK, Morrow JE, Welcsh PL, Leon PE, King MC (1997) Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science 278:1315–1318
Young TL, Ives E, Lynch E, Person R, Snook S, MacLaren L, Cator T, Griffin A, Fernandez B, Lee MK et al (2001) Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum Mol Genet 10:2509–2514
Bespalova IN, Van Camp G, Bom SJ, Brown DJ, Cryns K, DeWan AT, Erson AE, Flothmann K, Kunst HP, Kurnool P et al (2001) Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet 10:2501–2508
Takeda K, Inoue H, Tanizawa Y, Matsuzaki Y, Oba J, Watanabe Y, Shinoda K, Oka, Y (2001) WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 10:477–484
Hofmann S, Philbrook C, Gerbitz KD, Bauer MF (2003) Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet 12:2003–2012
Zhang MKG, Urutia R, Kalinec F (2001) Dial proteins participate in the regulation of outer hair cell motility by acteylcholine. Presented at the Assoc Res Otolaryngol, St. Petersburg Beach, Fla.
Vahava O, Morell R, Lynch ED, Weiss S, Kagan ME, Ahituv N, Morrow JE, Lee MK, Skvorak AB, Morton CC et al (1998) Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 279:1950–1954
Xia J, Deng H, Feng Y, Zhang H, Pan Q, Dai H, Long Z, Tang B, Chen Y, Zhang R et al (2002) A novel locus for autosomal dominant nonsyndromic hearing loss identified at 5q31.1–32 in a Chinese pedigree. J Hum Genet 47:635–640
Kammen-Jolly K, Scholtz AW, Kreczy A, Gluckert R, Thalmann I, Thalmann R, Schrott-Fischer A (2002) OCP2 immunoreactivity in the human fetal cochlea at weeks 11:17:20, and 28, and the human adult cochlea. Hear Res 167:102–109
Henzl MT, O’Neal J, Killick R, Thalmann I, Thalmann R (2001) OCP1, an F-box protein, co-localizes with OCP2/SKP1 in the cochlear epithelial gap junction region. Hear Res 157:100–111
Usami S, Abe S, Weston MD, Shinkawa H, Van Camp G, Kimberling WJ (1999) Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet 104:188–192
Li XC, Everett LA, Lalwani AK, Desmukh D, Friedman TB, Green ED., Wilcox ER (1998) A mutation in PDS causes non-syndromic recessive deafness. Nat Genet 18:215–217
Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB, Galer CE, Karniski LP, Sheffield VC, Smith RJ (2001) Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat 17:403–411
Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED (2001) Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 10:153–161
Huang L, Gitschier J (1997) A novel gene involved in zinc transport is deficient in the lethal milk mouse. Nat Genet 17:292–297
Dixon MJ, Gazzard J, Chaudhry SS, Sampson N, Schulte BA, Steel KP (1999) Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum Mol Genet 8:1579–1584
Wangemann P (2002) K+ cycling and the endocochlear potential. Hear Res 165:1–9
Everett LA, Morsli H, Wu DK., Green ED (1999) Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci USA 96:9727–9732
Superti-Furga A, Rossi A, Steinmann B, Gitzelmann R (1996) A chondrodysplasia family produced by mutations in the diastrophic dysplasia sulfate transporter gene: genotype/phenotype correlations. Am J Med Genet 63:144–147
Shafqat S, Velaz-Faircloth M, Henzi VA, Whitney KD, Yang-Feng TL, Seldin MF, Fremeau RT Jr (1995) Human brain-specific L-proline transporter: molecular cloning, functional expression, and chromosomal localization of the gene in human and mouse genomes. Mol Pharmacol 48:219–229
Sagne C, Agulhon C, Ravassard P, Darmon M, Hamon M, El Mestikawy S, Gasnier B, Giros B (2001) Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc Natl Acad Sci USA 98:7206–7211
Acknowledgements
We thank participating family members for their cooperation and the Molecular Genetics Facility at the University Hospital of Basel for technical support. This study was supported in part by grants from the Swiss National Science Foundation, the Novartis Foundation, and “Freiwillige Akademische Gesellschaft,” the latter both from Basel, Switzerland.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gürtler, N., Kim, Y., Mhatre, A. et al. DFNA54, a third locus for low-frequency hearing loss. J Mol Med 82, 775–780 (2004). https://doi.org/10.1007/s00109-004-0597-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-004-0597-1