
Abstract
Mass spectrometry coupled with gas chromatography is the ideal tool for structure elucidation of minute amounts of target components contained in complex mixtures. Derivatization of these targets and GC/MS analyses of the reaction products may provide decisive information about the parent compound. Removal of oxygen-containing functional groups may be particularly advantageous. Even when the transformation of a target into an appropriate derivative needs several steps, and corresponding reactions furnish low yields, derivatization is useful. Females of the shrimp Crangon crangon contain (S)-3-hydroxydecanoic acid amide and 3-hydroxy-8-methyldecanoic acid amide. Ketones in the desert locust Schistocerca gregaria proved to be 3,7-dimethylheptacosan-2-one and 3,7,15-trimethylheptacosan-2-one. Double bond positions in unsaturated macrocyclic lactones, which occur in halictine bees, can be easily assigned upon GC/MS after the addition of dimethyldisulfide even when they occur as mixtures of isomers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
During recent years, tremendous progress has been made in the methodological developments of analytical techniques. This is true for the separation of both volatile and non-volatile compounds by gas chromatography and HPLC, respectively, including enantioselective techniques. At present, the structure elucidation of natural products by NMR spectroscopy can be achieved with only nanogram amounts of material. Despite this enormous progress, the sensitivity of mass spectrometry remains unequalled, and gas chromatography coupled to mass spectrometry (GC/MS) remains the method of choice for the analysis of minute amounts of multicomponent mixtures. Supported by commercially available libraries and databases (Adams 2007; Joulain and König 2001; McLafferty and Stauffer 1989), mass spectrometry is an ideal tool for the detection and quantitation of already known substances, especially when authentic reference compounds are available. In contrast, structure elucidation of unknowns (new natural products), solely based on mass spectrometric techniques, can be extremely difficult if not impossible. This is because the fragmentation of a certain substance by electron impact, i.e., the formation of detectable ions and their relative abundance under experimental conditions can frequently not be predicted. Though there are some general rules in mass spectrometry, NMR spectra can be much better calculated on the basis of given structural parameters. This is the reason why MS is called mass spectrometry, while NMR is a spectroscopic method. Actually, to gain extreme sensitivity, much less (specific) analytical data have to be accepted (a kind of Heisenberg’s uncertainty principle in analytical chemistry: for a good X-ray analysis, the highest amounts of material are necessary). Recent developments in the analytical chemistry of natural products have been briefly reviewed (Francke and Schulz 2010).
Structure elucidation of an unknown (new) target compound, based on GC/MS analysis, will be largely facilitated by principal knowledge on the fragmentation pattern of the corresponding class of compounds (Francke and Schröder 1999; Francke and Kitching 2001; Franke et al. 2009). In addition, derivatization and GC/MS investigations of the respective reaction products may provide further information. Corresponding techniques have been carefully compiled (Attygalle 1998). Even when the transformation of a target compound needs several steps, application of derivatization reactions is useful as the mass spectrum of the derivative may provide decisive information. Even when yields of the procedure are low (e.g., reduction of the tosylate of a secondary alcohol), the extreme sensitivity of GC/MS will insure a useful set of data. The present study deals with the structure elucidation of minute amounts of some carbonyl compounds (embedded in complex matrices) by GC/MS and by micro-reactions followed by GC/MS analyses of the reaction products.
Results and discussion
Mass spectra of oxygen-containing compounds are frequently characterized by most specific, highly intense signals. These are formed by McLafferty rearrangements in ketones or esters and amides or by α-cleavage in ethers, etc. While these diagnostic fragments largely facilitate the assignment of functional groups or even their position in the target compound, their dominance largely hampers the detection of branchings along the chain. In a most elegant approach, Schulz solved the problem by transforming oxygen-containing functional groups into homologous nitriles (Schulz 1997, 1999; Bagnères et al. 1997).
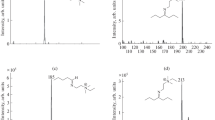
As shown in Fig. 1, short sequences of micro-reactions lead from acids, esters or methyl ethers to nitriles, the mass spectra of which show diagnostic signals that can be attributed to the formation of fragments upon α-cleavage at the methyl branching points along a branched carbon chain. During our investigations on volatile constituents of the shrimp Crangon crangon, we came across a group of β-hydroxycarboxylic acid amides (Fittschen 2001). The major component could be identified to be the new straight chain (S)-3-hydroxdecanoic acid amide; however, a homolog (11 carbon atoms, M = 201 upon CI-GC/MS) proved to show a branched chain because of its relatively low retention index. While the two most abundant fragments in the spectrum at m/z 88 (α-cleavage) and m/z 59 (McLafferty rearrangement) revealed the nature of the functional groups (Fig. 2a), the branching point along the chain could not be assigned as no corresponding diagnostic signal could be detected.

Formation of nitriles to facilitate location of methyl branchings in long chain mono functionalized aliphatics upon EI-GC/MS analyses (after Schulz 1999)

70 eV EI-mass spectra of 3-hydroxy-8-methyldecanoic acid amide (a) and 3-acetoxy-8-methyldecanoic acid nitrile (b) [Mass Spectrometer VG 70-250 SE, Vacuum Generators, Manchester, UK]
Heating of the natural extract with acetic anhydride in the presence of pyridine (60°C, 2 days, screw cap vial) transformed the target compound into the corresponding 3-acetoxynitrile. The mass spectrum of this derivative exhibited key fragments at m/z 136 (M+-AcOH-29) and m/z 108 (M+-AcOH-57) that clearly revealed a methyl branching at position 8 (Fig. 2b). Thus, the natural product was shown to be 3-hydroxy-8-methyl decanoic acid amide, and the structure was proven by independent synthesis. It should be noted that, in contrast to the amides, the enantiomers of 3-acetoxynitriles were particularly well resolved upon enantioselective gas chromatography using modified cyclodextrins.
Following the ideas of Schulz (Fig. 1), a successful approach to circumvent the formation of highly dominating oxygen-containing fragments in the mass spectra of carbonyl compounds is the removal of oxygen. The carbonyl group may be converted into a methylene group by either a short sequence of reduction steps via tosylates of corresponding alcohols (Fig. 3 includes the reduction of a secondary alcohol as the starting material) or via a tosyl hydrazone.

Transformation of ketones and secondary alcohols to deuterated hydrocarbons
The use of lithium alumina tetradeuteride furnishes a labeled hydrocarbon that can be easily distinguished from non-labeled congeners present in the natural extract. As the fragmentation pattern of branched hydrocarbons is well investigated (see Doolittle et al. 1995 and references cited therein), branching positions in the formed hydrocarbons can be easily assigned. In surface extracts of the desert locust Schistocerca gregaria, traces of two oxygen-containing components could be detected among vast amounts of straight chain and branched hydrocarbons (Fig. 4).

Gas chromatogram of a surface pentane extract of the locust Schistocerca gregaria showing hydrocarbons and two oxygen-containing components. Declaration: 23 = tricosane etc., 3-Me-26 = 3-methylhexacosane etc., br 34 = branched tetratriacontane, Tri-Me-33 = trimethyltritriacontane (30 m, 0.25 mm id fused silica capillary, DB5, temperature progr. 150–300°C, 3°C/min, He)
Because of the dominating signal at m/z 72 (McLafferty fragment) in the EI-mass spectra and relatively low abundant fragments at m/z 57 (acyl ion), the structures of the two compounds were suggested to be 3-methyl-2-alkanones (and not 3-alkanones). The spectrum of the natural C30 product (Fig. 5a) did not provide reliable hints for additional branching. In contrast, the 2,2-dideuterio-hydrocarbon, formed after micro-scale reduction of the tosylhydrazone according to Fischer et al. (1965), proved the formed hydrocarbon to be 2,2-dideuterio-3,7,15-trimethylheptacosane because of the presence of diagnostic signals at m/z 393 (M+-31 as expected), m/z 323, indicating a branching at C7, and m/z 255, revealing another branching at C15 (which was supported by the presence of the complementary signal at m/z 197/196) (Fig. 5b).

70 eV EI-mass spectra of 3,7,15-trimethylnonacosan-2-one (a) and 2,2-dideuterio-3,7,15-trimethylnonacosane (b) [Mass Spectrometer VG 70-250 SE, Vacuum Generators, Manchester, UK]
Consequently, the structure of the parent ketone was assigned to be 3,7,15-trimethylheptacosan-2-one. The C29-ketone was shown to be 3,7-dimethylheptacosan-2-one (Brunnemann 1996). In addition, very small amounts of 3,7-dimethylpentacosan-2-one could be detected. The structures were verified by independent synthesis; however, the configuration at the stereogenic centers are yet unknown. It should be noted that the reaction could be successfully carried out using the natural extract. It is interesting to compare the structures of these ketones with those known from the cockroach Blatella germanica: 3,11-dimethylhepacosan-2-one and 3,11-dimethylnonacosan-3-one, respectively, which are components of a female-specific contact pheromone (Schal et al. 1990). At this point, relations between cockroaches and locusts become evident.
As already mentioned, esters form another class of compounds providing distinct EI-mass spectra: methyl and ethyl esters of straight chain saturated fatty acid esters show pronounced McLafferty fragments at m/z 74 resp. m/z 88 as well as signals at m/z 87 resp. m/z 101 resulting from cleavage between C3 and C4 of the chain (Fig. 6a).

The two different ways of McLafferty rearrangements of carboxylic acid esters
While in esters of short chain alcohols the McLafferty rearrangement takes place predominantly at the acid side, in wax-type esters migration of the hydrogen involved in the McLafferty rearrangement occurs from the alcohol side (Fig. 6b) (Francke et al. 2000). As a result, wax-type esters show diagnostic fragments comprising the protonated acid, the corresponding acylium ion and an even-numbered alkene fragment representing the alcohol part (Fig. 7a). In saturated, straight chain esters, the signal representing the protonated acid is more abundant than that of the acylium ion. However, proportions may be reverse in esters of methyl carbinols when the alcohol shows a medium chain length (Fig. 7b). In addition, the mass spectrum may show a pronounced signal of the acid. It should be noted that wax-type esters such as octyl tetradecanoate (Fig. 7a) and its homolog, 1-methyloctyl tetradecanoate, (Fig. 7b) co-elute on several GC columns.

70 eV EI-mass spectra of octyl tetradecanoate (a) and 1-methyloctyl tetradecanoate (b) [Mass Spectrometer VG 70-250 SE, Vacuum Generators, Manchester, UK]
While fragmentation patterns of unsaturated fatty acid esters have been described (Francke et al. 2000), structural assignments of unsaturated macrocyclic lactones still need to be discussed. Macrolides form characteristic mixtures, showing ontogenetic changes in eusocial halictine bees (Ayasse et al. 1990, 1993). A typical EI-mass spectrum of docosanolide is shown in Fig. 8a. The spectrum is characterized by a pronounced signal for the molecular ion, a rather intense one at M+-18 and a less abundant, but significant, fragment at M+-60.

70 eV EI-mass spectra of the macrocyclic lactones docosanolide (a) and docosenolide (b) [Mass Spectrometer VG 70-250 SE, Vacuum Generators, Manchester, UK]
The spectrum of docosenolide (Fig. 8b) is even less specific and, similar to alkenes, there is no information about the position of the double bond. This picture changes dramatically after the addition of dimethyl disulfide (Lübke 1990). Methylthiolation proved to be a decisive tool for the determination of double bond positions in many natural products (see Attygalle 1998 and references cited therein). The EI-mass spectrum of the adduct of dimethyldisulfide to an unsaturated macrolide (Fig. 9) is characterized by two highly abundant diagnostic signals. Similar to a wax-type ester made up of a fatty acid and an unsaturated alcohol (Francke et al. 2000), the product obtained from the reaction of an unsaturated macrolide and dimethyldisulfide undergoes a McLafferty rearrangement involving the hydrogen at the β-position to the ring-oxygen. After cleavage between the two thiomethyl groups, the compound yields two fragments that are stabilized upon loss of methlysulfide and a hydrogen from the “alcohol side” and loss of water, methylsulfide and hydrogen from the “acid side”, respectively.

70 eV EI-mass spectrum of the adduct of dimethyldisulfide to 13-docosenolide, 13,14-di(methylthio)docosanolide [Mass Spectrometer VG 70-250 SE, Vacuum Generators, Manchester, UK]
The depicted spectrum is produced by the dimethyldisulfide adduct of docos-13-enolide giving rise to m/z 192 (alcohol fragment) and m/z 122 (acid fragment). Shifting of the double bond by two positions toward the carboxylic carbon (not unusual in rows of acetogenins) will shift m/z 122 down to m/z 94 and m/z 192 up to m/z 220. On the other hand, docos-15-enolide would yield the diagnostic fragments m/z 150 (acid fragment) and m/z 164 (alcohol fragment). This characteristic fragmentation pattern enables the reliable determination of double bond positions even in mixtures of unsaturated macrolides. It should be noted that an authentic reference compound will be needed when the stereochemistry of the double bond in an unsaturated macrolide must be determined. The gas chromatogram depicted in Fig. 10 shows a typical pattern of macrolides contained in the Dufour’s gland of the halictine bee, Lasioglossum malachurum, and its quantitative composition.

Gas chromatogram of a surface pentane extract of the halictine bee Lasioglossum malachurum showing a typical macrolide pattern. Note that the unsaturated macrolides (indicated by e.g., “C20-enolide”) represent at least two unsaturated macrolides (25 m, 0.25 mm id fused silica capillary CP-Sil 8, temperature progr. 120–300°C, 5°C/min, He)
References
Adams RP (2007) Identification of essential oil components by gas chromatography/mass spectrometry, 4th edn. Allured Publishing Corporation, Carol Stream
Attygalle AB (1998) Microchemical techniques In: Millar JG, Haynes KF (eds) Methods in chemical ecology I. Chemical methods. Kluwer, Dordrecht, The Netherlands
Ayasse M, Engels W, Hefetz A, Lübke G, Francke W (1990) Ontogenetic patterns in amounts and proportions of Dufour’s gland volatile secretions in virgin and nesting queens of Lasioglossum malachurum (Hymenotera: Halictidae). Z Naturforsch 45C:709–714
Ayasse M, Engels W, Hefetz A, Tengö J, Lübke G, Francke W (1993) Ontogenetic patterns of volatiles identified in Dufour’s gland extracts from queens and workers of the primitive eusocial halictine bee, Lasioglossum malachurum (Hymenoptera: Halictidae). Insectes Soc 40:41–58
Bagnères A-G, Trabalon M, Blomquist G, Schulz S (1997) Waxes of the social spider Anelosimus eximus (Aranae, Theridiidae): abundance of novel n-propyl esters of long-chain methyl-branched fatty acids. Arch Insect Biochem Physiol 36:295–314
Brunnemann U (1996) Identifzierung und Synthese von Heuschreckenpheromonen, PhD-thesis, University of Hamburg, 145 pp
Doolittle RE, Proveaux AT, Alborn HT, Heath RR (1995) Quadrupole storage mass spectrometry of mono- and dimethylalkanes. J Chem Ecol 21:1677–1695
Fischer M, Pelah Z, Williams DH, Djerassi C (1965) Mechanismus der Reduktion von Tosylhydrazonenen mit komplexen Metallhydriden. Chem Ber 98:3236–3250
Fittschen UEA (2001) Identifizierung von Naturstoffprofilen aus der Nordseekrabbe Crangon crangon. PhD-thesis, University of Hamburg, 228 pp
Francke W, Kitching W (2001) Spiroacetals in insects. Curr Org Chem 5:233–251
Francke W, Schröder W (1999) Bicyclic acetals in systems of chemical communication. Curr Org Chem 3:407–443
Francke W, Schulz S (2010) Pheromones of terrestrial invertebrates. In: Mander L, Liu H-W (eds) Comprehensive natural products chemistry II, Vol 4. Elsevier, Oxford (Mori K guest ed), pp 153–224
Francke W, Lübke G, Schröder W, Reckziegel A, Imperatriz-Fonseca V, Kleinert A, Engels E, Hartfelder K, Radke R, Engels W (2000) Identification of oxygen containing volatiles in cephalic secretions of workers of Brazilian stingless bees. J Braz Chem Soc 11:562–571
Franke S, Ibarra F, Schulz CM, Twele R, Poldy J, Barrow RA, Peakall R, Schiestl FP, Francke W (2009) The discovery of 2,5-dialkylcyclohexan-1,3-diones as a new class of natural products. Proc Natl Acad Sci USA 106:8877–8882
Joulain D, König WA (2001) The Atlas of spectral data of sesquiterpene hydrocarbons. Verlag, Hamburg
Lübke G (1990) Untersuchungen flüchtiger Inhaltsstoffe sozialer Bienen und Wespen. PhD-thesis, University of Hamburg, 131 pp
McLafferty FW, Stauffer DB (1989) The Wiley/NBS registry of mass spectral data. Wiley, NY
Schal C, Burns EL, Jurenka RA, Blomquist G (1990) A new component of the female sex pheromone of Blattella germanica (L.) (Dictyoptera: Blatellidae) and interaction with other pheromone components. J Chem Ecol 16:1997–2008
Schulz S (1997) Mass spectrometric determination of methyl group positions in long chain methyl esters and alcohols via nitriles. J Chem Soc Chem Commun 10:969–970
Schulz S (1999) Structural diversity of surface lipids from spiders. In: Diederichsen U, Lindhorst TK, Westermann B, Wessjohann LA (eds) Bioorganic chemistry. Wiley, Weinheim, pp 1–7
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Francke, W. Structure elucidation of some naturally occurring carbonyl compounds upon coupled gas chromatography/mass spectrometry and micro-reactions. Chemoecology 20, 163–169 (2010). https://doi.org/10.1007/s00049-010-0048-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00049-010-0048-0