
Abstract
Pancreatic islet β-cell death occurs in type 1 and 2 diabetes mellitus, leading to absolute or relative insulin deficiency. β-cell death in type 1 diabetes is due predominantly to autoimmunity. In type 2 diabetes β-cell death occurs as the combined consequence of increased circulating glucose and saturated fatty acids together with adipocyte secreted factors and chronic activation of the innate immune system. In both diabetes types intra-islet inflammatory mediators seem to trigger a final common pathway leading to β-cell apoptosis. Therefore anti-inflammatory therapeutic approaches designed to block β-cell apoptosis could be a significant new development in type 1 and 2 diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes is caused by absolute insulin deficiency due to destruction of the pancreatic β-cells. The majority of type 1 diabetes cases are considered to be due to immune mediated β-cell destruction, leaving a small proportion of idiopathic cases in which immune markers cannot be detected, and which are caused by other pathogenetic mechanisms such as rare genetic syndromes, β-cell lytic virus infections, or environmental toxins [1]. As described below, immune-mediated type 1 diabetes can be considered to be an inflammatory disease of the pancreatic islet [2]. The histopathology in recent-onset type 1 diabetic patients resembles a delayed type 4 hypersensitivity reaction, i.e., mononuclear cell infiltration into the islets and selective β-cell destruction (insulitis) [3]. Interaction between antigen presenting cells and T-cells leads to prolonged presence in high local concentrations of inflammatory mediators, for example cytokines, chemokines, reactive oxygen species (ROS), and other inflammatory products [4]. Thus all islet cells are exposed to the same inflammatory environment, and although β-cell destruction may not be exclusively specific (there is also evidence of damage to peri-islet Schwann cells [5] and some α-cell damage), inherent features of the β-cell sensitize the β-cell to the destructive abilities of the inflammatory mediators (see below).
Type 2 diabetes mellitus manifests itself in individuals who loose the ability to produce sufficient quantities of insulin to maintain normoglycemia in the face of insulin resistance [6] (Fig. 1). The contribution of a relative insulin deficiency to the establishment of overt diabetes is now widely accepted [7, 8, 9, 10]. The ability to secrete adequate amounts of insulin depends on β-cell function and mass. The endocrine pancreas has a remarkable capacity to adapt to conditions of increased insulin demand, such as in obesity, pregnancy, cortisol and growth hormone excess, by increasing its functional β-cell mass; only 10–20% of individuals fail to adapt and become diabetic with time [10]. Long-term adaptation of the β-cell mass to conditions of increased demand involves a balance of β-cell replication and apoptosis as well as development of new islets from exocrine pancreatic ducts [10, 11]. There is controversy whether β-cell mass, and not only β-cell function, is decreased in type 2 diabetes [6, 12, 13, 14, 15]. These discrepancies are in part due to difficulty in procuring well-preserved pancreas tissue from humans. However, two recent and important studies may end the controversy [16, 17]. In particular, Butler et al. [16] have studied a large sample of well-preserved pancreases obtained from humans at autopsy. In nondiabetic controls obesity resulted in an increase in relative β-cell volume. In contrast, humans with impaired fasting glucose and type 2 diabetes had a deficit in relative β-cell volume compared to nondiabetic weight matched cases. Moreover, the frequency of β-cell apoptosis was increased in type 2 diabetes. Taken together these data imply a central role for a deficit of β-cell mass in the pathophysiology of type 2 diabetes and suggest that the underlying mechanism is increased β-cell apoptosis.

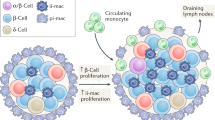
Proposed scheme for the initiation of type 2 diabetes. The ability to secrete adequate amounts of insulin depends on the pancreatic β-cell mass (green spheres). Insulin resistance (stop sign) increases insulin demand, leading to β-cell proliferation and increased β-cell mass. When β-cell expansion is offset by concomitant apoptosis, a relative insulin deficiency occurs, leading to diabetes
Increasing evidence links type 1 and 2 diabetes [18, 19, 20, 21, 22, 23]. Pancreatic β-cell demise by apoptosis is a common cellular denominator in both diseases in humans. Although types 1 and 2 diabetes may have etiological and pathogenetic differences, β-cell destruction eventually occurs in both cases, leading to clinical manifestation of absolute or relative insulin deficiency. Insulin dependence occurs rapidly in type 1 and following a prolonged time in type 2 diabetes. This reflects a difference in the rapidity of β-cell destruction but not in the hallmark of the diseases: β-cell apoptosis [21]. This may be compared to cardiac failure: several underlying and precipitating heart diseases cause the same clinical manifestation of heart failure. This contribution reviews the mechanisms leading to decreased β-cell functional mass in type 1 and 2 diabetes with a focus on inflammatory mediators as a possible final common pathway.
Inflammation is defined as the local physiological response to tissue injury. It is characterized by cell invasion and local metabolic and circulatory alterations, sometimes accompanied by functional or structural damage of the invaded tissue. It is not in itself a disease but rather a manifestation of disease. Inflammation has beneficial effects such as preventing spread of infections. Equally, it may produce disease by tissue destruction due to inflammatory mediators, ROS and complement components. In this review we describe the involvement of inflammatory mediators and the signaling pathways activated by these mediators in pancreatic β-cells to illustrate the concept that immunological and metabolic stressors may converge on common molecular mechanisms leading to β-cell apoptosis in the two main forms of diabetes.
Inflammatory mediators in type 1 diabetes
Type 1 diabetes is, as noted above, caused by immune mediated β-cell destruction triggered by environmental factors in genetically predisposed individuals [24].
Etiology
The following observations support the importance of the environmental factors in the etiology of type 1 diabetes [24, 25]:
-
The concordance rate of type 1 diabetes in monozygotic twin pairs is 30–50%, underlining that at least 50% of the etiology are not explained by genetic factors.
-
Low-risk populations acquire a higher diabetes risk when migrating to higher incidence areas.
-
Rapid increases in diabetes incidence within relatively few years in nonmigratory populations.
-
Seasonal variations in diabetes incidence.
-
Incidence differences between genetically comparable populations.
Apart from the congenital Rubella syndrome, where individuals exposed to Rubella infection in vitro develop type 1 diabetes later in life [26], it is not at present possible to pinpoint one particular type of virus as a causal factor. Much interest has focused on Coxsackie-B virus infection, particularly in early childhood and in utero. At present, however, there are no intervention studies to support antiviral strategies in the prevention of type 1 diabetes apart from the Rubella program implemented in most countries. β-cell toxic substances in certain foods such as nitrosamines have been accused of predisposing to type 1 diabetes, but this has not been universally confirmed. Other nutritional factors such as early exposure to cow's milk protein, gluten, or vitamin D are under investigation as causal factors. Thus, although environmental factors undoubtedly contribute to the etiology of type 1 diabetes, there is no ubiquitous factor to the targeted as intervention.
Type 1 diabetes is a polygenic disease [25]. Approximately 50% of the genetic susceptibility can be explained by alleles in the HLA class II region, in particular certain DQ alleles. More than 95% of type 1 diabetic patients carry these predisposing alleles, but the occurrence of these alleles in the background population is high, approximately 50%. It is believed that the diabetes predisposing DQ antigens have a shape of the antigen presenting groove of the molecule that leads to more efficient presentation of β-cell associated autoantigens. Genome-wide scans have identified approximately 15 other loci linked to type 1 diabetes with only five other loci than the HLA region found to be associated in three or more of these genome-wide scans. It has been possible as yet to identify the nature of only one of these four additional susceptibility genes, IDDM2, a 5′ variable number of tandem repeats of the insulin gene. This variation may code for altered insulin expression in the thymus leading to reduced central tolerance against insulin, believed to be one of the β-cell autoantigens. The nature of the remaining genetic loci associated with type 1 diabetes has not been identified. Large international consortia are currently searching for additional non-HLA diabetes disposing regions and attempting to identify the candidate genes in these regions and their function. In summary, type 1 diabetes is a polygenic disease, the genetic predisposition of which is carried mostly by DQ antigens of the HLA class II region. Diabetes predisposing alleles are common variations in normally functioning genes making antenatal diagnosis and gene therapy unlikely as curative strategies.
Pathogenesis of type 1 diabetes
Apart from being associated with immune response genes the disease is also associated with the occurrence of other organ specific autoimmune diseases, which occur three to six times more commonly in type 1 diabetic patients. These observations support that these diseases constitute a family of immune mediated disorders. Both cell-mediated and humoral autoimmunity have been detected in type 1 diabetic patients, and, as noted, recent-onset type 1 diabetic patients exhibit a mononuclear cell infiltrate into the pancreatic islets and β-cell destruction entitled insulitis. More direct evidence for the involvement of the immune system comes from large placebo-controlled intervention studies using the T-cell immunosuppressant cyclosporine A [27, 28]. These studies showed that immunosuppression induces and maintains remission and preserves β-cell function throughout the treatment period with the immunosuppressant. The studies confirm (a) that the immune system is directly involved in the β-cell destruction and not the consequence of secondary immune activation to β-cell destruction caused by other primary factors, and (b) that β-cell mass can be preserved by intervening in immune functions.
Unfortunately, due to side effects to cyclosporine and other currently available potent immunosuppressants the use of immunosuppressive therapy is not clinically feasible. Immunoregulatory defects have been identified in animal models and in patients and these defects may contribute to the loss of self-tolerance and lead to autoimmune activation. Recent studies using short-term therapy with anti-T-cell intervention (nondepleting anti-CD3 antibodies) have shown positive results [29] that are now under evaluation in randomized clinically controlled trials. Unfortunately, primary prevention studies using either nicotinamide as a β-cell protectant or low-dose subcutaneous intermediate-acting insulin [30] to reduce β-cell stress have failed to show any benefits in individuals at risk of developing type 1 diabetes. Therefore much more needs to be known about the basic mechanisms leading to β-cell destruction.
More detailed studies of the pathogenetic mechanisms leading to β-cell destruction in humans have been hindered by the inaccessibility of the human islet tissue from type 1 diabetic patients. The insulitis lesion can be detected in most recent onset type 1 diabetic patients in childhood and adolescents, but it is much more difficult to detect in older individuals. The insulitis lesion is not synchronized, and the difficulty in demonstrating insulitis in older type 1 diabetic patients may be due to a slower progressing, more desynchronized β-cell destruction. Interestingly many patients, particularly those with older age at onset, have some residual β-cell function left even many years after the diagnosis. The mechanisms sparing those β-cells are completely unknown, but may be due to (a) selection of resistant β-cell populations, (b) a lack of stimulation of the immune response at a certain threshold of β-cell antigenic load, (c) immunoregulatory events relating to diabetes duration or therapy. In support of the latter is the fact that intensive insulin therapy seems to lead to preserved β-cell function [31]. Since residual β-cell function is being associated with improved metabolic control and lower incidence of acute and late diabetic complications [32], even intervention that would rescue the remaining β-cell mass at diagnosis would be valuable.
For the reasons above, intensive research is being invested into the molecular mechanisms of β-cell destruction with the aim of developing novel pharmacological targets.
Effector mechanisms
In humans the insulitis infiltrate carries most types of inflammatory cells, i.e., antigen-presenting cells, especially macrophages, T-helper cells, cytotoxic T-cells, B-lymphocytes, and natural killer cells [24]. In animal models it has been possible to study the sequence of events, and the first cells to infiltrate the islet are antigen-presenting cells, followed by T-cells [33]. Although B-lymphocytes may act as antigen-presenting cells and may be required for diabetes development in the nonobese diabetic (NOD) mouse model, diabetes has been observed even in B-lymphocyte deficient patients, confirming that B-lymphocytes are not crucial for development of type 1 diabetes in humans [34]. Antibodies are not considered to be pathogenetic effectors but rather markers of disease due to poly-clonal immune activation. There are two main schools of thought regarding effectors in type 1 diabetes: (a) β-cell destruction is mediated by cytotoxic T-cells via the T-cell effector mechanisms Fas/Fas-ligand, membrane-bound TNF, or the perforin/granzyme system. (b) β-cell destruction is caused mainly by inflammatory mediators such as cytokines, which lead to the induction of apoptosis in β-cells via mechanisms described below.
Neither of the two mechanisms alone can be responsible for complete β-cell destruction. The following observations argue that T-cells are not sufficient. (a) Destruction of syngenic islet grafts by disease occurrence in animal models is independent of cytotoxic T-cells [35, 36]. (b) In the NOD mouse cytotoxic T-cells are required only for the first 14 weeks of the pathogenetic process whereas the later islet destruction is T-cell independent [37]. (c) A CD4+ helper T-cells clone from insulitis infiltrate of NOD mice is necessary and sufficient to transfer disease whereas only very unique T-cell receptor transgenic T-cell clones can transfer disease independently of CD4+ [38]. Similarly cytokines are probably not the sole mediators of β-cell destruction. (a) Many systemic cytokine knock-outs in the NOD model do not have reduced diabetes incidence or only partially reduced diabetes incidence [4, 33]. (b) Local overexpression of a number of cytokines have been insufficient individually to cause islet cell destruction in diabetes [4, 33]. Thus most likely both inflammatory mediators and the T-cell system contribute to β-cell destruction in type 1 diabetes.
It has been shown that cytokines are a prerequisite for β-cell induction of Fas, thereby sensitizing the β-cell to T-cell mediated destruction [39]. Cytokines either individually or in combinations cause apoptosis in human and rodent pancreatic β-cells [4, 33]. Since cytokines therefore seem to be central in both pathogenetic mechanisms described above we deal in detail with cytokine signaling and molecular effector pathways in β-cells below (see "Signaling and molecular effector pathways").
Inflammatory mediators in type 2 diabetes
Chronic inflammation has received increasing attention in recent years as an important pathophysiological mechanism in various diseases. Increased release and action of proinflammatory cytokines are thought to be involved in insulin resistance and atherosclerosis [40, 41, 42, 43, 44, 45]. Similar mechanisms could also occur in islets of type 2 diabetics. Indeed, as discussed in the "Introduction," a progressive decline in β-cell function and mass characterizes not only type 1 but also type 2 diabetes. Inflammatory mediators may induce both impaired β-cell function and death. Therefore the chronic increase in inflammatory mediators observed in type 2 diabetes might affect not only insulin-sensitive tissues and blood vessel walls but could also affect pancreatic β-cells. If so, what are the causes of this increase in inflammatory mediators?
Etiology
Type 2 diabetes is influenced genetically, and it occurs in identical twins with almost total concordance [46]. In most cases there is evidence that multiple gene defects influence overall susceptibility to type 2 diabetes [47]. Single gene defects leading to type 2 diabetes have been identified only in a subgroup of at the most 5%. The best described monogenic forms are the maturity-onset diabetes of the young. Interestingly, all genes for maturity-onset diabetes of the young that have been identified to date alter glucose sensing or intracellular signaling events involved in insulin secretion [48, 49, 50], in line with the concept of a central role for the functional β-cell mass (see above). In addition to genetic factors, development of type 2 diabetes is strongly influenced by environmental factors, including decreased physical activity, nutrition and obesity. This promotes the following factors, which are possible mediators of an inflammatory process.
Adipocyte-secreted factors
Adipose tissue was long considered a passive tissue whose role was limited to the storage of fat. Several observations, however, have now uncovered the endocrine activity of adipocytes. Locally produced hormones and cytokines possess important auto-/paracrine properties. Some are also released into the circulation and have endocrine effects. In particular, leptin, TNF-α, interleukin (IL) 6, and IL-1 receptor antagonist (IL-1Ra) are produced and secreted by fat tissue [51, 52, 53, 54, 55]. Interestingly, expression levels of these factors are increased in human obesity and have been causally linked to insulin resistance. Nevertheless, these factors may affect not only insulin-sensitive tissues but also act on other cells including the pancreatic β-cells.
Increased cell nutrients
Obesity is associated with changes in the plasma levels of cell nutrients. Plasma free fatty acid (FFA) levels are permanently increased in obesity. Moreover, insulin resistance diminishes glucose uptake, resulting in transient postprandial hyperglycemic excursions. This mild hyperglycemia could act on the β-cells even before diabetes manifests itself or at the very early stages of the disease. Therefore both nutrients, FFAs and glucose, may interfere with β-cell turnover and function influencing the onset and course of diabetes.
Innate immune system
Innate immunity is considered to provide rapid host defenses until the slower adaptive immune response develops [56, 57, 58]. These responses comprise the release of acute-phase proteins such as C-reactive protein, haptoglobin, fibrinogen, plasminogen activator inhibitor and serum amyloid A. A number of studies have reported increased acute-phase proteins in type 2 diabetes [44, 45, 57, 59, 60, 61, 62, 63]. It is unclear why the innate immune system is activated in type 2 diabetics. Possibly it is induced by overnutrition, altered nutrition, and insulin deficiency and facilitated by genetic predisposition.
Autoimmunity
In 1997 the Expert Committee of the American Diabetes Association introduced a new classification of diabetes based on etiological considerations [1]. The class termed type 1 diabetes includes the vast majority of cases that are due primarily to pancreatic islet β-cell destruction, in particular the cases attributable to an autoimmune process. However, 10–15% of subjects originally diagnosed with type 2 diabetes are cases of "cryptic" type 1 diabetes or evolve with time to a type 1 state and exhibit anti-β-cell autoimmunity [18, 19, 20, 21, 22, 23], referred to as latent autoimmune diabetes in adults [64, 65]. Furthermore, and as discussed above, an ongoing process of β-cell destruction by apoptosis is not limited to type 1 diabetes but has also been described in animal models of type 2 diabetes and in human type 2 diabetics [16, 66]. In turn, apoptotic cells can provoke an immune response [67, 68]. Moreover, innate immunity may determine to which antigens the acquired immune system responds [69]. Therefore the activation of the innate immune system observed in type 2 diabetics (see above) may predispose to islet cell autoimmunity. Finally, hyperglycemia by itself induces β-cell-expression of several molecules involved in immunological processes, for example, IL-1β and Fas [22, 23]. Therefore the innate immune system may serve functions outside the classical immune framework as determinants of diseases not traditionally conceived to be autoimmune, i.e., atherosclerosis, osteoporosis, and type 2 diabetes. Thus many cells other than immune cells produce inflammatory mediators.
Mediators
Based on the above, secretory products of the adipocytes, FFA, glucose, the innate and adaptive immune system may all be mediators of the ongoing process of β-cell destruction occurring in type 2 diabetes. We now discuss the potential individual contribution of each of these factors in this process.
Adipocyte-secreted factors
Leptin is expressed primarily in the adipose tissue and therefore represents the most obvious exponent of the adipocyte. In rodent islets leptin induces β-cell proliferation and protects from FFA-induced β-cell apoptosis [70, 71, 72, 73]. On the other hand, chronic exposure of human islets to leptin leads to β-cell apoptosis (K. Maedler and M.Y. Donath, manuscript in preparation). Thus it remains to be clarified whether leptin predominantly links obesity to islet hyperplasia or to β-cell apoptosis. TNFα, in combination with other cytokines, accelerates dysfunction and destruction of the β-cells [4, 74]. However, it is unclear whether adipose tissue releases sufficient amounts of TNFα into the circulation [53]. In contrast, IL-6 released by adipocytes may be responsible for the increases in plasma IL-6 concentrations observed in obesity [75], and, at least in combination with other cytokines, IL-6 has cytotoxic effects on β-cells [76] and synergizes with IL-1 in this respect [77].
Increased cell nutrients
Increased FFA levels per se are known to be toxic for β-cells, leading to the concept of lipotoxicity [78, 79, 80, 81]. Depending on the food consumed, the ratio of saturated to unsaturated FFA varies, leading to distinct effects. Thus, saturated FFA are highly toxic whereas unsaturated FFA may prevent these deleterious effects [82, 83, 84, 85, 86]. The toxic effect of FFA is mediated via formation of ceramide, increased nitric oxide production and activation of the apoptotic mitochondrial pathway [85, 86, 87]. Thus, lipotoxicity may play an important role in the process of β-cell destruction, but it does not seem to involve an inflammatory process. Elevated glucose concentrations induce β-cell apoptosis in cultured islets from diabetes-prone Psammomys obesus, an animal model of type 2 diabetes [66], in human islets [23, 88] and at higher concentrations in rodent islets [66, 85, 89]. In human islets glucose-induced β-cell apoptosis and dysfunction are mediated by β-cell production and secretion of IL-1β [22]. Furthermore, chronic hyperglycemia increases production of ROS, which may cause oxidative damage in β-cells [90, 91, 92, 93, 94]. Both IL-1β and ROS activate the transcription factor nuclear transcription factor (NF) κB, which plays a critical role in mediating inflammatory responses.
Innate immune system
In addition to the endocrine activity of the adipocytes described above, macrophages and endothelium may contribute to increase serum levels of IL-1β, IL-6, and TNFα in type 2 diabetic patients [57]. These cytokines induce the liver to produce acute-phase proteins. Similarly, these cytokines may act on the pancreatic islets and impair β-cell secretory function.
Autoimmunity
Apoptotic cells can provoke an immune response under appropriate conditions, for example, when present in high enough numbers or when apoptosis is the consequence of exposure to cytokines such as IL-1β and TNF-α [18, 67, 68]. Moreover, a pronounced activation of the acute-phase response is associated with islet cell autoantibodies in patients with type 2 diabetes [19]. Following glucose- and FFA-induced β-cell apoptosis it is conceivable that depending on age and on genetic and/or environmental factors, some type 2 diabetics may show mobilization of T cells reactive to β-cells antigens, culminating in autoimmune destruction of β-cells similar to that observed at earlier stages in "classical" type 1 diabetics. This response may be so discrete and desynchronized in time and space that it has evaded detection in earlier autopsy studies. Clearly, additional studies need to be conducted to evaluate this notion.
Effector pathways
As described above and illustrated in Fig. 2, the number of potential β-cell-aggressors resulting from obesity is impressive. However, it is probable that most of the above-mentioned cell death mediators have also physiological effects depending on concentrations and duration of exposure. This is certainly true for glucose and FFA. In addition to their role as cell nutrients, elevated glucose concentrations and FFA have a dual effect on β-cell turnover. Depending on duration of exposure to glucose or FFA and on the genetic background of the islets, glucose and FFA may induce or impair β-cell proliferation and have pro- or antiapoptotic effects [10, 23, 66, 85, 86, 88, 89, 95]. The expression level of pivotal intracellular factors may explain these dual effects. For example, the naturally occurring caspase-8 inhibitor referred to as Fas-associated death domain-like interleukin-1β converting enzyme inhibitory protein (FLIP) switches glucose signaling in human pancreatic β-cells from apoptosis to cell replication [96]. In turn, the proportion of saturated vs. unsaturated fatty acids determines the distinct effects of FFA on the mitochondrial apoptotic pathway [85, 86]. Similar dual effects have been observed for IL-1β, NF-κB, and NO [4, 74, 97]. This illustrates the complexity of the intracellular signaling pathway. On the other hand, several signaling pathways, relevant for type 1 and 2 diabetes, converge toward common effectors. This is true for glucose-induced IL-1β, for TNFα, Fas, NF-κB, and caspase activation.

Inflammatory mediators in type 2 diabetes. Cell nutrients (glucose and FFA) have direct and indirect effects on β-cells. Elevated glucose concentrations induce β-cell production of IL-1β leading to β-cell apoptosis. Increased FFA concentrations may affect the viability of the β-cells directly or via obesity, i.e., adipocyte secreted cytokines (TNFα, IL-6, and leptin) may act directly on the β-cells or activate the innate immune system. The precise role of the innate and acquired immune system in the ongoing process of β-cell demise in type 2 diabetics remains to be investigated
Signaling and molecular effector pathways
As noted above, evidence favors that apoptosis is the dominant form of β-cell death in both animal models of diabetes and in humans. Two principal apoptotic pathways exist: the "intrinsic" pathway (initiated by the mitochondria) and the "extrinsic" pathway (initiated by cell surface receptors; Fig. 3). It is likely that both of these apoptotic pathways are involved in the demise of β-cells in type 1 and 2 diabetes, although the relative contribution of each is not clear. As outlined above, pro-inflammatory cytokines, and in particular IL-1β, are thought to be important pathogenic effectors responsible for the induction β-cell apoptosis in both types of diabetes.

Overview of cytokine signaling leading to β-cell apoptosis. Following IL-1β binding to IL-1R1 and docking of IL-1AcP, MyD88 is recruited to the receptor complex. MyD88 interacts with IRAK, allowing the binding of TRAF6 to IRAK. TRAF6 causes activation of the MAP/SAPK pathways through ECSIT, and activation of the NFκB pathway via TAK1-mediated activation of IKK. IL-1β also stimulates activation of PKCδ possibly through phospholipase C generation of diacylglycerol. IFNγ binding to IFN receptor 1 leads to recruitment of IFN receptor 2. Both IFN receptor types are associated with JAKs, which are activated by auto- and transphosphorylation upon formation of receptor-ligand complexes. JAK2-mediated phosphorylation of STAT1 leads to STAT1 homodimerization and translocation of the dimers to the nucleus. TNFα signals through trimerized p60 receptors which via the DD in the receptors interact with TRADD. FADD is then recruited via TRADD thus allowing binding of receptor-interacting protein (RIP) and TRAF2 to the receptor complex. TRAF2 activates NFκB through NFκB-inducing kinase (NIK)-IKK and activates the JNK/p38 pathways. The figure highlights the importance of the NFκB and the MAP/SAPK signaling pathways as key components in mediating cytokine-induced β-cell apoptosis
Three main cytokines most likely act in synergy during the immune infiltration of the pancreas to induce β-cell damage and apoptosis in type 1 diabetes: IL-1β, TNFα, and interferon (IFN) γ. IL-1β is secreted by activated macrophages and, paradoxically, under some circumstances by β-cells [22, 98]. TNFα is solely produced and secreted by macrophages, whereas IFNγ is secreted by T-helper cells. In vitro, IL-1β is the most β-cell cytotoxic cytokine sufficient to cause inhibition of β-cell function and often sufficient to promote an apoptotic response. However, massive induction of apoptosis in β-cells usually requires a combination of IL-1β plus IFNγ and/or TNFα. Whether IL-1β alone is sufficient to evoke apoptosis in human β-cells is controversial. Nevertheless, several studies have pointed to the fact that IL-1β alone does induce apoptotic death of human β-cells [22, 23, 99, 100, 101, 102].
IL-1 signaling
IL-1β signal transduction is initiated by ligand binding to type 1 IL-1 receptor (IL-1R1) allowing docking of the IL-1R accessory protein (IL-1AcP). Following this, IL-1R activated kinase (IRAK) is recruited to the receptor complex via the adaptor protein MyD88. IRAK then interacts with and activates TNF receptor associated factor (TRAF) 6 [103]. Signaling events downstream TRAF6 include the activation of the evolutionary conserved signaling intermediate in Toll/IL-1 pathways (ECSIT), which activates mitogen-activated protein (MAP) kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase kinase (MEKK) 1. MEKK-1 activation can lead to activation of the group of MAP/stress-activated protein kinases (MAP/SAPK) as well as NFκB [104]. MAP/SAPKs, which comprise ERK, p38, and c-jun N-terminal kinase (JNK), are activated in three-kinase modules or cascades which are often controlled by the action of scaffold proteins. Activated MAP/SAPKs are able to phosphorylate a broad spectrum of cellular proteins including transcription factors of the activator protein 1 family. Active TRAF6 also causes activation of inhibitory κB protein (IκB) kinase (IKK) through TAB-1 and transforming growth factor β-activated kinase 1 (TAK-1) [105, 106]. This leads to phosphorylation of IκB, a cytosolic inhibitor of NFκB. Phosphorylation targets IκB for ubiquitination and degradation, thus freeing NFκB to translocate to the nucleus and regulate the transcription of target genes [103]. In addition to activation of these two main and general IL-1β-activated pathways, both of which are strongly activated by IL-1β in β-cells, IL-1β also activates protein kinase C (PKC) δ in β-cells [107]. As detailed below, the MAP/SAPK, NFκB, and PKC-δ pathways have individually been linked to the apoptotic response in β-cells.
One IL-1β-induced (late) event that has gained much attention is the induction of expression of inducible nitric oxide synthase (iNOS). IL-1β stimulation of this enzyme occurs after approx. 4–6 h and results in massive production of reactive NO. Although the precise role and contribution of NO in β-cell killing remain unclear, it is generally believed that NO accounts for some of the deleterious effects of IL-1β in rodent β-cells, whereas NO is dispensable for cytokine-induced human β-cell apoptosis [4, 108]. NFκB seems to be an absolute requirement for cytokine-induced iNOS expression in both rodent and human β-cells [109, 110, 111, 112], but the promoter for iNOS contains multiple potential binding sites for other transcription factors including activator protein 1 and signal transducer and activator of transcription (STAT, see below), suggesting that a number of different pathways regulate iNOS transcription. In support of this is that iNOS cannot be induced by IL-1β alone in human islets, but requires a cocktail of cytokines [22, 76, 110, 113, 113].
IFNγ signaling
IFNγ signaling seems more linear and simple than IL-1 signaling. IFNγ binds to IFNγ receptor 1 which leads to receptor dimerization and subsequent recruitment of two membrane-associated accessory proteins, the IFNγ receptor 2. On the cytoplasmic side the IFNγ receptors are associated with the Janus tyrosine kinases (JAK) 1 and 2. When brought into proximity following receptor complex formation, JAK1/2 are activated by auto- and transphosphorylation. Following this, STAT1 molecules bind to the IFNγ receptor 1 and are subsequently phosphorylated by JAK2. Phosphorylated, activated STAT1 then homodimerizes, translocates to the nucleus and binds to DNA at γ-activated sites. Further, STAT1 binds and activates members of the interferon regulatory factor (IRF) family of transcription factors [114, 115]. IFNγ activation of STAT1 has been demonstrated in both insulin-secreting cells and primary islet cells [116, 117]. Further, it has been shown that IFNγ induces upregulation of IRF-1 expression in insulin-producing cells [118], and this in combination with the observation that IFNγ-induced mRNA expression of IL-1 converting enzyme (caspase-1) is abrogated in IRF-1−/− mouse islets [119] suggests that IRF-1 plays an important role in IFNγ signaling in β-cells. IFNγ neither stimulates nuclear translocation and DNA binding of NFκB in β-cells nor affects NFκB activation by IL-1β [118]. In some cells IFNγ activates ERK MAPK through JAK [114], but this does not seem to be the case in β-cells. In fact, IFNγ may even lead to decreased constitutive MAP/SAPK activities in rat islets [120].
TNFα signaling
TNF signals through two different receptors, p60 and p80. The p60 receptor is expressed on all cell types, whereas expression of the p80 receptor is restricted primarily to cells of the immune system and endothelial cells. The two receptors have similar extracellular domains but dissimilar intracellular domains. The intracellular part of the p60 receptor contains a so-called death domain (DD), which lacks the p80 receptor [121]. It is well established that TNF activates multiple signaling pathways. The DD of the activated p60 receptor interacts with the DD-containing protein TNF receptor associated death domain (TRADD), which in turn recruits Fas-associated death domain (FADD). This leads to binding of TRAF2 and a receptor-interacting protein to the p60/TRADD/FADD complex. Down-stream signals from these early signaling events include activation of phospholipases and sphingomyelinases (resulting in generation of arachidonic acid/diacylglycerol and ceramide, respectively), activation of NFκB through NFκB-inducing kinase, and stimulation of the JNK and p38 MAP/SAPK pathways. In addition to these signaling pathways that are also activated by IL-1β, TNF is capable of directly activating the apoptotic execution program by activating a caspase cascade triggered by FADD activation of caspase-8 [121]. However, caspase activation by TNFα does not seem to be induced in β-cells, as TNFα alone fails to cause impairment of β-cell function [122, 123]. In contrast, TNFα stimulation of MAP/SAPK activation in rat islets [120] and induction of NFκB in insulin-producing cells have been demonstrated [124].
The effector pathways
MAP/SAPKs
Studies on insulin-secreting cells and primary β-cells have revealed that IL-1β is a potent activator of the MAP/SAPKs ERK, p38, and JNK [125, 126, 127]. The functional roles of ERK and p38 in IL-1β signaling in β-cells have been investigated using pharmacological inhibitors. These studies showed that both ERK and p38 are required for IL-1β-induced expression of iNOS [125]. Subsequent studies revealed that blocking ERK or p38 partially (approx. 25 and 40%, respectively) decreased cytokine-induced apoptosis in primary β-cells [112, 128]. Thus, although it is not clear how ERK and p38 are involved in promoting iNOS expression and apoptosis, these two MAPKs are involved at least to some degree in mediating β-cell apoptosis upon cytokine exposure. More convincing results have been obtained on the role of JNK in controlling IL-1β-mediated apoptosis. Transfection experiments and the use of cell-permeable peptide inhibitors have demonstrated that inhibiting the JNK pathway confers almost full protection against apoptosis induced by IL-1β in insulin-secreting cells [129, 130, 131]. The protective effect of blocking JNK awaits to be confirmed in primary β-cells. However, a recent study suggested that JNK is also a critical component in oxidative stress-induced suppression of insulin gene transcription in primary islet cells [132]. The same study reported that transplantation of streptozotocin-induced diabetic nude mice with islets infected with dominant negative JNK expressing adenovirus preserved the insulin gene expression in islet grafts. Further, in these animals hyperglycemia was ameliorated compared with control mice. Hence together these findings support an important role for JNK in the regulation of β-cell function and death.
Given the fact that IFNγ and TNFα strongly potentiate the cytotoxic effects of IL-1β on β-cells, it is of note that these two cytokines synergistically augment IL-1β induced signaling via MAP/SAPKs in rat islets [120]. This observation may provide at least a partial explanation at the signaling level for the synergistic toxic effects of cytokines on β-cells.
What makes the β-cell so sensitive to proinflammatory cytokines? Transfection of a glucagon-producing rat cell line with the pancreatic duodenal homeobox factor (Pdx) 1 transcription factor leading to an insulin-producing β-cell phenotype results in higher sensitivity to cytokine toxicity [133] and is correlated with higher MAP/SAPK activation upon treatment with IL-1β [129]. This suggests that enhanced susceptibility to proapoptotic stimuli and signaling is a direct consequence of β-cell differentiation, and this is potentially the answer why β-cells are so vulnerable to cytotoxic stimuli and in particular cytokines. Interestingly, while Pdx-1 seems to "code" for higher proapoptotic JNK signaling, JNK at the same time is able to inhibit Pdx-1 DNA binding upon oxidative stress [132], which would lead to suppression of β-cell functions. This further supports a strong relationship between Pdx-1 (and thus the β-cell phenotype) and JNK in β-cells. Hence JNK is possibly a key mediator and regulator of β-cell fate and may therefore represent an attractive therapeutic target for preservation of β-cell function and mass in diabetes.
Recently it was demonstrated that JNK activity is abnormally elevated in liver, fat, and muscle in dietary and genetic mouse models of obesity [134]. Also, JNK1−/− mice on a high fat diet had lower body weight, blood glucose, and plasma insulin levels than control (JNK1+/+) mice on high fat diet which were obese, hyperinsulinemic, and hyperglycemic. Hence in diabetes JNK may not only be a crucial factor in the β-cell but also play a significant role in mediating obesity and insulin resistance.
NFκB
The involvement of NFκB in cytokine-induced β-cell death was recently elucidated by adenoviral gene transfer of a nonphosphorylatable and thus a nondegradable form of IκB, the so-called IκB superrepressor. Infection of primary purified rat β-cells with adenovirus containing the IκB superrepressor resulted in decreased apoptotic (and necrotic) cell death induced by a combination of IL-1β and IFNγ [135]. Similarly, experiments with human islets have shown that NFκB inhibition by the IκB superrepressor protects against IL-1β-stimulated, Fas-triggered apoptosis [136]. Hence there is no doubt that NFκB plays an important role in cytokine-induced β-cell apoptosis. What are the target genes of NFκB in β-cells? The use of novel high-density oligonucleotide arrays has provided an important tool to investigate the expression pattern of thousands of genes in parallel. Of the approx. 200 genes whose expression is altered by 24-h exposure to IL-1β plus IFNγ [137] 66 have been found to be regulated by NFκB in cytokine-treated in primary rat β-cells. Among these are genes encoding transcription factors, glucose transporters, and proteins involved in signal transduction [109]. The relative importance of each of the NFκB-regulated genes is not clear, but it is likely that it is the sum of the up- and downregulation of the multiple genes that drives the β-cell into an apoptotic response.
PKC-δ
Recent work has provided evidence for a role of PKC-δ in IL-1β-mediated β-cell death. IL-1β was shown to induce rapid PKC-δ, but not PKC-α, translocation to the plasma membrane in insulin-secreting cells [107]. Using both a pharmacological PKC inhibitor and overexpression of a kinase dead PKC-δ mutant it was shown that IL-1β stimulated iNOS expression and NO production were drastically inhibited. The effect of PKC-δ on iNOS expression was seemingly due to PKC-δ dependent stabilization of iNOS mRNA [107]. A subsequent study observed that cells containing kinase dead PKC-δ were protected against IL-1β-induced apoptosis [138].
Calcium
As in other cell types, the role of Ca2+ in the apoptotic process in β-cells has gained attention, and a number of studies have demonstrated that Ca2+ is a key player in β-cell apoptosis. When β-cells are incubated with serum from type 1 diabetes patients, they undergo apoptosis in a manner dependent on Ca2+ signaling via L-type voltage-gated channels [139]. Further, β-cell apoptosis induced by high glucose or a potassium channel inhibitor (tolbutamide) can be prevented by blocking L-type Ca2+ channels [89]. Cytokine-induced apoptosis also seems to involve Ca2+. It has been shown that IL-1β following a 2-h exposure stimulates a cellular net-uptake of Ca2+ via L-type voltage-gated Ca2+ channels in rat islets [140], and blocking L-type Ca2+ channels suppresses IL-1β-mediated islet cell apoptosis [141]. Further, chronic exposure of mouse islets to a combination of IL-1β and IFNγ induces an increase in the activity of T-type Ca2+ channels, resulting in a threefold higher sustained plateau of cytosolic Ca2+, which is associated with apoptosis [142]. Interestingly, β-cells from NOD mice have been found to have abnormally higher activity of T-type Ca2+ channels and higher cytosolic free Ca2+ concentration than control mouse β-cells [143]. In further support of a role of Ca2+ in mediating cytokine-induced apoptosis is the finding that calbindin-D28k, a cytosolic Ca2+-binding protein (which at the mRNA level is downregulated by cytokines [109]), protects insulin-secreting cells from apoptosis induced by a mixture of IL-1β, IFNγ, and TNFα [144]. Thus, a substantial amount of evidence suggests a role for Ca2+ in β-cell apoptosis, although the molecular mechanisms behind Ca2+-mediated β-cell apoptosis remain to be described. We have recently explored the role of Ca2+ in the regulation of proapoptotic IL-1β signaling and found that Ca2+ entering through L-type Ca2+ channels regulates IL-1β activation of MAP/SAPKs (J. Størling and T. Mandrup-Poulsen, manuscript in preparation). Therefore one (but probably not the only) role of Ca2+ could be to amplify and prolong proapoptotic MAP/SAPK signaling.
Another event which is related to cellular handling of Ca2+ and which is associated with apoptosis in β-cells is the depletion of Ca2+ from the endoplasmic reticulum (ER). Inhibition of sarco-/endoplasmic reticulum Ca2+ ATPase (SERCA), which results in release of Ca2+ from the ER, induces apoptosis in insulin-producing cells [145]. The effect of SERCA inhibition on apoptosis does not seems to be caused by an increase in cytosolic Ca2+, but is likely to be due to the sustained depletion of ER Ca2+ [145]. Such a depletion of ER Ca2+, which perturbs the function of the ER, is well known in many cell types to give rise to a condition known as ER stress. Interestingly, it has recently been suggested that the ER is a target of NO in β-cells as NO was found to cause depletion of ER Ca2+ [146, 147]. Further, cytokines have been shown to cause downregulation of SERCA at the mRNA level in β-cells [109, 137]. Together, these observations suggest that β-cells exposed to cytokines are impaired in their ability to maintain an adequate level of Ca2+ within the ER leading to ER stress. Although the transcription factor C/EBP homologous protein is induced by ER stress and shown to be involved in mediating ER stress-induced β-cell apoptosis [146], we have data to suggest that the JNK signaling pathway is also induced by ER stress in insulin-secreting cells (J. Størling and T. Mandrup-Poulsen, manuscript in preparation).
Fas
The apoptosis-inducing receptor Fas (also known as CD95 or APO-1) is a member of the TNF receptor superfamily. As with the TNF p60 receptor, Fas has a DD in the intracellular part of the receptor that is responsible for the transduction of the apoptotic signal. Activation of Fas is triggered by Fas ligand (FasL), which exists both in a soluble and in a membrane-bound form [148], the former being a weak activator. FasL stimulation of Fas induces trimerization of the Fas receptor which through the DD recruits caspase-8 via the FADD/mediator of receptor induced toxicity (MORT) 1 adaptor. This causes activation of caspase-8, which in turn triggers a cascade of down-stream caspases resulting in proteolytic cleavage of critical cellular components including lamins and actin [148]. Normally β-cells do not express Fas but do so after exposure to cytokines rendering them susceptible to FasL-induced apoptosis [39, 100, 149, 150]. Also, exposure of human β-cells to high concentrations of glucose induces upregulation of Fas expression and causes apoptosis by Fas receptor interaction with constitutively expressed FasL on neighboring β-cells [23]. However, Fas receptor signaling may not only be connected to apoptosis but also implicated in proliferative signals. Thus proliferation of human T-cells induced by T-cell receptor activation is augmented by FasL [151] and T-cells expressing dominant negative FADD/MORT1 have inhibited T-cell receptor induced proliferation [152]. What determines whether Fas signaling leads to apoptosis or proliferation? FLIP seems to direct Fas signaling from apoptosis into survival/proliferation. In human β-cells FLIP is constitutively expressed but is downregulated by high glucose. Overexpression of FLIP switches glucose-induced, Fas-mediated β-cell apoptosis into proliferation in a manner totally dependent on Fas signaling [96], indicating that Fas signaling in the presence of FLIP is connected with cell replication and survival in β-cells. The molecular link(s) between FLIP and cell proliferation have been investigated in T-cells. In these cells FLIP has been shown to stimulate the NFκB and the ERK MAPK pathways, which leads to production of the T-cell growth factor IL-2 [151]. The mechanism(s) underlying Fas-induced, caspase-8/FLIP-mediated proliferative signaling in β-cells is currently not known but may potentially involve altered NFκB and/or MAP/SAPK signaling. However, regardless the mechanisms involved, it appears that Fas is not simply a death receptor but a key regulator of cell cycle which may induce cell death or proliferation depending on intracellular cofactors.
Inflammatory mediators in type 1 and 2 diabetes: how can we extinguish the flame burning the β-cells?
The first indirect indication that diabetes mellitus has inflammatory components was reported in a contribution in 1876 to the predecessor of the Journal of Molecular Medicine, the Berliner Klinische Wochenschrift, in which Professor Ebstein observed that high doses of sodium salicylate improves glucosuria in diabetic patients [153, 154]. Salicylate prevents the activation of NF-κB, a mediator of inflammation and apoptosis [155]. Another clinical hint to the role of inflammatory mediators in the pathogenesis of type 2 diabetes has been recently reported [156]. A prospective study showed that a specific pattern of cytokines is associated with an increased risk of developing type 2 diabetes. In particular, participants with a elevation in both IL-6 and IL-1β had a threefold increased risk of developing diabetes compared to the reference group. Interestingly, participants with elevated levels of IL-6 alone and undetectable levels of IL-1β had no increase in diabetes risk, suggesting the central role of IL-1β. It is likely that part of those observations are related to changes in insulin resistance and not only in β-cell function [157, 158]. Nevertheless, based on current thinking, modulation of the intra-islet inflammatory mediators in type 1 and 2 diabetes appears as a promising approach. The progressive decline in functional β-cell mass observed in diabetic patients may thus be prevented and even reversed. Several drugs are in use for the management of diabetics with the primary aim to prevent its complications, for example, statins, but not to improve β-cell function. One can speculate that the strong anti-inflammatory effects of these drugs are partially responsible for the improved outcome of diabetic patients, which cannot be explained solely by the lipid-lowering effects [159]. However, it will probably take several years until drugs are available with the primary aim of preventing the inflammatory process of islets. In the meantime, implementing insulin therapy at early stages of not only type 1 but also type 2 diabetes appears reasonable. Insulin has strong antiapoptotic effects and seems to lead to preserved β cell function (see above). Although current evidence does not support that insulin treatment in risk individuals of type 1 diabetes development is of benefit [30], it does not rule out that other approaches with different timing, doses and sites of application could be successful. Until then, understanding that the functional β-cell mass is, relatively or absolutely, decreased in most diabetic patients justifies the replacement of what is missing: insulin!
Abbreviations
- DD :
-
Death domain
- ECSIT :
-
Evolutionary conserved signaling intermediate in Toll/IL-1 pathways
- ER :
-
Endoplasmic reticulum
- ERK :
-
Extracellular signal-regulated kinase
- FADD :
-
Fas-associated death domain
- FasL :
-
Fas ligand
- FFA :
-
Free fatty acid
- FLIP :
-
Fas-associated death domain-like IL-1β converting enzyme inhibitory protein
- IFN :
-
Interferon
- IKK :
-
IκB kinase
- IL :
-
Interleukin
- IL-1R1 :
-
IL-1 type 1 receptor
- IL-1Ra :
-
IL-1 receptor antagonist
- iNOS :
-
Inducible nitric oxide synthase
- IRAK :
-
IL-1R activated kinase
- IRF :
-
Interferon regulatory factor
- IκB :
-
Inhibitory κB protein
- JAK :
-
Janus tyrosine kinases
- JNK :
-
c-jun N-terminal kinase
- MAP :
-
Mitogen-activated protein
- MAPK :
-
Mitogen-activated protein kinase
- MEKK :
-
MAPK/ERK kinase kinase
- MORT :
-
Mediator of receptor induced toxicity
- NF :
-
Nuclear transcription factor
- NOD :
-
Nonobese diabetic
- Pdx :
-
Pancreatic duodenal homeobox factor
- PKC :
-
Protein kinase C
- ROS :
-
Reactive oxygen species
- SAPK :
-
Stress-activated protein kinases
- SERCA :
-
Sarco-/endoplasmic reticulum Ca2+ ATPase
- STAT :
-
Signal transducer and activator of transcription
- TAK :
-
Transforming growth factor β-activated kinase
- TNF :
-
Tumor necrosis factor
- TRADD :
-
TNF receptor associated death domain
- TRAF :
-
TNF receptor associated factor
References
Anonymous (1997) Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 20:1183–1197
Bergholdt R, Heding P, Nielsen K, Nolsøe R, Sparre T, Størling J, et al (2002) Type 1 diabetes mellitus, an inflammatory disease of the islet. In: Eisenbarth GS (ed) Type 1 diabetes: molecular, cellular and clinical immunology.http://www.uchscedu/misc/diabetes/bdc.html
Gepts W (1965) Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14:619–633
Eizirik DL, Mandrup-Poulsen T (2001) A choice of death-the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 44:2115–2133
Winer S, Tsui H, Lau A, Song A, Li X, Cheung RK, Sampson A, Afifiyan F, Elford A, Jackowski G, Becker DJ, Santamaria P, Ohashi P, Dosch HM (2003) Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat Med 9:198–205
Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU (1985) Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res 4:110–125
Cerasi E (1995) Insulin deficiency and insulin resistance in the pathogenesis of NIDDM: is a divorce possible? Diabetologia 38:992–997
Taylor SI, Accili D, Imai Y (1994) Insulin resistance or insulin deficiency. Which is the primary cause of NIDDM? Diabetes 43:735–740
Gerich JE (2000) Insulin resistance is not necessarily an essential component of type 2 diabetes. J Clin Endocrinol Metab 85:2113–2115
Bonner-Weir S (2000) Islet growth and development in the adult. J Mol Endocrinol 24:297–302
Finegood DT, Scaglia L, Bonner-Weir S (1995) Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 44:249–256
Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, Turner RC (1988) Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res 9:151–159
Ritzel RA, Sultana C, Butler PC (2001) Preferential apoptosis of actively dividing cells induced by human islet amyloid polypeptide (abstract). Diabetes 50 [Suppl 2]:A32
Gepts W, Lecompte PM (1981) The pancreatic islets in diabetes. Am J Med 70:105–115
Guiot Y, Sempoux C, Moulin P, Rahier J (2001) No decrease of the beta-cell mass in type 2 diabetic patients. Diabetes 50 [Suppl 1]:S188
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type-2 diabetes mellitus. Diabetes 52:102–110
Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S (2002) Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 45:85–96
Mathis D, Vence L, Benoist C (2001) Beta-cell death during progression to diabetes. Nature 414:792–798
Pietropaolo M, Barinas-Mitchell E, Pietropaolo SL, Kuller LH, Trucco M (2000) Evidence of islet cell autoimmunity in elderly patients with type 2 diabetes. Diabetes 49:32–38
Rowley MJ, Mackay IR, Chen QY, Knowles WJ, Zimmet PZ (1992) Antibodies to glutamic acid decarboxylase discriminate major types of diabetes mellitus. Diabetes 41:548–551
Wilkin TJ (2001) The accelerator hypothesis: weight gain as the missing link between type I and type II diabetes. Diabetologia 44:914–922
Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY (2002) Glucose-induced beta-cell production of interleukin-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Maedler K, Spinas GA, Lehmann R, Sergeev P, Weber M, Fontana A, Kaiser N, Donath MY (2001) Glucose induces beta-cell apoptosis via upregulation of the Fas-receptor in human islets. Diabetes 50:1683–1690
Atkinson MA, Eisenbarth GS (2001) Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 358:221–229
Pociot F, Karlsen AE, Mandrup-Poulsen T (2002) Etiology and pathogenesis of insulin-dependent diabetes mellitus. In: Bertagna X, Fischer J, Groop L, Schoemaker J, Serio M, Wass J (eds) Endocrinology and metabolism. McGraw-Hill International, London, pp 593–606
Dahlquist GG (1997) Viruses and other perinatal exposures as initiating events for beta-cell destruction. Ann Med 29:413–417
The Canadian-European Randomized Control Trial Group. Cyclosporin-induced remission of IDDM after early intervention. Association of 1 yr of cyclosporin treatment with enhanced insulin secretion. Diabetes 37:1574–15821988
Feutren G, Papoz L, Assan R, Vialettes B, Karsenty G, Vexiau P, Du RH, Rodier M, Sirmai J, Lallemand A (1986) Cyclosporin increases the rate and length of remissions in insulin-dependent diabetes of recent onset. Results of a multicentre double-blind trial. Lancet II:119–124
Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA (2002) Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 346:1692–1698
Anonymous (2002) Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 346:1685–1691
Shah SC, Malone JI, Simpson NE (1989) A randomized trial of intensive insulin therapy in newly diagnosed insulin-dependent diabetes mellitus. N Engl J Med 320:550–554
Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
Mandrup-Poulsen T (1996) The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia 39:1005–1029
Martin S, Wolf-Eichbaum D, Duinkerken G, Scherbaum WA, Kolb H, Noordzij JG, Roep BO (2001) Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Engl J Med 345:1036–1040
Nomikos IN, Prowse SJ, Carotenuto P, Lafferty KJ (1986) Combined treatment with nicotinamide and desferrioxamine prevents islet allograft destruction in NOD mice. Diabetes 35:1302–1304
Weringer EJ, Like AA (1985) Immune attack on pancreatic islet transplants in the spontaneously diabetic BioBreeding/Worcester (BB/W) rat is not MHC restricted. J Immunol 134:2383–2386
DiLorenzo TP, Graser RT, Ono T, Christianson GJ, Chapman HD, Roopenian DC, Nathenson SG, Serreze DV (1998) Major histocompatibility complex class I-restricted T cells are required for all but the end stages of diabetes development in nonobese diabetic mice and use a prevalent T cell receptor alpha chain gene rearrangement. Proc Natl Acad Sci USA 95:12538–12543
Peterson JD, Pike B, McDuffie M, Haskins K (1994) Islet-specific T cell clones transfer diabetes to nonobese diabetic (NOD) F1 mice. J Immunol 153:2800–2806
Yamada K, Takane-Gyotoku N, Yuan X, Ichikawa F, Inada C, Nonaka K (1996) Mouse islet cell lysis mediated by interleukin-1-induced Fas. Diabetologia 39:1306–1312
Dandona P, Aljada A (2002) A rational approach to pathogenesis and treatment of type 2 diabetes mellitus, insulin resistance, inflammation, and atherosclerosis. Am J Cardiol 90:27G–33G
Esch T, Stefano G (2002) Proinflammation: a common denominator or initiator of different pathophysiological disease processes. Med Sci Monit 8:HY1–HY9
Ludewig B, Zinkernagel RM, Hengartner H (2002) Arterial inflammation and atherosclerosis. Trends Cardiovasc Med 12:154–159
Marette A (2002) Mediators of cytokine-induced insulin resistance in obesity and other inflammatory settings. Curr Opin Clin Nutr Metab Care 5:377–383
Muller S, Martin S, Koenig W, Hanifi-Moghaddam P, Rathmann W, Haastert B, Giani G, Illig T, Thorand B, Kolb H (2002) Impaired glucose tolerance is associated with increased serum concentrations of interleukin 6 and co-regulated acute-phase proteins but not TNF-alpha or its receptors. Diabetologia 45:805–812
Syed MA, Barinas-Mitchell E, Pietropaolo SL, Zhang YJ, Henderson TS, Kelley DE, Korytkowski MT, Donahue RP, Tracy RP, Trucco M, Kuller LH, Pietropaolo M (2002) Is type 2 diabetes a chronic inflammatory/autoimmune disease? Diabetes Nutr Metab 15:68–83
Barnett AH, Eff C, Leslie RD, Pyke DA (1981) Diabetes in identical twins. A study of 200 pairs. Diabetologia 20:87–93
Elbein SC (2002) Perspective: the search for genes for type 2 diabetes in the post-genome era. Endocrinology 143:2012–2018
Byrne MM, Sturis J, Clement K, Vionnet N, Pueyo ME, Stoffel M, Takeda J, Passa P, Cohen D, Bell GI (1994) Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest 93:1120–1130
Byrne MM, Sturis J, Menzel S, Yamagata K, Fajans SS, Dronsfield MJ, Bain SC, Hattersley AT, Velho G, Froguel P, Bell GI, Polonsky KS (1996) Altered insulin secretory responses to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY3 on chromosome 12. Diabetes 45:1503–1510
Byrne MM, Sturis J, Menzel S, Yamagata K, Fajans SS, Dronsfield MJ, Bain SC, Hattersley AT, Velho G, Froguel P, Bell GI, Polonsky KS (1996) Altered insulin secretory responses to glucose in diabetic and nondiabetic subjects with mutations in the diabetes susceptibility gene MODY3 on chromosome 12. Diabetes 45:1503–1510
Fried SK, Bunkin DA, Greenberg AS (1998) Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab 83:847–850
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259:87–91
Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95:2409–2415
Meier CA, Bobbioni E, Gabay C, Assimacopoulos-Jeannet F, Golay A, Dayer JM (2002) IL-1 receptor antagonist serum levels are increased in human obesity: a possible link to the resistance to leptin? J Clin Endocrinol Metab 87:1184–1188
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Pickup JC, Mattock MB, Chusney GD, Burt D (1997) NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40:1286–1292
Pickup JC, Crook MA (1998) Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 41:1241–1248
Pickup JC, Chusney GD, Mattock MB (2000) The innate immune response and type 2 diabetes: evidence that leptin is associated with a stress-related (acute-phase) reaction. Clin Endocrinol (Oxf) 52:107–112
Watts GF, Mandalia S, Brunt JN, Slavin BM, Coltart DJ, Lewis B (1993) Independent associations between plasma lipoprotein subfraction levels and the course of coronary artery disease in the St. Thomas' Atherosclerosis Regression Study (STARS). Metabolism 42:1461–1467
Bastard JP, Pieroni L, Hainque B (2000) Relationship between plasma plasminogen activator inhibitor 1 and insulin resistance. Diabetes Metab Res Rev 16:192–201
Ganrot PO, Gydell K, Ekelund H (1967) Serum concentration of alpha-2-macroglobulin, haptoglobin and alpha-1-antitrypsin in diabetes mellitus. Acta Endocrinol (Copenh) 55:537–544
Jonsson A, Wales JK (1976) Blood glycoprotein levels in diabetes mellitus. Diabetologia 12:245–250
McMillan DE (1989) Increased levels of acute-phase serum proteins in diabetes. Metabolism 38:1042–1046
Tuomi T, Groop LC, Zimmet PZ, Rowley MJ, Knowles W, Mackay IR (1993) Antibodies to glutamic acid decarboxylase reveal latent autoimmune diabetes mellitus in adults with a non-insulin-dependent onset of disease. Diabetes 42:359–362
Zimmet PZ, Tuomi T, Mackay IR, Rowley MJ, Knowles W, Cohen M, Lang DA (1994) Latent autoimmune diabetes mellitus in adults (LADA): the role of antibodies to glutamic acid decarboxylase in diagnosis and prediction of insulin dependency. Diabet Med 11:299–303
Donath MY, Gross DJ, Cerasi E, Kaiser N (1999) Hyperglycemia-induced beta-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes 48:738–744
Bellone M, Iezzi G, Rovere P, Galati G, Ronchetti A, Protti MP, Davoust J, Rugarli C, Manfredi AA (1997) Processing of engulfed apoptotic bodies yields T cell epitopes. J Immunol 159:5391–5399
Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT (2000) Neonatal beta-cell apoptosis: a trigger for autoimmune diabetes? Diabetes 49:1–7
Fearon DT, Locksley RM (1996) The instructive role of innate immunity in the acquired immune response. Science 272:50–53
Islam MS, Sjoholm A, Emilsson V (2000) Fetal pancreatic islets express functional leptin receptors and leptin stimulates proliferation of fetal islet cells. Int J Obes Relat Metab Disord 24:1246–1253
Okuya S, Tanabe K, Tanizawa Y, Oka Y (2001) Leptin increases the viability of isolated rat pancreatic islets by suppressing apoptosis. Endocrinology 142:4827–4830
Shimabukuro M, Wang MY, Zhou YT, Newgard CB, Unger RH (1998) Protection against lipoapoptosis of beta cells through leptin-dependent maintenance of Bcl-2 expression. Proc Natl Acad Sci U S A 95:9558–9561
Tanabe K, Okuya S, Tanizawa Y, Matsutani A, Oka Y (1997) Leptin induces proliferation of pancreatic beta cell line MIN6 through activation of mitogen-activated protein kinase. Biochem Biophys Res Commun 241:765–768
Mandrup-Poulsen T (2001) beta-cell apoptosis: stimuli and signaling. Diabetes 50 [Suppl 1]:S58–S63
Mohamed-Ali V, Flower L, Sethi J, Hotamisligil G, Gray R, Humphries SE, York DA, Pinkney J (2001) beta-Adrenergic regulation of IL-6 release from adipose tissue: in vivo and in vitro studies. J Clin Endocrinol Metab 86:5864–5869
Eizirik DL, Sandler S, Welsh N, Cetkovic-Cvrlje M, Nieman A, Geller DA, Pipeleers DG, Bendtzen K, Hellerstrom C (1994) Cytokines suppress human islet function irrespective of their effects on nitric oxide generation. J Clin Invest 93:1968–1974
Wadt KA, Larsen CM, Andersen HU, Nielsen K, Karlsen AE, Mandrup-Poulsen T (1998) Ciliary neurotrophic factor potentiates the beta-cell inhibitory effect of IL-1beta in rat pancreatic islets associated with increased nitric oxide synthesis and increased expression of inducible nitric oxide synthase. Diabetes 47:1602–1608
LeRoith D (2002) Beta-cell dysfunction and insulin resistance in type 2 diabetes: role of metabolic and genetic abnormalities. Am J Med 113 [Suppl 6A]:3S–11S
McGarry JD, Dobbins RL (1999) Fatty acids, lipotoxicity and insulin secretion. Diabetologia 42:128–138
Randle PJ, Garland PB, Newsholme EA, Hales CN (1965) The glucose fatty acid cycle in obesity and maturity onset diabetes mellitus. Ann N Y Acad Sci 131:324–333
Unger RH (1995) Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 44:863–870
Vries JE de, Vork MM, Roemen TH, de Jong YF, Cleutjens JP, van der Vusse GJ, van Bilsen M (1997) Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J Lipid Res 38:1384–1394
Hardy S, Langelier Y, Prentki M (2000) Oleate activates phosphatidylinositol 3-kinase and promotes proliferation and reduces apoptosis of MDA-MB-231 breast cancer cells, whereas palmitate has opposite effects. Cancer Res 60:6353–6358
Dyntar D, Eppenberger-Eberhardt M, Maedler K, Pruschy M, Eppenberger HM, Spinas GA, Donath MY (2001) Glucose and palmitic acid induce degeneration of myofibrils and modulate apoptosis in rat adult cardiomyocytes. Diabetes 50:2105–2113
Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY (2001) Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 50:69–76
Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY (2003) Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 52:726–733
Shimabukuro M, Zhou YT, Levi M, Unger RH (1998) Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci U S A 95:2498–2502
Federici M, Hribal M, Perego L, Ranalli M, Caradonna Z, Perego C, Usellini L, Nano R, Bonini P, Bertuzzi F, Marlier LN, Davalli AM, Carandente O, Pontiroli AE, Melino G, Marchetti P, Lauro R, Sesti G, Folli F (2001) High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: a potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes 50:1290–1301
Efanova IB, Zaitsev SV, Zhivotovsky B, Kohler M, Efendic S, Orrenius S, Berggren PO (1998) Glucose and tolbutamide induce apoptosis in pancreatic beta-cells. A process dependent on intracellular Ca2+ concentration. J Biol Chem 273:33501–33507
Evans JL, Goldfine ID, Maddux BA, Grodsky GM (2003) Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 52:1–8
Hunt JV, Dean RT, Wolff SP (1988) Hydroxyl radical production and autoxidative glycosylation. Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes mellitus and ageing. Biochem J 256:205–212
Kaneto H, Fujii J, Myint T, Miyazawa N, Islam KN, Kawasaki Y, Suzuki K, Nakamura M, Tatsumi H, Yamasaki Y, Taniguchi N (1996) Reducing sugars trigger oxidative modification and apoptosis in pancreatic beta-cells by provoking oxidative stress through the glycation reaction. Biochem J 320:855–863
Laybutt DR, Kaneto H, Hasenkamp W, Grey S, Jonas JC, Sgroi DC, Groff A, Ferran C, Bonner-Weir S, Sharma A, Weir GC (2002) Increased expression of antioxidant and antiapoptotic genes in islets that may contribute to beta-cell survival during chronic hyperglycemia. Diabetes 51:413–423
Matsuoka T, Kajimoto Y, Watada H, Kaneto H, Kishimoto M, Umayahara Y, Fujitani Y, Kamada T, Kawamori R, Yamasaki Y (1997) Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J Clin Invest 99:144–150
Hoorens A, Van dC, Kloppel G, Pipeleers D (1996) Glucose promotes survival of rat pancreatic beta cells by activating synthesis of proteins which suppress a constitutive apoptotic program. J Clin Invest 98:1568–1574
Maedler K, Fontana A, Ris F, Sergeev P, Toso C, Oberholzer J, Lehmann R, Bachmann F, Tasinato A, Spinas GA, Halban PA, Donath MY (2002) FLIP switches Fas-mediated glucose signaling in human pancreatic beta cells from apoptosis to cell replication. Proc Natl Acad Sci USA 99:8236–8241
Spinas GA, Mandrup-Poulsen T, Molvig J, Baek L, Bendtzen K, Dinarello CA, Nerup J (1986) Low concentrations of interleukin-1 stimulate and high concentrations inhibit insulin release from isolated rat islets of Langerhans. Acta Endocrinol (Copenh) 113:551–558
Heitmeier MR, Arnush M, Scarim AL, Corbett JA (2001) Pancreatic β-cell damage mediated by beta-cell production of IL-1: a novel mechanism for virus-induced diabetes. J Biol Chem 276:11151–11158
Giannoukakis N, Rudert WA, Ghivizzani SC, Gambotto A, Ricordi C, Trucco M, Robbins PD (1999) Adenoviral gene transfer of the interleukin-1 receptor antagonist protein to human islets prevents IL-1beta-induced beta-cell impairment and activation of islet cell apoptosis in vitro. Diabetes 48:1730–1736
Loweth AC, Williams GT, James RF, Scarpello JH, Morgan NG (1998) Human islets of Langerhans express Fas ligand and undergo apoptosis in response to interleukin-1beta and Fas ligation. Diabetes 47:727–732
Rabinovitch A, Sumoski W, Rajotte RV, Warnock GL (1990) Cytotoxic effects of cytokines on human pancreatic islet cells in monolayer culture. J Clin Endocrinol Metab 71:152–156
Stassi G, De Maria R, Trucco G, Rudert W, Testi R, Galluzzo A, Giordano C, Trucco M (1997) Nitric oxide primes pancreatic beta cells for Fas-mediated destruction in insulin-dependent diabetes mellitus. J Exp Med 186:1193–1200
Dunne A, O'Neill LA (2003) The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE re3
Kopp E, Medzhitov R, Carothers J, Xiao C, Douglas I, Janeway CA, Ghosh S (1999) ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev 13:2059–2071
Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K (1999) The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398:252–256
Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K (1996) TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science 272:1179–1182
Carpenter L, Cordery D, Biden TJ (2001) Protein kinase Cdelta activation by interleukin-1beta stabilizes inducible nitric-oxide synthase mRNA in pancreatic beta-cells. J Biol Chem 276:5368–5374
Eizirik DL, Flodstrom M, Karlsen AE, Welsh N (1996) The harmony of the spheres: inducible nitric oxide synthase and related genes in pancreatic beta cells. Diabetologia 39:875–890
Cardozo AK, Heimberg H, Heremans Y, Leeman R, Kutlu B, Kruhoffer M, Orntoft T, Eizirik DL (2001) A comprehensive analysis of cytokine-induced and NF-kB dependent genes in primary rat pancreatic β-cells. J Biol Chem 276:48879–48886
Flodstrom M, Welsh N, Eizirik DL (1996) Cytokines activate the nuclear factor kappa B (NF-kappa B) and induce nitric oxide production in human pancreatic islets. FEBS Lett 385:4–6
Kwon G, Corbett JA, Rodi CP, Sullivan P, McDaniel ML (1995) Interleukin-1 beta-induced nitric oxide synthase expression by rat pancreatic beta-cells: evidence for the involvement of nuclear factor kappa B in the signaling mechanism. Endocrinology 136:4790–4795
Saldeen J (2000) Cytokines induce both necrosis and apoptosis via a common Bcl-2-inhibitable pathway in rat insulin-producing cells. Endocrinology 141:2003–2010
Chen MC, Proost P, Gysemans C, Mathieu C, Eizirik DL (2001) Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic NOD mice and in interleukin-1 beta-exposed human and rat islet cells. Diabetologia 44:325–332
Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD (1998) How cells respond to interferons. Annu Rev Biochem 67:227–264
Tau G, Rothman P (1999) Biologic functions of the IFN-gamma receptors. Allergy 54:1233–1251
Karlsen AE, Ronn SG, Lindberg K, Johannesen J, Galsgaard ED, Pociot F, Nielsen JH, Mandrup-Poulsen T, Nerup J, Billestrup N (2001) Suppressor of cytokine signaling 3 (SOCS-3) protects beta-cells against interleukin-1beta-and interferon-gamma-mediated toxicity. Proc Natl Acad Sci USA 98:12191–12196
Heitmeier MR, Scarim AL, Corbett JA (1999) Double-stranded RNA inhibits beta-cell function and induces islet damage by stimulating beta-cell production of nitric oxide. J Biol Chem 274:12531–12536
Flodstrom M, Eizirik DL (1997) Interferon-gamma-induced interferon regulatory factor-1 (IRF-1) expression in rodent and human islet cells precedes nitric oxide production. Endocrinology 138:2747–2753
Karlsen AE, Pavlovic D, Nielsen K, Jensen J, Andersen HU, Pociot F, Mandrup-Poulsen T, Eizirik DL, Nerup J (2000) Interferon-gamma induces interleukin-1 converting enzyme expression in pancreatic islets by an interferon regulatory factor-1-dependent mechanism. J Clin Endocrinol Metab 85:830–836
Andersen NA, Larsen CM, Mandrup-Poulsen T (2000) TNFalpha and IFNgamma potentiate IL-1beta induced mitogen activated protein kinase activity in rat pancreatic islets of Langerhans. Diabetologia 43:1389–1396
Rath PC, Aggarwal BB (1999) TNF-induced signaling in apoptosis. J Clin Immunol 19:350–364
Mandrup-Poulsen T, Bendtzen K, Dinarello CA, Nerup J (1987) Human tumor necrosis factor potentiates human interleukin 1-mediated rat pancreatic beta-cell cytotoxicity. J Immunol 139:4077–4082
Pukel C, Baquerizo H, Rabinovitch A (1988) Destruction of rat islet cell monolayers by cytokines. Synergistic interactions of interferon-gamma, tumor necrosis factor, lymphotoxin, and interleukin 1. Diabetes 37:133–136
Bernal-Mizrachi E, Wen W, Shornick M, Permutt MA (2002) Activation of nuclear factor-kappaB by depolarization and Ca (2+) influx in MIN6 insulinoma cells. Diabetes 51 [Suppl 3]:S484–S488
Larsen CM, Wadt KA, Juhl LF, Andersen HU, Karlsen AE, Su MS, Seedorf K, Shapiro L, Dinarello CA, Mandrup-Poulsen T (1998) Interleukin-1beta-induced rat pancreatic islet nitric oxide synthesis requires both the p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J Biol Chem 273:15294–15300
Welsh N (1996) Interleukin-1 beta-induced ceramide and diacylglycerol generation may lead to activation of the c-Jun NH2-terminal kinase and the transcription factor ATF2 in the insulin-producing cell line RINm5F. J Biol Chem 271:8307–8312
Major CD, Wolf BA (2001) Interleukin-1beta stimulation of c-Jun NH (2)-terminal kinase activity in insulin-secreting cells: evidence for cytoplasmic restriction. Diabetes 50:2721–2728
Pavlovic D, Andersen NA, Mandrup-Poulsen T, Eizirik DL (2000) Activation of extracellular signal-regulated kinase (ERK)1/2 contributes to cytokine-induced apoptosis in purified rat pancreatic beta-cells. Eur Cytokine Netw 11:267–274
Ammendrup A, Maillard A, Nielsen K, Aabenhus AN, Serup P, Dragsbaek MO, Mandrup-Poulsen T, Bonny C (2000) The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 49:1468–1476
Bonny C, Oberson A, Steinmann M, Schorderet DF, Nicod P, Waeber G (2000) IB1 reduces cytokine-induced apoptosis of insulin-secreting cells. J Biol Chem 275:16466–16472
Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF (2001) Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50:77–82
Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC (2002) Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 277:30010–30018
Nielsen K, Karlsen AE, Deckert M, Madsen OD, Serup P, Mandrup-Poulsen T, Nerup J (1999) Beta-cell maturation leads to in vitro sensitivity to cytotoxins. Diabetes 48:2324–2332
Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Heimberg H, Heremans Y, Jobin C, Leemans R, Cardozo AK, Darville M, Eizirik DL (2001) Inhibition of cytokine-induced NF-kappaB activation by adenovirus-mediated expression of a NF-kappaB super-repressor prevents beta-cell apoptosis. Diabetes 50:2219–2224
Giannoukakis N, Rudert WA, Trucco M, Robbins PD (2000) Protection of human islets from the effects of interleukin-1beta by adenoviral gene transfer of an Ikappa B repressor. J Biol Chem 275:36509–36513
Cardozo AK, Kruhoffer M, Leeman R, Orntoft T, Eizirik DL (2001) Identification of novel cytokine-induced genes in pancreatic beta-cells by high-density oligonucleotide arrays. Diabetes 50:909–920
Carpenter L, Cordery D, Biden TJ (2002) Inhibition of protein kinase C delta protects rat INS-1 cells against interleukin-1beta and streptozotocin-induced apoptosis. Diabetes 51:317–324
Juntti-Berggren L, Larsson O, Rorsman P, Ammala C, Bokvist K, Wahlander K, Nicotera P, Dypbukt J, Orrenius S, Hallberg A, Berggren PO (1993) Increased activity of L-type Ca2+ channels exposed to serum from patients with type I diabetes. Science 261:86–90
Borg LA, Eizirik DL (1990) Short-term exposure of rat pancreatic islets to human interleukin-1 beta increases cellular uptake of calcium. Immunol Lett 26:253–258
Zaitsev SV, Appelskog IB, Kapelioukh IL, Yang SN, Kohler M, Efendic S, Berggren PO (2001) Imidazoline compounds protect against interleukin 1beta-induced beta-cell apoptosis. Diabetes 50 [Suppl 1]:S70–S76
Wang L, Bhattacharjee A, Zuo Z, Hu F, Honkanen RE, Berggren PO, Li M (1999) A low voltage-activated Ca2+ current mediates cytokine-induced pancreatic beta-cell death. Endocrinology 140:1200–1204
Wang L, Bhattacharjee A, Fu J, Li M (1996) Abnormally expressed low-voltage-activated calcium channels in beta-cells from NOD mice and a related clonal cell line. Diabetes 45:1678–1683
Rabinovitch A, Suarez-Pinzon WL, Sooy K, Strynadka K, Christakos S (2001) Expression of calbindin-D (28 k) in a pancreatic islet beta-cell line protects against cytokine-induced apoptosis and necrosis. Endocrinology 142:3649–3655
Zhou YP, Teng D, Dralyuk F, Ostrega D, Roe MW, Philipson L, Polonsky KS (1998) Apoptosis in insulin-secreting cells. Evidence for the role of intracellular Ca2+ stores and arachidonic acid metabolism. J Clin Invest 101:1623–1632
Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada I, Akira S, Araki E, Mori M (2001) Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA 98:10845–10850
Oyadomari S, Araki E, Mori M (2002) Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 7:335–345
Nagata S (1997) Apoptosis by death factor. Cell 88:355–365
Suarez-Pinzon W, Sorensen O, Bleackley RC, Elliott JF, Rajotte RV, Rabinovitch A (1999) Beta-cell destruction in NOD mice correlates with Fas (CD95) expression on beta-cells and proinflammatory cytokine expression in islets. Diabetes 48:21–28
Stassi G, Todaro M, Richiusa P, Giordano M, Mattina A, Sbriglia MS, Lo MA, Buscemi G, Galluzzo A, Giordano C (1995) Expression of apoptosis-inducing CD95 (Fas/Apo-1) on human beta-cells sorted by flow-cytometry and cultured in vitro. Transplant Proc 27:3271–3275
Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, Burns K, Hahne M, Kennedy N, Kovacsovics M, Tschopp J (2000) The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol 10:640–648
Newton K, Harris AW, Bath ML, Smith KG, Strasser A (1998) A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J 17:706–718
Ebstein, W. Zur Therapie des Diabetes Mellitus, insbesondere über die Anwendung des Salicylsauren Natron bei demselben. Berliner Klin Wochenschr 24:337–340:1876
Shoelson S (2002) Invited comment on W. Ebstein: on the therapy of diabetes mellitus, in particular on the application of sodium salicylate. J Mol Med 80:618–619
Kopp E, Ghosh S (1994) Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 265:956–959
Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, Boeing H, Pfeiffer AF (2003) Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam study. Diabetes 52:812–817
Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, Shoelson SE, Shulman GI (2001) Prevention of fat-induced insulin resistance by salicylate. J Clin Invest 108:437–446
Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE (2001) Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikebeta. Science 293:1673–1677
Ridker PM, Rifai N, Clearfield M, Downs JR, Weis SE, Miles JS, Gotto AM Jr (2001) Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 344:1959–1965
Acknowledgements
This work was supported in part by the Juvenile Diabetes Research Foundation International Grant # 4-2002-457, the Danish Diabetes Association and Novo Nordisk (J.S., T.M.P.) and by the Swiss National Science Foundation Grants #3200-067049.01 and PP00B-68874/1 and by the Max Cloetta Foundation (M.Y.D.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Donath, M.Y., Størling, J., Maedler, K. et al. Inflammatory mediators and islet β-cell failure: a link between type 1 and type 2 diabetes. J Mol Med 81, 455–470 (2003). https://doi.org/10.1007/s00109-003-0450-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-003-0450-y