
Abstract
Modification of daunorubicin on its NH2 moiety is a well-established synthetic approach to obtain novel derivatives of this compound. Moreover, the daunosamine moiety of this antibiotic is considered to be the most sensitive part of a molecule in terms of biological response to chemical transformations. Using simple and effective synthetic techniques, namely, alkylation under phase-transfer catalysis and an amine addition across an activated multiple bond, a series of daunorubicin derivatives retaining the amine functionality have been obtained, which could also be used as potential precursors for further transformations.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Daunorubicin, like other anthracycline antibiotics, is presently the first-line drug in the therapy of oncological diseases, particularly leukemias and solid tumors [1]. All such drugs exhibit very high antimitotic activity which is unfortunately accompanied by a low selectivity, high cardiotoxicity as well as immunosuppressive, embryotoxic, mutagenic, and teratogenic effects [2]. To reduce their adverse action, studies on synthetic modification of these compounds began as soon as they were developed in the 1960s. By now, hundreds of derivatives of daunorubicin and its analogs had been synthesized, however the problem of the drug selectivity has not yet been solved.
There is a plethora of reviews devoted to synthetic approaches to modifying anthracycline antibiotics and, above all, daunorubicin as the most available and low-cost of them, and often the methods of modification of daunomycine and daunosamine parts of the molecule differ significantly, resulting in the need to consider these approaches separately [3]. Chemical modification of daunorubicin and its derivatives is rather challenging and in some cases requires carrying out hydrolysis of the parent molecule to give aglycon and daunosamine, synthetic transformations of both fragments and their final assembly into the target molecule [4]. Such way is rather laborious and includes 6–8 chemical steps, thus affecting significantly the yields of the desired compounds, and does not provide an access to large libraries of derivatives to perform a rapid primary screening and identify lead compounds among them.
In the present study, we propose simple methods for the modification of the molecule of daunorubicin on the amine nitrogen atom, allowing rapid and effective synthesis of novel anthracycline antibiotics not only with the aim of assessing their primary cytotoxicity, but also of using them as starting materials (building blocks) for further transformations. All compounds obtained by us comprise the amine nitrogen atom in the daunosamine moiety, i.e., the amine functionality remains unchanged, which is crucial to retaining high antiproliferative activity and some other important properties of the cancerolytic agents [5].
The importance of retaining the amine functionality in the structure of anthracycline antibiotics stems from the mechanism of action of such cancerolytic agents on a tumor cell. Intercalation of daunorubicin into the DNA strand implies the incorporation of its aglycon residue between pairs of nitrogenous bases such as cytosine–guanine and formation of hydrogen bonds between the hydroxyl group of daunomycine and guanine, thereby changing the conformation of the aminoglycoside moiety and providing the close fit of the intercalator to the DNA right minor groove [1]. And finally, electrostatic interaction between positively charged amino group and the oxygen atom of the DNA phosphate moiety proves to be critical to stabilization of daunorubicin–DNA duplex [6].
The amino group also plays an important role in the membrane transport of a molecule due to the ability of the latter to bind to negatively charged membrane phospholipids [7]. Blocking of the amine functionality significantly reduces the cytotoxicity of compounds and their ability to bind to DNA as confirmed, e.g., for the known daunorubicin amides [8]. Studies of the influence of the NH2 moiety modifications on alleviating mutagenicity of compounds without changing their high antiproliferative activity have also been conducted [9, 10].
As it has been shown earlier by us and our colleagues, functionalization of the amino group of daunorubicin in order to obtain N-alkylated derivatives is a well-established method for modifying anthracyclines [11,12,13].
Results and discussion
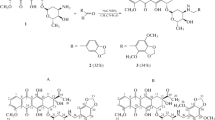
We found that NH2 group of daunorubicin can be alkylated with active halogenated reagents in a similar way as it had been described earlier for twinning anthracyclines with p-xylylenedibromide [14, 15]. In our case, we used compounds comprising an activated halogen atom, e.g., propargyl bromide (derivatives 1a, b), allyl bromide (derivatives 1c, d), 3-bromo-1-phenyl-1-propene (derivative 1e), as well as bromoacetic acid ethyl ester (derivative 1f). With 2-bromo-1-phenylacetone and 2-bromoethanol the reaction proceeded so vigorously that only products of anthracycline degradation instead of the desired compounds were observed (Scheme 1).

Synthesis of compounds 1a–1f
As in most cases of alkylation of the primary amine nitrogen atom with haloalkanes, daunorubicin also produced a mixture of mono- and dialkylated products. The products of the reaction with propargyl- and allyl bromides can be easily separated by chromography to give two desired building blocks in the ratio of mono- and dialkylated compounds ~5: 1.
It should be noted that the optimal reagent ratio is daunorubicin: halogenated derivative = 1: 1.2 ÷ 1.3. Any shift in this ratio reduces the yields of both mono- and dialkylated products; increasing or decreasing the proportion of the halogenated derivative does not result in a corresponding increase or decrease of the proportion of dialkylaminodaunorubicin.
When using bromoacetate acid ester, only mono-derivative 1f is obtained, the di-product might also be formed, but is lost in chromatographical purification. The similar situation is observed for the product formed from 3-bromo-1-phenyl-1-propene, but in this case only di-product 1e can be isolated.
Individual monoalkylated products can be used in the alkylation reaction over again with the same or another alkyl halide; in this way, the mixed product 2 was obtained (Scheme 2).

Synthesis of compound 2
All alkylation reactions of daunorubicin and its derivatives are carried out under phase-transfer catalysis (PTC) conditions using freshly calcined potassium carbonate as a base and in the absence of a phase-transfer catalyst. This reaction is very sensitive to the quality of the base in use: potassium carbonate must necessarily be freshly calcined, otherwise the product yields will decrease dramatically. Replacing potassium carbonate with sodium carbonate or cesium carbonate, or its use in larger amounts than specified have a similar result. Heating the reaction mixture in order to accelerate the process dramatically decreases the yields of the products right up to their complete resinification. The low yield of compounds 1b and 1d can be explained by their ability to react with alkyl bromides with to formation of quaternary ammonium salts.
Another approach to obtain derivatives of daunorubicin functionalized on the nitrogen atom comprises the aza-Michael reaction [16], i.e., the addition of anthracycline across an activated multiple bond. Such approach had been proposed earlier to obtain conjugates of daunorubicin with cyclic ketones containing conjugated C=C, C=O bonds [17]. We found that the amino group of the cancerolytic agent readily reacted with acrylonitrile (derivative 3a), methacrylate (derivative 3b), dimethylacetylenedicarboxylate (derivative 3c), and ethylpropiolate (derivative 3d).
The addition of the daunorubicin base across the activated multiple bonds was carried out under standard conditions at 20 °C in the methanol (derivative 3a–3c) or ethanol (derivative 3d) solution in the absence of a catalyst. The daunorubicin free base was obtained according to the previously reported procedure [17] immediately prior to the reaction from its hydrochloride, since the free base undergoes degradation upon storage. More than 2 mol of an electrophile per 1 mol of anthracycline were used in the reaction (with equimolar ratio of reactants the yields of the products decreased markedly) (Scheme 3).

Synthesis of compounds 3a–3d
Only in the case of ethylpropiolate we managed to isolate the di-addition product 3d, while the other derivatives were the products of monoaddition.
Compounds 1–3 were tested in the lung carcinoma (А549), rhabdomyosarcoma (RD), colorectal carcinoma (HCT116), and embryonal renal epithelium (HEK293) cell lines. The results of the primary screening of cytotoxicity are summarized in Table 1. Also, the cytotoxicity data for the starting daunorubucin are given, which was a reliable reference compounds in our case.
As can be seen from the above data, almost all of the compounds show moderate toxicity values. The adduct 1e of 3-bromo-1-phenyl-1-propene and daunorubicin was found to have especially low toxicity towards tumor cells. It had low affinity for tumor tissues compared to the healthy ones. This is probably due to the steric hindrances caused by phenyl groups, which hinder the ability the derivative to bind to DNA. Indeed, for anthracycline analogs bearing bulky substituents at the nitrogen atom, the mechanism of inhibition of topoisomerase II involved in the DNA cleavage processes differ significantly from that described, e.g., for daunorubicin [18].
Among the products obtained, compound 1c is the most promising in terms of studying its biological activity. The compound derived from allyl bromide and anthracycline exhibits higher efficiency towards rhabdomyosarcoma cells, namely, 1.3 times higher than that of the starting daunorubicin, and also high potency towards colorectal carcinoma cells, which is 8 times higher than in the case of the starting cancerolytic drug. This can be explained by the structure of the substituent in the nitrogen atom. In the case of compounds 1c, are likely to play two factors, these are the smallest steric obstacles, as well as the presence of a double bond. These factors probably facilitate the incorporation of compound 1c into the DNA-topoisomerase complex and prevent further DNA synthesis. Further studies of acute toxicity of adduct 1c will make it possible to reach an unequivocal conclusion about the prospects of this compounds for the treatment of the colorectal tumors.
Conclusion
All compounds obtained by us are secondary or tertiary amines bearing various functionalities and represent building blocks for subsequent synthesis of daunorubicin derivatives retaining the amine moiety. The proposed methods for functionalizing daunorubicin open the way to the synthesis of entire libraries of its novel derivatives, which can be readily obtained from the above compounds, for instance, via the addition of azides or nitrile oxides across multiple bonds, metathesis reaction, etc. according to the known procedures.
Experimental/Materials and methods
NMR spectra were recorded on a Bruker AV-400 spectrometer in CDCl3 solutions using residual proton signals of the deuterated solvent as an internal reference (1H, 13C). 13C NMR spectra were recorded in JMODECHO mode, the signals of carbon atoms with even and odd number of protons have opposite polarity. Reactions were monitored by TLC on alumina TLC plates w/UV254. The chromatographic purification of compounds was carried out on a Macherey-Nagel silica gel (MN Kieselgel 60, 70–230 mesh) using the solvent systems CHCl3: MeOH and CHCl3: MeOH: NH3(aq). IR samples were measured as KBr pellets with FT-IR Spectrometer (InfraRed Bruker Tensor 37) in the 400–4000 cm−1 range with 2 cm−1 resolution, 32 scans. High resolution mass spectra (HR MS) were measured on a Bruker micrOTOF II instrument using electrospray ionization (ESI) [19]. The measurements were done in a positive ion mode (interface capillary voltage −4500 V); mass range from m/z 50 to m/z 3000; external or internal calibration was done with ESI Tuning Mix, Agilent. A syringe injection was used for solutions in acetonitrile (flow rate 4 μL/min). Nitrogen was applied as a dry gas; interface temperature was set at 180 or 200 °C. HR MS were recorded in the Department of Structural Studies of Zelinsky Institute of Organic Chemistry, Moscow. The biological testing of anticancer activity was performed in the Institute of Physiologically Active Compounds, Russian Academy of Sciences, Chernogolovka. Daunorubicin hydrochloride was purchased from Aldrich. Commercially available starting materials were used without further purification. All obtained daunorubicin derivatives are decomposed on heating above 200 °C.
General procedure for the alkylation of the daunorubicin
Daunorubicin hydrochloride (100 mg, 0.18 mmol) or compounds 1a, 1c (100 mg, 0.18 mmol) were dissolved in a mixture of N,N-dimethylformamide (DMF) and CH2Cl2 (1:1 v/v; 3 mL). Then, a powder of K2CO3 (49 mg, 0.36 mmol) and an appropriate bromide (0.22 ÷ 0.23 mmol) were added. The reaction mixture was stirred at room temperature for 20–28 h. The progress of the reaction was monitored by TLC (CHCl3: MeOH: NH3(aq) at v/v ratios 85: 14: 1). After the reaction was complete, the reaction mixture was diluted with CH2Cl2 (50 mL) and poured into water (50 mL). The organic layer was separated and washed with water until neutral pH, dried over anhydrous Na2SO4, and evaporated under reduced pressure. The crude products were purified by column chromatography, eluted with CHCl3: MeOH at v/v ratios from 100: 1 to 10: 1 (compounds 1c, 1d, 1e, 2) or CHCl3: MeOH: NH3(aq) at v/v ratios from 100: 1: 0.1 to 10: 1: 0.1 (compounds 1a, 1b, 1f).
(8S,10S)-8-Acetyl-6,8,11-trihydroxy-10-{[(2R,4S,5S,6S)-5-hydroxy-6-methyl-4-(prop-2-yn-1-ylamino)tetrahydro-2H-pyran-2-yl]oxy}-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione (1a): red solid, yield 81%; IR (KBr) ν = 3480 (br, m) (νOH), 3283 (br, m) (νNH), 2972 (sh, m), 2934 (m) and 2843 (sh, m) (all three νCH), 2249 (vw) and 2120 (vw) (both νC≡CH), 1716 (s) (νC=O), 1617 (s) and 1578 (s) (both νC=C), 1413 (s) and 1352 (s) (both δOH), 1286 (vs), 1232 (s) and 1208 (vs) (all three νC–O), 1118 (s), 984 (vs) (δC=C), 816 (m), 792 (m), 464 (w) cm−1; 1H NMR (CDCl3, 400 MHz,):δ = 14.05 (1H, br. s, OH-6), 13.67 (1H, br. s, OH-11), 7.98 (1H, d, J = 7.6 Hz, H-1), 7.77 (1H, t, J = 8.0 Hz, H-2), 7.38 (1H, d, J = 8.0 Hz, H-3), 5.53 (1H, br. s, H-1′), 5.27 (1H, br. s, H-7), 4.69 (1H, br. s, OH-9), 4.08 (4H, br. s, H-15, H-5′), 3.66 (1H, br. s, H-4′), 3.42 (2H, d, J = 4.0 Hz, N–CH2), 3.16 (1H, d, J = 18.8 Hz, H-10), 3.13–3.09 (1H, m, H-3′), 2.87 (1H, d, J = 18.8 Hz, H-10), 2.43 (3H, s, H-14), 2.36 (1H, d, J = 14.6 Hz, H-8), 2.20 (1H, t, J = 2.2 Hz, C≡CH), 2.10 (1H, dd, J = 4.0, 14.6 Hz, H-8), 1.82–1.67 (2H, m, H-2′), 1.38 (3H, d, J = 8.0 Hz, H-6′);13C NMR (CDCl3, 100 MHz,):δ = 211.77 (C, C-13), 186.71 (C, C-12), 186.38 (C, C-5), 160.82 (C, C-4), 156.23 (C, C-6), 155.61 (C, C-11), 135.53 (CH, C-3), 135.24 (C, C-12a), 134.28 (C, C-6a), 134.06 (C, C-4a), 120.62 (C, C-10a), 119.58 (CH, C-2), 118.23 (CH, C-1), 111.15 (C, C-11a), 111.01 (C, C-5a), 100.55 (CH, C-1′), 81.05 (C, C≡CH), 76.91 (C, C-9), 71.98 (CH, C≡CH), 69.51 (CH, C-7), 66.49 (CH, C-4′, C-5′), 56.49 (CH3, C-15), 51.09 (CH, C-3′), 34.70 (CH2, NCH2), 34.32 (CH2, C-10), 33.14 (CH2, C-8), 29.79 (CH2, C-2′), 24.67 (CH3, C-14), 16.92 (CH3, C-6′); HRMS (ESI) m/z found 566.2025, 588.1841, 604.1585; calculated for C30H31NO10 566.2021 (M + H+), 588.1840 (M + Na+), 604.1580 (M + K+).
(8S,10S)-8-Acetyl-6,8,11-trihydroxy-10-{[(2R,4S,5S,6S)-5-hydroxy-6-methyl-4-(di(prop-2-yn-1-yl)amino)tetrahydro-2H-pyran-2-yl]oxy}-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione (1b): red solid, yield 15%; IR (KBr) ν = 3482 (br, m) (νOH), 3280 (br, m) (νNH), 2974 (sh, m), 2936 (m) and 2842 (sh, m) (all three νCH), 2283 (vw), 2250 (vw), 2120 (vw) and 2107 (vw) (all four νC≡CH), 1716 (s) and 1669 (m) (both νC=O), 1618 (s) and 1578 (s) (both νC=C), 1414 (s) and 1352 (s) (both δOH), 1286 (vs), 1232 (s) and 1210 (vs) (all three νC–O), 1124 (s), 985 (vs) (δC=C), 792 (m), 662 (w) 464 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 13.89 (1H, br. s, OH-6), 13.13 (1H, br. s, OH-11), 7.92 (1H, d, J = 7.8 Hz, H-1), 7.73 (1H, t, J = 7.8 Hz, H-2), 7.35 (1H, d, J = 8.0 Hz, H-3), 5.55 (1H, br. s, H-1′), 5.24 (1H, br. s, H-7), 4.66 (1H, br. s, OH-9), 4.05 (4H, br. s, H-15, H-5′), 3.71 (1H, br. s, H-4′), 3.57–3.48 (4H, m, N–CH2), 3.11 (1H, d, J = 17.6 Hz, H-10), 2.86–2.83 (1H, m, H-3′), 2.80 (1H, d, J = 18.0 Hz, H-10), 2.40 (3H, s, H-14), 2.32 (1H, d, J = 14.8 Hz, H-8), 2.19 (2H, br. s, C≡CH), 2.07 (1H, dd, J = 4.0, 14.8 Hz, H-8), 1.92–1.83 (2H, m, H-2′), 1.37 (3H, d, J = 8.0 Hz, H-6′); 13C NMR (CDCl3, 100 MHz,): δ = 211.70 (C, C-13), 186.54 (C, C-12), 186.22 (C, C-5), 160.72 (C, C-4), 156.16 (C, C-6), 155.45 (C, C-11), 135.46 (CH, C-3), 135.08 (C, C-12a), 134.22 (C, C-6a), 134.00 (C, C-4a), 120.46 (C, C-10a), 119.47 (CH, C-2), 118.18 (CH, C-1), 111.01 (C, C-11a), 110.91 (C, C-5a), 100.12 (CH, C-1′), 78.02 (C, C≡CH), 76.53 (C, C-9), 73.55 (CH, C≡CH), 69.15 (CH, C-7), 66.64 (CH, C-4′, C-5′), 56.42 (CH3, C-15), 54.70 (CH, C-3′), 38.45 (CH2, NCH2), 34.61 (CH2, C-10), 33.09 (CH2, C-8), 29.76 (CH2, C-2′), 24.61 (CH3, C-14), 16.90 (CH3, C-6′); HRMS (ESI) m/z found 604.2174, 626.1982; calculated for C33H33NO10 604.2177 (M + H+), 626.1997 (M + Na+).
(8S,10S)-8-Acetyl-6,8,11-trihydroxy-10-{[(2R,4S,5S,6S)-5-hydroxy-6-methyl-4-(prop-2-en-1-ylamino)tetrahydro-2H-pyran-2-yl]oxy}-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione (1c): red solid, yield 80%; IR (KBr) ν = 3480 (br, m) (νOH), 3327 (br, m) (νNH), 2974 (sh, m), 2933 (m) and 2843 (sh, m) (all three νCH), 1716 (s) (νC=O), 1618 (s) and 1578 (s) (both νC=C), 1414 (s) and 1352 (s) (both δOH), 1286 (vs), 1232 (s) and 1209 (vs) (all three νC–O), 1119 (s), 985 (vs) (δC=C), 817 (m), 792 (m), 464 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 14.01 (1H, br. s, OH-6), 13.32 (1H, br. s, OH-11), 8.05 (1H, d, J = 7.7 Hz, H-1), 7.81 (1H, t, J = 8.0 Hz, H-2), 7.42 (1H, d, J = 8.4 Hz, H-3), 5.91 (1H, dd, J = 10.3, 16.9 Hz, CH=CH2), 5.55 (1H, br. d, J = 3.4 Hz, H-1′), 5.31 (1H, br. s, H-7), 5.20 (2H, dd, J = 10.1, 17.2 Hz, CH=CH2), 4.64 (1H, br. s, OH-9), 4.15–4.06 (4H, br. s, H-15, H-5′), 3.77 (1H, br. s, H-4′), 3.44 (2H, d, J = 6.0 Hz, NCH2), 3.25 (1H, d, J = 18.8 Hz, H-10), 3.05 (1H, d, J = 12.4 Hz, H-3′) 2.99 (1H, d, J = 18.8 Hz, H-10), 2.44 (3H, s, H-14), 2.37 (1H, d, J = 14.9 Hz, H-8), 2.13 (1H, dd, J = 4.1, 14.8 Hz, H-8), 1.94 (1H, dd, J = 4.0, 14.1 Hz, H-2′), 1.76 (1H, dd, J = 4.7, 13.0 Hz, H-2′), 1.38 (3H, d, J = 6.6 Hz, H-6′); 13C NMR (CDCl3, 100 MHz,): δ = 211.73 (C, C-13), 186.76 (C, C-12), 186.39 (C, C-5), 160.85 (C, C-4), 156.33 (C, C-6), 155.65 (C, C-11), 135.53 (CH, C-3), 135.28 (C, C-12a), 134.37 (CH, CH=CH2), 134.29 (C, C-6a), 134.17 (C, C-4a), 120.65 (C, C-10a), 119.59 (CH, C-2), 118.24 (CH, C-1), 117.80 (CH2, CH=CH2), 111.14 (C, C-11a), 111.03 (C, C-5a), 100.86 (CH, C-1′), 77.24 (C, C-9), 69.63 (CH, C-7), 66.63 (CH, C-4′), 66.27 (CH, C-5′), 56.51 (CH3, C-15), 54.72 (CH, C-3′), 51.12 (CH2, NCH2), 34.67 (CH2, C-10), 33.15 (CH2, C-8), 28.27 (CH2, C-2′), 24.67 (CH3, C-14), 17.06 (CH3, C-6′); HRMS (ESI) m/z found 568.2169; calculated for C30H33NO10 568.2177 (M + H+).
(8S,10S)-8-Acetyl-6,8,11-trihydroxy-10-{[(2R,4S,5S,6S)-5-hydroxy-6-methyl-4-(di(prop-2-en-1-yl)amino)tetrahydro-2H-pyran-2-yl]oxy}-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione (1d): red solid, yield 16%; IR (KBr) ν = 3477 (br, m) (νOH), 2975 (sh, m), 2936 (m) and 2841 (sh, m) (all three νCH), 1717 (s) (νC=O), 1618 (s) and 1579 (s) (both νC=C), 1414 (s) and 1352 (s) (both δOH), 1287 (vs), 1232 (s) and 1210 (vs) (all three νC–O), 1126 (s), 987 (vs) (δC=C), 811 (m), 792 (m), 463 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 13.95 (1H, br. s, OH-6), 13.23 (1H, br. s, OH-11), 8.01 (1H, d, J = 7.9 Hz, H-1), 7.78 (1H, t, J = 8.0 Hz, H-2), 7.39 (1H, d, J = 8.5 Hz, H-3), 5.91 (2H, dd, J = 10.0, 17.1 Hz, CH=CH2), 5.56 (1H, br. d, J = 3.4 Hz, H-1′), 5.27 (1H, br. s, H-7), 5.12 (4H, br. d, J = 15.3 Hz, CH=CH2), 4.71 (1H, br. s, OH-9), 4.09 (3H, s, H-15), 4.03 (1H, q, J = 6.4 Hz, H-5′), 3.71 (1H, br. s, H-4′), 3.22 (4H, br. d, J = 6.4 Hz, NCH2), 3.17 (1H, d, J = 19.0 Hz, H-10), 2.96 (1H, d, J = 12.0 Hz, H-3′) 2.87 (1H, d, J = 18.9 Hz, H-10), 2.42 (3H, s, H-14), 2.35 (1H, d, J = 14.6 Hz, H-8), 2.09 (1H, dd, J = 4.0, 14.8 Hz, H-8), 1.86–1.80 (2H, m, H-2′), 1.38 (3H, d, J = 6.6 Hz, H-6′); 13C NMR (CDCl3, 100 MHz,): δ = 211.75 (C, C-13), 186.77 (C, C-12), 186.42 (C, C-5), 160.85 (C, C-4), 156.27 (C, C-6), 155.64 (C, C-11), 135.56 (CH, C-3), 135.27 (C, C-12a), 135.17 (CH, CH=CH2), 134.28 (C, C-6a), 134.09 (C, C-4a), 120.65 (C, C-10a), 119.61 (CH, C-2), 118.26 (CH, C-1), 117.04 (CH2, CH=CH2), 111.17 (C, C-11a), 111.04 (C, C-5a), 100.68 (CH, C-1′), 76.69 (C, C-9), 69.68 (CH, C-7), 66.63 (CH, C-4′), 66.51 (CH, C-5′), 56.52 (CH3, C-15), 51.93 (CH, C-3′), 48.28 (CH2, NCH2), 34.72 (CH2, C-10), 33.13 (CH2, C-8), 29.73 (CH2, C-2′), 24.69 (CH3, C-14), 16.94 (CH3, C-6′); HRMS (ESI) m/z found 608.2488; calculated for C33H37NO10 608.2490 (M + H+).
(8S,10S)-8-Acetyl-6,8,11-trihydroxy-10-{[(2R,4S,5S,6S)-5-hydroxy-6-methyl-4-(di(3-phenylprop-2-en-1-yl)amino)tetrahydro-2H-pyran-2-yl]oxy}-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione (1e): red solid, yield 78%; IR (KBr) ν = 3479 (br, m) (νOH), 3024 (sh, m), 2973 (sh, m), 2935 (m) and 2840 (sh, m) (all four νCH), 1717 (s) (νC=O), 1617 (s) and 1578 (s) (both νC=C), 1414 (s) and 1352 (s) (both δOH), 1286 (vs), 1232 (s) and 1209 (vs) (all three νC–O), 1125 (s), 987 (vs) (δC=C), 793 (m), 764 (m), 694 (m), 464 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 13.97 (1H, br. s, OH-6), 13.22 (1H, br. s, OH-11), 8.01 (1H, d, J = 7.6 Hz, H-1), 7.78 (1H, t, J = 8.1 Hz, H-2), 7.38 (1H, d, J = 8.4 Hz, H-3), 7.26 (4H, d, J = 7.3 Hz, o-H-Ph), 7.21 (4H, t, J = 7.6 Hz, m-H-Ph), 7.17–7.13 (2H, m, p-H-Ph), 6.45 (2H, d, J = 15.9 Hz, CH=CHPh),6.16 (2H, dt, J = 6.5, 15.8 Hz, CH=CHPh), 5.59 (1H, br. s, H-1′), 5.28 (1H, br. s, H-7), 4.68 (1H, br. s, OH-9), 4.10–4.07 (4H, m, H-15, H-5′), 3.82 (1H, br. s, H-4′), 3.44 (4H, d, J = 6.4 Hz, NCH2), 3.16 (1H, d, J = 19.1 Hz, H-10), 3.01–2.98 (1H, m, H-3′) 2.87 (1H, d, J = 18.8 Hz, H-10), 2.41 (3H, s, H-14), 2.37 (1H, br. s, H-8), 2.11 (1H, dd, J = 4.0, 14.8 Hz, H-8), 2.01–1.96 (2H, m, H-2′), 1.42 (3H, d, J = 6.6 Hz, H-6′); 13C NMR (CDCl3, 100 MHz,): δ = 211.57 (C, C-13), 186.08 (C, C-12), 185.80 (C, C-5), 160.48 (C, C-4), 156.00 (C, C-6), 155.15 (C, C-11), 136.50 (C, ipso-C-Ph), 135.23 (CH, C-3), 134.86 (C, C-12a), 133.93 (C, C-6a), 133.87 (C, C-4a),132.62 (CH, CH=CHPh), 128.07 (CH, m-C-Ph), 127.04 (CH, p-C-Ph), 125.98 (CH, CH=CHPh), 125.87 (CH, o-C-Ph), 120.20 (C, C-10a), 119.27 (CH, C-2), 117.98 (CH, C-1), 110.73 (C, C-11a), 110.64 (C, C-5a), 100.82 (CH, C-1′), 76.44 (C, C-9), 69.63 (CH, C-7), 66.74 (CH, C-4′), 66.67 (CH, C-5′), 56.21 (CH3, C-15), 54.73 (CH, C-3′), 50.64 (CH2, NCH2), 34.47 (CH2, C-10), 32.81 (CH2, C-8), 28.08 (CH2, C-2′), 24.46 (CH3, C-14), 16.94 (CH3, C-6′); HRMS (ESI) m/z found 760.3109; calculated for C45H45NO10 760.3116 (M + H+).
Ethyl 2-[(6-[{(1S,3S)-3-acetyl-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl}oxy](2S,3S,4S,6R)-3-hydroxy-2-methyltetrahydro-2H-pyran-4-yl)amino]acetate (1f): red solid, yield 76%; IR (KBr) ν = 3482 (br, m) (νOH), 3342 (br, m) (νNH), 2975 (sh, m), 2936 (m) and 2843 (sh, m) (all three νCH), 1739 (s) and 1718 (s) (both νC=O), 1618 (s) and 1578 (s) (both νC=C), 1414 (s) and 1352 (s) (both δOH), 1287 (vs), 1232 (s) and 1209 (vs) (all three νC–O), 1122 (s), 1009 (sh, s) and 985 (vs) (both δC=C), 818 (m), 793 (m), 764 (m), 731 (m), 464 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 13.96 (1H, br. s, OH-6), 13.28 (1H, br. s, OH-11), 8.04 (1H, d, J = 7.5 Hz, H-1), 7.79 (1H, t, J = 7.8 Hz, H-2), 7.40 (1H, d, J = 8.5 Hz, H-3), 5.52 (1H, br. d, J = 3.2 Hz, H-1′), 5.29 (1H, br. s, H-7), 4.71 (1H, br. s, OH-9), 4.15 (1H, q, J = 7.1 Hz, H-5′), 4.14–4.04 (5H, m, H-15, NHCH2), 3.51 (1H, br. s, H-4′), 3.39 (2H, br. s, COOCH2), 3.21 (1H, d, J = 18.9 Hz, H-10), 2.93–2.79 (2H, m, H-10, H-3′), 2.41 (3H, s, H-14), 2.36 (1H, d, J = 14.8 Hz, H-8), 2.09 (1H, dd, J = 3.9, 14.7 Hz, H-8), 1.83 (1H, dd, J = 3.8, 13.0 Hz, H-2′), 1.69 (1H, dd, J = 4.3, 13.0 Hz, H-2′), 1.37 (3H, d, J = 6.5 Hz, H-6′), 1.23 (3H, t, J = 7.1 Hz, CH2CH3); 13C NMR (CDCl3, 100 MHz,): δ = 211.67 (C, C-13), 186.92 (C, C-12), 186.58 (C, C-5), 172.65 (C, COOCH2), 160.90 (C, C-4), 156.29 (C, C-6), 155.73 (C, C-11), 135.55 (CH, C-3), 135.37 (C, C-12a), 134.21 (C, C-6a), 134.15 (C, C-4a), 120.81 (C, C-10a), 119.62 (CH, C-2), 118.26 (CH, C-1), 111.27 (C, C-11a), 111.13 (C, C-5a), 100.75 (CH, C-1′), 76.72 (C, C-9), 69.60 (CH, C-7), 66.81 (CH, C-4′), 66.45 (CH, C-5′), 61.01 (CH2,CH2COO), 56.54(CH3, C-15), 52.97(CH, C-3′),47.49 (CH2, NHCH2), 34.76 (CH2, C-10), 33.19 (CH2, C-8), 30.09 (CH2, C-2′), 24.64 (CH3, C-14), 16.99 (CH3, C-6′), 13.97 (CH3, CH2CH3); HRMS (ESI) m/z found 614.2231, 636.2041; calculated for C31H35NO12 614.2232 (M + H+), 636.2051 (M + Na+).
(8S,10S)-8-Acetyl-6,8,11-trihydroxy-10-{[(2R,4S,5S,6S)-5-hydroxy-6-methyl-4-(prop-2-en-1-yl(prop-2-yn-1-yl)amino)tetrahydro-2H-pyran-2-yl]oxy}-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione (2): red solid, yield 75%; IR (KBr) ν = 3479 (br, m) (νOH), 3275 (br, m) (νNH), 2975 (sh, m), 2935 (m) and 2842 (sh, m) (all three νCH), 2250 (vw) and 2105 (vw) (both νC≡CH), 1716 (s) and 1671 (s) (both νC=O), 1620 (s) and 1587 (s) (both νC=C), 1415 (s) and 1352 (s) (both δOH), 1286 (vs), 1231 (s) and 1208 (vs) (all three νC–O), 1125 (s), 1018 (sh, s) and 983 (vs) (both δC=C), 813 (m), 793 (m), 764 (m), 732 (m), 463 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 13.95 (1H, br. s, OH-6), 13.22 (1H, br. s, OH-11), 7.99 (1H, d, J = 7.7 Hz, H-1), 7.76 (1H, t, J = 7.9 Hz, H-2), 7.38 (1H, d, J = 8.4 Hz, H-3), 6.24–6.14 (1H, m, CH=CH2), 5.79–5.69 (2H, m, CH=CH2), 5.58 (1H, br. d, J = 3.0 Hz, H-1′), 5.26 (1H, br. s, H-7), 4.73 (1H, br. s, OH-9), 4.08–4.02 (4H, m, H-15, H-5′), 3.73 (1H, br. s, H-4′), 3.49 (1H, d, J = 17.9 Hz, CH2C≡CH), 3.36 (1H, d, J = 17.8 Hz, CH2C≡CH), 3.27–3.14 (3H, m, CH2CH=CH2, H-10), 2.92–2.83 (2H, m, H-3′, H-10), 2.43 (3H, s, H-14), 2.36 (1H, d, J = 14.8 Hz, H-8), 2.12–2.05 (1H, m, H-8), 1.86–1.80 (2H, m, H-2′), 1.39 (3H, d, J = 6.4 Hz, H-6′); 13C NMR (CDCl3, 100 MHz,): δ = 211.61 (C, C-13), 186.59 (C, C-12), 186.25 (C, C-5), 160.66 (C, C-4), 156.11 (C, C-6), 155.47 (C, C-11), 135.36 (CH, C-3), 135.12 (C, C-12a), 134.45 (CH, CH=CH2), 134.18 (C, C-6a), 133.99 (C, C-4a), 120.52 (C, C-10a), 119.41 (CH, C-2), 118.30 (CH2, CH=CH2), 118.06 (CH, C-1), 110.98 (C, C-11a), 111.88 (C, C-5a), 100.16 (CH, C-1′),77.06 (C, C≡CH), 76.91 (C, C-9), 74.85 (CH, C≡CH), 69.06 (CH, C-7),66.44 (CH, C-4′), 65.83 (CH, C-5′), 56.33 (CH3, C-15), 54.50 (CH, C-3′), 51.55 (CH2, CH2CH=CH2),37.11 (CH2, CH2C≡CH), 34.52 (CH2, C-10), 33.08 (CH2, C-8), 27.83 (CH2, C-2′), 24.52 (CH3, C-14), 16.84 (CH3, C-6′); HRMS (ESI) m/z found 606.2327; calculated for C33H35NO10 606.2334 (M + H+).
General procedure for the Aza-Michael reaction
A mixture of amine of daunorubicin (100 mg, 0.19 mmol) and alkene or alkyne (0.47 mmol) in MeOH (compounds 3a–3c) or EtOH (compound 3d) (5 mL) was stirred at room temperature for 20–28 h. The progress of the reaction was monitored by TLC (CHCl3: MeOH at v/v ratios 85: 15). After the reaction was complete, the solvent was evaporated under reduced pressure. The crude products were purified by column chromatography, eluted with CHCl3: MeOH at v/v ratios from 100: 1 to 10: 1.
3-[(6-[{(1S,3S)-3-Acetyl-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl}oxy](2S,3S,4S,6R)-3-hydroxy-2-methyltetrahydro-2H-pyran-4-yl)amino]propanenitrile (3a): red solid, yield 63%; IR (KBr) ν = 3475 (br, m) (νOH), 2967 (sh, m), 2935 (m) and 2844 (sh, m) (all three νCH), 2248 (w) (νC≡N), 1706 (s) (νC=O), 1617 (s) and 1577 (s) (both νC=C), 1414 (s) and 1353 (s) (both δOH), 1292 (vs), 1262 (s), 1228 (sh, s), 1210 (vs) and 1192 (sh, s) (all five νC–O), 1114 (s), 1018 (sh, s) and 982 (vs) (both δC=C), 839 (m), 792 (m), 765 (m), 731 (m), 463 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 14.01 (1H, br. s, OH-6), 13.31 (1H, br. s, OH-11), 8.05 (1H, d, J = 7.6 Hz, H-1), 7.81 (1H, t, J = 8.1 Hz, H-2), 7.42 (1H, d, J = 8.4 Hz, H-3), 5.54 (1H, br. s, H-1′), 5.31 (1H, br. s, H-7), 4.64 (1H, br. s, OH-9), 4.11 (4H, br. s, H-15, H-5′), 3.64 (1H, br. s, H-4′), 3.25 (1H, dd, J = 1.5, 18.9 Hz, H-10), 2.97 (1H, d, J = 18.8 Hz, H-10), 2.95–2.92 (3H, m, N–CH2, H-3′), 2.52 (2H, t, J = 6.6 Hz, CH2CN),2.44 (3H, s, H-14), 2.38 (1H, d, J = 14.8 Hz, H-8), 2.14 (1H, dd, J = 4.0, 14.8 Hz, H-8), 1.84–1.73 (2H, m, H-2′), 1.38 (3H, d, J = 6.6 Hz, H-6′); 13C NMR (CDCl3, 100 MHz,): δ = 211.57 (C, C-13), 186.72 (C, C-12), 186.39 (C, C-5), 160.83 (C, C-4), 156.22 (C, C-6), 155.56 (C, C-11), 135.57 (CH, C-3), 135.21 (C, C-12a), 134.14 (C, C-6a), 133.98 (C, C-4a), 120.59 (C, C-10a), 119.60 (CH, C-2), 118.27 (C, C≡N), 118.21 (CH, C-1), 111.17 (C, C-11a), 111.03 (C, C-5a), 100.74 (CH, C-1′), 76.66 (C, C-9), 69.84 (CH, C-7), 67.23 (CH, C-4′), 66.59 (CH, C-5′), 56.49 (CH3, C-15), 52.17 (CH, C-3′), 41.33 (CH2, NCH2), 34.71 (CH2, C-10), 33.06 (CH2, C-8), 30.12 (CH2, C-2′), 24.64 (CH3, C-14), 19.07 (CH2, CH2C≡N), 16.89 (CH3, C-6′); HRMS (ESI) m/z found 581.2128, 603.1947, 619.1683; calculated for C30H32N2O10 581.2130 (M + H+), 603.1949 (M + Na+), 619.1689 (M + K+).
Methyl 3-[(6-[{(1S,3S)-3-acetyl-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl}oxy](2S,3S,4S,6R)-3-hydroxy-2-methyltetrahydro-2H-pyran-4-yl)amino]propanoate (3b): red solid, yield 67%; IR (KBr) ν = 3475 (br, m) (νOH), 2971 (sh, m), 2935 (m) and 2845 (sh, m) (all three νCH), 1734 (s) and 1718 (s) (both νC=O), 1618 (s) and 1578 (s) (both νC=C), 1414 (s) and 1352 (s) (both δOH), 1287 (vs), 1232 (s) and 1209 (vs) (all three νC–O), 1120 (s), 1007 (sh, s) and 983 (vs) (both δC=C), 817 (m), 793 (m), 764 (m), 464 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 14.00 (1H, br. s, OH-6), 13.33 (1H, br. s, OH-11), 8.06 (1H, d, J = 7.4 Hz, H-1), 7.81 (1H, t, J = 8.0 Hz, H-2), 7.42 (1H, d, J = 8.3 Hz, H-3), 5.54 (1H, br. d, J = 3.4 Hz, H-1′), 5.31 (1H, br. s, H-7), 4.71 (1H, br. s, OH-9), 4.11–4.07 (4H, m, H-15, H-5′), 3.71 (1H, br. s, H-4′), 3.68 (3H, s, COOCH3), 3.25 (1H, d, J = 18.8 Hz, H-10), 3.02–2.90 (4H, m, H-10, H-3′, NHCH2), 2.55 (2H, br. s, CH2COO), 2.45 (3H, s, H-14), 2.38 (1H, d, J = 14.9 Hz, H-8), 2.13 (1H, dd, J = 4.0, 14.8 Hz, H-8), 1.83 (1H, dd, J = 3.8, 12.9 Hz, H-2′), 1.71 (1H, dd, J = 4.3, 13.4 Hz, H-2′), 1.39 (3H, d, J = 6.5 Hz, H-6′); 13C NMR (CDCl3, 100 MHz,): δ = 211.69 (C, C-13), 186.59 (C, C-12), 186.27 (C, C-5), 172.81 (C, COOCH3), 160.77 (C, C-4), 156.20 (C, C-6), 155.54 (C, C-11), 135.48 (CH, C-3), 135.16 (C, C-12a), 134.16 (C, C-6a), 134.09 (C, C-4a), 120.54 (C, C-10a), 119.54 (CH, C-2), 118.22 (CH, C-1), 111.07 (C, C-11a), 110.92 (C, C-5a), 100.85 (CH, C-1′), 76.60 (C, C-9), 69.72 (CH, C-7), 66.44 (CH, C-4′, C-5′), 56.45 (CH, C-3′), 52.12 (CH3, C-15), 51.53 (CH3, COOCH3), 40.79 (CH2, NHCH2), 34.66 (CH2, C-10), 34.38 (CH2, CH2COO),33.02 (CH2, C-8), 30.07 (CH2, C-2′), 24.64 (CH3, C-14), 16.96 (CH3, C-6′); HRMS (ESI) m/z found 614.2230; calculated for C31H35NO12 614.2232 (M + H+).
Dimethyl 2-[(6-[{(1S,3S)-3-acetyl-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl}oxy](2S,3S,4S,6R)-3-hydroxy-2-methyltetrahydro-2H-pyran-4-yl)amino]fumarate (3c): red solid, yield 71%; IR (KBr) ν = 3483 (br, m) (νOH), 3319 (br, m) (νNH), 2978 (sh, m), 2947 (m) and 2845 (sh, m) (all three νCH), 1745 (s) and 1719 (s) (both νC=O), 1668 (s), 1619 (s) and 1578 (s) (all three νC=C), 1434 (s) and 1414 (s) (both δOH), 1286 (vs), 1252 (vs) and 1209 (vs) (all three νC–O), 1107 (s), 1033 (sh, s), 1019 (s) and 991 (s) (all three δC=C), 820 (m), 775 (m), 462 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 13.97 (1H, br. s, OH-6), 13.24 (1H, br. s, OH-11), 8.46 (1H, d, J = 5.0 Hz, C=CH), 8.01 (1H, d, J = 7.4 Hz, H-1), 7.79 (1H, t, J = 8.2 Hz, H-2), 7.40(1H, d, J = 8.4 Hz, H-3), 5.55 (1H, br. d, J = 3.4 Hz, H-1′), 5.26 (1H, br. s, H-7), 4.45 (1H, br. s, OH-9), 4.34–4.30 (1H, m, H-5′), 4.08 (3H, s, H-15), 3.83–3.70 (2H, m, H-4′, H-3′), 3.66 (3H, s, COOCH3), 3.20 (1H, d, J = 18.8 Hz, H-10), 2.93 (1H, d,J = 18.8 Hz, H-10), 2.41 (3H, s, H-14), 2.32 (1H, d, J = 14.8 Hz, H-8), 2.17 (1H, br. s, H-8), 2.03–1.98 (1H, m, H-2′), 1.88–1.82 (1H, m, H-2′), 1.43(3H, d, J = 6.6 Hz, H-6′); 13C NMR (CDCl3, 100 MHz,): δ = 211.11 (C, C-13), 186.85 (C, C-12), 186.58 (C, C-5), 170.02 (C, COOCH3), 160.93 (C, C-4), 156.14 (C, C-6), 155.47 (C, C-11), 142.38 (C, C=CH), 135.65 (CH, C-3), 135.25 (C, C-12a), 133.93 (C, C-6a), 133.59 (C, C-4a), 120.64 (C, C-10a), 119.63 (CH, C-2), 118.36 (CH, C-1), 111.36 (C, C-11a), 111.21 (C, C-5a), 100.17 (CH, C-1′), 90.56 (C, C=CH), 76.44 (C, C-9), 70.11 (CH, C-7), 65.46 (CH, C-4′, C-5′), 56.53 (CH3, C-15), 50.87 (CH, C-3′), 43.00 (CH3, COOCH3), 34.87 (CH2, C-10), 33.08 (CH2, C-8), 32.23 (CH2, C-2′), 24.47 (CH3, C-14), 15.85 (CH3, C-6′); HRMS (ESI) m/z found 670.2125; calculated for C33H35NO14 670.2130 (M + H+).
Diethyl 3,3′-[(6-[{(1S,3S)-3-acetyl-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl}oxy](2S,3S,4S,6R)-3-hydroxy-2-methyltetrahydro-2H-pyran-4-yl)azanediyl]diacrylate (3d): red solid, yield 61%; IR (KBr) ν = 3483 (br, m) (νOH), 2979 (sh, m), 2936 (m) and 2845 (sh, m) (all three νCH), 1715 (s) and 1663 (s) (both νC=O), 1618 (s) and 1588 (s) (both νC=C), 1414 (s) and 1380 (s) (both δOH), 1287 (vs) and 1209 (vs) (both νC–O), 1115 (s), 1033 (sh, s), 1016 (s) and 990 (s) (all three δC=C), 850 (m), 792 (m), 764 (m), 463 (w) cm−1; 1H NMR (CDCl3, 400 MHz,): δ = 14.01 (1H, br. s, OH-6), 13.28 (1H, br. s, OH-11), 8.03 (1H, d, J = 7.6 Hz, H-1), 7.80 (1H, t, J = 8.2 Hz, H-2), 7.41 (1H, d, J = 8.5 Hz, H-3), 7.20 (2H, d, J = 13.6 Hz, NH–CH = CH), 5.96 (2H, d, J = 15.6 Hz, CH=CH-COOEt), 5.54 (1H, br. d, J = 3.2 Hz, H-1′), 5.28 (1H, br. s, H-7), 4.43 (1H, br. s, OH-9),4.13 (4H, q, J = 7.1 Hz, OCH2), 4.09 (4H, br. s, H-15, H-5′), 3.71 (1H, br. s, H-4′), 3.25 (1H, d, J = 18.8 Hz, H-10), 2.93(1H, dd, J = 5.5, 18.8 Hz, H-10), 2.45–2.43 (4H, m, H-14, H-3′), 2.35 (1H, d, J = 14.9 Hz, H-8), 2.18 (1H, dd, J = 3.9, 14.8 Hz, H-8), 2.05 (1H, dd, J = 3.5, 11.2 Hz, H-2′), 1.88 (1H, dd, J = 4.7, 13.5 Hz, H-2′), 1.33(3H, d, J = 7.0 Hz, H-6′), 1.38–1.27 and 1.26–1.21 (6H, m, CH2CH3); 13C NMR (CDCl3, 100 MHz,): δ = 211.40 (C, C-13), 186.93 (C, C-12), 186.62 (C, C-5), 168.61 (C, COOCH2), 160.94 (C, C-4), 156.20 (C, C-6), 155.58 (C, C-11), 143.01 (CH, NH–CH=CH), 135.66 (CH, C-3), 135.31 (C, C-12a), 133.99 (C, C-6a), 133.68 (C, C-4a), 120.63 (C, C-10a), 119.67 (CH, C-2), 118.33 (CH, C-1), 111.26 (C, C-11a), 111.12 (C, C-5a), 108.14 (CH, CH=CH-COOEt), 100.45 (CH, C-1′), 76.58 (C, C-9), 70.19 (CH, C-7), 69.81 (CH, C-4′), 66.90 (CH, C-5′), 59.78 and 59.47 (CH2, OCH2), 56.54 (CH3, C-15), 55.06 (CH, C-3′), 34.85 (CH2, C-10), 33.15 (CH2, C-8), 31.28 (CH2, C-2′), 24.62 (CH3, C-14), 16.55 (CH3, C-6′), 14.31 (CH3, CH2CH3); HRMS (ESI) m/z found 724.2595, 746.2417; calculated for C37H41NO14 724.2600 (M + H+), 746.2419 (M + Na+).
Biological studies
Antiproliferative properties of the claimed compounds were determined by the MTT test. Human A549 cell cultures (ATCC® CCL-185 ™), RD (ATCC® CC-136 ™) and HCT116 (ATCC® CCL-247 ™) are grown in DMEM (NLP PanEko). In growth medium 10% fetal calf serum (HyClone®, Thermo Scientific), 2 mmol l-glutamine (NLP PanEco), 1% gentamicin (JSC Biochemist) were added as antibiotic and incubated at 37 °C in an atmosphere of 5% CO2 and 95% air. Cells were sown in a 96-well plate (Costar®) in an amount of 1 × 104 cells/200 μl and cultured at 37 °C in a humid environment containing 5% CO2. After 24 h of incubation, solutions of different concentrations (from 100 to 0.0012 μmol/l) were added to the cell culture and then the cells were cultured under the same conditions for 72 h. For each concentration, the experiments were performed in triplicate. All compounds were dissolved in DMSO (PANREAC QUIMICA S.L.U). The final concentration of DMSO in the well did not exceed 0.1% and was not toxic to the cells. The control wells were added to the solvent in an amount of 0.1%. After incubation, 20 μl of a solution of 5 mg/ml of MTT [bromide 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium] (Sigma Aldrich) in PBS (Phosphate Buffered Saline) and additionally incubated for 2 h. Then the medium was removed and 100 μL DMSO was added to each well to dissolve formed formazan crystals. Using a BioTek Instruments Cytation 3 Imager plate analyzer, the optical density at 536 nm was determined. The concentration value, which causes a 50% inhibition of cell population growth (IC 50), was estimated on the basis of dose-dependent curves using the software OriginPro 9.0.
References
Tan C, Tasaka H, Yu KP, Murphy ML, Karnofsky DA. Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease. Clinical evaluation with special reference to childhood leukemia. Cancer. 20;333–53. https://doi.org/10.1002/1097-0142(1967)20:33.0.CO;2-K
Cortes-Funes H, Coronado C. Role of anthracyclines in the era of targeted therapy. Cardiovasc Toxicol. 2007;7:56–60. https://doi.org/ 10.1007/s12012-007-0015-3
Artyushin OI, Brel VK, Moiseeva AA. Anthracycline derivatives and their anticancer activity. INEOS OPEN. 2019;2:9–18. Moscow University Chemistry Bulletin. https://doi.org/10.32931/io1902r.
Zhang YS, Zhang G, Zhang W, Luo H, Qiu L, Liu Q, et al. Synthesis and biological activities of a 3′-azido analogue of doxorubicin against drug-resistant cancer cells. Int J Mol Sci. 2012;13:3671–84. https://doi.org/10.3390/ijms13033671
Zunino F, Pratesi G, Perego P. Role of the sugar moiety in the pharmacological activity of anthracyclines: development of a novel series of disaccharide analogs. Biochem Pharm. 2001;61:933–8. https://doi.org/10.1016/s0006-2952(01)00522-6
Burke TG, Morin MJ, Sartorelli AC, Lane PE, Tritton TR. Function of the anthracycline amino group in cellular transport and cytotoxicity. Mol Pharm. 1987;31:552–6. PMID: 3472065.
Burke TG, Sartorelli AC, Tritton TR. Selectivity of the anthracyclines for negatively charged model membranes: role of the amino group. Cancer Chemother Pharm. 1988;21:274–80. https://doi.org/10.1007/BF00264191
Zunino F, Gambetta R, Di Marco A, Zaccara A. Interaction of daunomycin and its derivatives with DNA. Biochim Biophys Acta. 1972;277:489–98. https://doi.org/10.1016/0005-2787(72)90092-5
Marzin D, Jasmin C, Maral R, Mathe G. Mutagenicity of eight anthracycline derivatives in five strains of Salmonella typhimurium. Eur J Cancer Clin Oncol. 1983;19:641–7. https://doi.org/10.1016/0277-5379(83)90180-3
Umezawa K, Haresaku M, Muramatsu M, Matsushima T. Mutagenicity of anthracycline glycosides and bleomycins in Salmonella assay system. Biomed Pharmacother. 1987;41:214–8. https://doi.org/10.1002/tcm.1770060307
Moiseeva AA, Artyushin OI, Anikina LV, Brel VK. Synthesis and antitumor activity of daunorubicin conjugates with of 3,4-methylendioxybenzaldehyde. Bioorg Med Chem Lett. 2019;29:126617. https://doi.org/10.1016/j.bmcl.2019.08.021
Tong GL, Wu HY, Smith TH, Henry DW. Adriamycin analogs. 3. Synthesis of N-alkylated anthracyclines with enhanced efficacy and reduced cardiotoxicity. J Med Chem. 1979;22:912–8. https://doi.org/10.1021/jm00194a005
Masquelier M, Tirzitis G, Peterson CO, Pålsson M, Amolins A, Plotniece M, et al. Plasma stability and cytotoxicity of lipophilic daunorubicin derivatives incorporated into low density lipoproteins. Eur J Med Chem. 2000;35:429–38. https://doi.org/10.1016/s0223-5234(00)00139-2
Chaires JB, Leng F, Przewloka T, Fokt I, Ling Y-H, Perez–Solarand R, et al. Structure-based design of a new bisintercalating anthracycline antibiotic. J Med Chem. 1997;40:261–6. https://doi.org/10.1021/jm9607414
Hu GG, Shui X, Leng F, Priebe W, Chaires JB, Williams LD. Structure of a DNA–bisdaunomycin complex. Biochem. 1997;36:5940–6. https://doi.org/10.1021/bi9705218
De K, Legros J, Crousse B, Bonnet-Delpon D. Solvent-promoted and -controlled Aza-Michael reaction with aromatic amines. J Org Chem. 2009;74:6260–5. https://doi.org/10.1021/jo9012699
Semakov AV, Anikina LV, Afanasyeva SV, Pukhov SA, Klochkov SG. Synthesis and antiproliferative activity of conjugates of anthracycline antibiotics with sesquiterpene lactones. Russ J Bioorg Chem. 2018;44:538–46. https://doi.org/10.1134/S1068162018040167
Gruber BM, Anuszewska EL, Bubko I, Goździk A, Fokt I, Priebe W. Effect of structural modification at the 4, 3’, and 2’ positions of doxorubicin on topoisomerase II poisoning, apoptosis, and cytotoxicity in human melanoma cells. Arch Immunol Ther Exp. 2007;55:193–8. https://doi.org/10.1007/s00005-007-0018-6
Tsedilin AM, Fakhrutdinov AN, Eremin DB, Zalesskiy SS, Chizhov AO, Kolotyrkina NG, et al. How sensitive and accurate are routine NMR and MS measurements? Mendeleev Comm. 2015;25:454–6. https://doi.org/10.1016/j.mencom.2015.11.019
Acknowledgements
This work was supported by RFBR (№ 18-03-00073) and the scholarship of the President of the Russian Federation for young scientists and postgraduates (Competition «SP-2019», No SP-2717.2019.4). NMR studies, spectral characterization, and elemental analysis were performed with the financial support from Ministry of Science and Higher Education of the Russian Federation using the equipment of Center for molecular composition studies of INEOS RAS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Brel, V.K., Moiseeva, A.A., Artyushin, O.I. et al. Simple methods of modification of daunorubicin on the daunosamine nitrogen atom. Med Chem Res 30, 564–573 (2021). https://doi.org/10.1007/s00044-020-02664-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-020-02664-8