
Abstract
A multicomponent reaction was used as a synthetic strategy to prepare organotin (IV) complexes, 2-amino-3-hydroxypyridine, saliciladehydes 5-R-substituted (H, CH3, OCH3, C(CH)3, F, Cl, Br, I, NO2), and dibutyltin(IV) oxide were used as starting materials. The complexes were analyzed by IR, UV–Visible, MS, NMR of 1H, 13C, 119Sn. The complex 3a was structurally identified by X-ray crystallography, which revealed a dimeric arrangement where the tin atom possess a distorted hexacoordinated environment in which the deprotonated phenolic oxygen atoms and the nitrogen atom of the azomethine from the ligand are coordinated to the metallic center, and one of the phenoxy oxygens bridges with the tin through an intermolecular interaction forming a planar Sn2O2 core. As strategy of molecular design, isosteric and bioisosteric replacement of halogens were employed. All organotin compounds were assessed for their in vitro cytotoxic activity on cancer cell lines K‐562 (chronic myelogenous leukemia), U‐251 (glioblastoma), HCT‐15 (human colorectal cancer), MCF‐7, MDA-MB-231 (human breast cancer), and SKLU‐1 (non‐small‐cell lung cancer). They evidenced an elevated cytotoxic activity, and the inhibitory percentage values stated higher activity than the cis-platin. The K-562 and MDA-MB-231 cells were more sensitive to organotin (IV) complexes than HCT-15 and MCF-7. The organotin (IV) compounds were also tested in vivo for brine shrimp lethality to examine their toxic properties.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Schiff base ligands are some of the most extensively used organic compounds in the biochemistry and chemistry fields. This is owing to their feasibility of synthesis, stability, and their catalytic and pharmacological applications. Additionally, these types of ligands have been employed as biological models to understand the structure of biomolecules and their bioactivity [1,2,3,4,5,6]. Schiff bases are simple and highly versatile ligands that also serve as building blocks for various organic substrates of interest in organic chemistry [6,7,8]. Schiff bases are also well known as excellent ligands to coordinate metal ions. Their structural diversity allows for the inclusion of different donors or acceptors that readily form a variety of molecular structures with various coordination geometries around metallic ions [9].
Metals possess unique properties such as redox states, Lewis acid character, variable geometries, and kinetic and hydrolytic stability features that are not usually observed in organic drugs. This makes them useful for designing therapeutic metal-based drugs or agents with an enhanced pharmacological response in comparison to the organic free ligand [10,11,12,13,14]. Imine ligands and their metal complexes are of paramount importance in coordination chemistry, not only because of the diversity of interesting biological activities but also for the formation of stable complexes with most metal ions which are of interest to study a variety of biological reactions catalyzed by enzymes [2]. Metal-based drugs offer the possibility of designing therapeutic agents with an increased therapeutic effect and major bioavailability and biodistribution [15].
Despite their toxicity, resistance limitations and high side effects, platinum-based drugs are undoubtedly the most widely studied for their potential use in oncology [16,17,18]. Since the success of cis-platin in cancer treatment, new metallodrugs are in clinical trials. Cis-platin has encouraged the search for novel metal species with exceptional anticancer activity, lower side effects, lower toxicity, and a specific antitumor mechanism [13, 19]. Numerous metal-based compounds have been synthesized with promising anticancer properties. Among these are organotin(IV) compounds that have attracted interest in view of their pharmacological response and apoptosis-inducing character. Furthermore, organotin complexes constitute an interesting option not only because of their remarkable cytostatic properties but also for their toxicological profiles in comparison to other drugs, including platinum-based drugs. Organotin(IV) complexes are also known for their potential biological applications which include antileishmanial, antifungal, antimicrobial, antioxidant, anti-tuberculosis, and anti-inflammatory applications [3, 20,21,22,23,24]. The pyridines are an important class of heterocyclic molecules able to form metallic complexes with several metals, and there is a wide range of applications for them such as in antibacterial, antimicrobial, and antifungal applications. Furthermore, it is well known that substituted pyridines are selective inhibitors of cyclooxygenase-2 [25]. Moreover, ligands containing a vitamin B6 moiety are the paramount importance for their tumor-targeting properties. Consequently, organotin(IV) containing pyridoxal and pyridoxamine Schiff base skeletons are highly cytotoxic agents on cancer cell lines such as U-251 (glioblastoma), PC-3 (prostate), K-562 (chronic myelogenous leukemia), HCT-15 (human colorectal), MCF-7 (human breast), SKLU-1 (non-small cell lung), and MDA-MB-231 (human breast) [26,27,28].
The present paper informs the synthesis and spectroscopic characterization of dibutyltin(IV) complexes derived from 2-amino-3-hydroxypyridine and 5-R-salicylaldehyde, the substituents bonded to the aromatic ring possess electron donating or electron releasing character. Also, the structural parameters and the effect of isosteric and biososteric replacement on the cytotoxic response on cancer cell lines were examined, as well as the toxicity to brine shrimp (Artemia Salina).
Materials and methods
Reactants and solvents were acquired from commercial suppliers and used without additional purification.
Melting points of all complexes were determined with a Fischer-Johns MEL-TEMP II and are uncorrected. The UV–Vis absorption measurements were performed in a Shimadzu UV-160U spectrometer, solutions at 2.045 × 10−5 M in methanol were prepared and the molar absorption coefficient calculated. Infrared (IR) spectra were conducted with a Bruker Tensor 27 spectrometer. The use of a Metrohm 644 instrument allowed us to measure molar conductivity using anhydrous methanol as the solvent. NMR spectra were acquired using chloroform-d or DMSO-d6 as solvent on a Bruker Advance III spectrometer, 2D NMR experiments (COSY, HSQC, and HMBC) allowed us unambiguous assignment of the 1H and 13C signals. FAB (fast atom bombardment) mass spectra were recorded with a JEOL-JMS-X103 spectrometer. The molecular structure of 3a was established by single crystal X-ray diffraction, suitable crystals were grown by slow evaporation of methanol, the data were collected at 150 (2) K, on a Bruker Smart Apex CCD diffractometer using with Mo radiation (λMo Kα = 0.71073 Å). Direct methods were used to solve the structure using all reflections and the refinement program SHELXS-2014. All non-hydrogen atoms were refined using anisotropic displacement. Refinements and structure solution and were fulfilled using SHELXL-2014 software [29]. All hydrogen atoms were inserted at calculated positions based upon hybridization and refining by means of the riding model. Crystallographic data of 3a were deposited with the Cambridge Crystallographic Data Center CCDC 1981242 number.
General procedure
2-amino-3-hydroxypyridine, dibutyltin oxide, and the corresponding salicylaldehyde-5-R-substituted were added to 30 mL of a mixture of solvents toluene:methanol (80:20) in a stoichiometric ratio. The reaction was stirred and refluxed for 12 h. Once the reaction was accomplished, the solvent was eliminated under vacuum to give dark red and orange solids, which were purified by crystallization with methanol.
10,10-Di-butyl-1,6-diaza-10-stanna-9,11-dioxatetracycle[8.7.0.01,80.12,17] heptadeca-1,3,5,7,13,15-heptaene (3a)
Compound 3a was synthetized from 0.1 ml (0.938 mmol) of salicylaldehyde, 0.103 g (0.938 mmol) of 2-amino-3-hydroxypyridine and 0.233 g (0.938 mmol) of dibutyltin oxide, affording 0.376 g (90%) of a dark orange solid; m.p. 90–92 °C (dec); molar conductance, ΛM (1 × 10−3 M, methanol): 65.8 μS cm−1 (non-electrolyte); UV–Vis [methanol, λmáx/nm (ε M−1 cm–1)]: 206 (16,232), 286 (6503) π → π* (Aromatic), 380 (3911) π → π* (C=N), 457 (8654) n → π* (C=N); IR (KBr) cm−1: 1606 ν(C=N), 1235 ν(C–O), 683, 591ν(Sn–C), 520 ν(Sn–O), 442 ν(Sn–N); 1H NMR (300.52 MHz, DMSO-d6) δ: 0.74 (6H, t, J = 7.5 Hz, H-δ), 1.22 (4H, sext, J = 7.2 Hz, H-γ), 1.34-1.51 (8H, m, H-α, H-β), 6.61 (1H, d, J = 6.3 Hz, H-3), 6.68 (1H, t, J = 8.1 Hz, H-5), 7.04 (1H, dd, J = 0.9 Hz, J = 7.8 Hz, H-11), 7.16 (1H, dd, J = 4.2 Hz, J = 7.8 Hz, H-10), 7.43 (1H, td, J = 2.1 Hz, J = 7.2 Hz, H-4), 7.59 (1H, dd, J = 1.2 Hz, J = 8.1 Hz, H-6), 7.70 (1H, dd, J = 0.9 Hz, J = 4.2 Hz, H-9) 9.47 (1H, s, 3J(1H-119/117Sn) = 47.5 Hz, H-7); 13C NMR (75.57 MHz, DMSO-d6) δ: 170.5 (C-2), 163.6 (C-7), 155.0 (C-12), 144.4 (C-8), 137.9 (C-4), 137.5 (C-6), 134.8 (C-9), 125.7 (C-10), 125.1 (C-11), 122.3 (C-5), 118.1 (C-1), 116.8 (C-3), 26.9 (3J119Sn–13C = 34.8 Hz, C-γ), 26.6 (C-β, 2J119/117Sn–13C = 89.2/84.6 Hz), 24.8 (C-α, 1J (119/117Sn–13C) = 607.6, 594.0 Hz, θ = 135.5°), 13.6 (C-δ); 119Sn NMR (112.07 MHz, CDCl3) δ: −194; 119Sn NMR (112.07 MHz, DMSO-d6) δ: −238; MS: (FAB+) [m/z] (%):[M+ + 1, 447] (100), [M+-2Bu, 333] (85), [M+-C5H3NO, 240] (25); HR-MS (FAB+) m/z: 447.1095 (Calc. for C20H26N2O2Sn), observed: 447.1096.
10,10-Dibutyl-5-methyl-1,6-diaza-10-stanna-9,11-dioxatetracycle [8.7.0.01,80.12,17] heptadeca-1,3,5,7,13,15-hexaene (3b)
Compound 3b was obtained from 0.109 g (0.803 mmol) of 5-methylsalicylaldehyde 0.088 g (0.803 mmol) de 2-amino-3-hydroxypyridine and 0.2 g (0.803 mmol) dibutyltin oxide, affording 0.313 g (85%) of a dark red solid; m. p. 37–39 °C; molar conductance, ΛM (1 × 10–3 M, methanol): 3.6 μS cm−1 (non-electrolyte); UV–Vis [methanol, λmáx/nm (ε M−1 cm−1)]: 223 (52,804), 254 (41,656), 288 (27,331) π → π* (Arom), 375 (16,868) π → π* (C=N), 468 (34,909) n → π* (C=N); IR (KBr) cm−1: 1587 ν(C=N), 1235 ν(C–OArom.), 683,570 ν(Sn–C), 539 ν(Sn–O), 466 ν(Sn–N); 1H NMR (300.52 MHz, DMSO-d6) δ: 0.75 (6H, t, J = 7.2 Hz, H-δ), 1.16–1.23 (4H, sext, J = 7.2 Hz, H-γ), 1.33–1.53 (8H, m, H-α, H-β), 2.20 (3H, s, C6H3–CH3), 6.62 (1H, d, J = 8.7 Hz, H-3), 7.04 (1H, dd, J = 1.2 Hz, J = 8.1 Hz, H-11), 7.14 (1H, dd, J = 4.5 Hz, J = 8.1 Hz, H-10), 7.26 (1H, dd, J = 2.1 Hz, J = 8.4 Hz, H-4), 7.34 (1H, s, H-6), 7.69 (1H, dd, J = 1.2 Hz, J = 4.5 Hz, H-9), 9.46 (1H, s, 3J(1H–119/117Sn) = 40.9 Hz, H-7); 13C NMR (75.57 MHz, DMSO-d6) δ: 168.8 (C-2), 163.3 (C-7), 154.9 (C-12), 144.5 (C-8), 139.5 (C-4), 136.3 (C-6), 134.8 (C-9), 125.5 (C-10), 125.2 (C-5), 125.0 (C-11), 122.2 (C-3), 117.5(C-1), 27.2 (C-γ, 3J(119Sn–13C) = 37.0 Hz), 26.2 (C-β, 2J(119Sn–13C) = 96.7 Hz), 24.5 (C-α, 1J(119/117Sn–13C) = 698.3/674.1 Hz, θ = 144.6°), 20.1 (C6H3–CH3), 13.9 (C-δ);119Sn NMR (112.07 MHz, CDCl3) δ: −193; 119Sn NMR (112.07 MHz, DMSO-d6) δ: −233; MS: (FAB+) [m/z] (%):[M+ + 1, 461] (100), [M+-2Bu, 347] (20), [M+-C5H3NO, 254] (5); MS-HR (FAB+) m/z: 461.1251 (Calc. for C21H29N2O2Sn), observed: 461.1260.
10,10-Di-butyl-5-tert-butyl-1,6-diaza-10-stanna-9,11-dioxatetracycle [8.7.0.01,80.12,17] heptadeca-1,3,5,7,13,15-heptaeno (3c)
Compound 3c was prepared from 0.143 g (0.803 mmol) of 5-tert-butylsalicylaldehyde, 0.088 g (0.803 mmol) of 2-amino-3-hydroxypyridine and 0.2 g (0.803 mmol) dibutytin oxide, affording 0.331 g (80% yield) of a dark red solid, m.p. 57–59 °C dec; molar Conductance, ΛM (1 × 10–3 M, methanol): 2.3 µS cm−1 (non-electrolyte); UV–Vis [methanol, λmáx/nm (ε M−1 cm–1)]: 221 (57,498), 290 (25,620) π → π* (Arom), 383 (17,308) π → π* (C=N), 466 (35,643) n → π* (C=N); IR (KBr) cm−1: 1618 ν(C=N), 1244 ν(C–OArom.), 681,570 ν(Sn–C), 511 ν(Sn–O), 450 ν(Sn–N); 1H NMR (300.52 MHz, DMSO-d6) δ: 0.74 (6H, t, H-δ), 1.20–1.22 (4H, m, H-γ), 1.33–1.52 (8H, m, H-α, H-β), 1.25 (9H, s, tert-Bu), 6.64 (1H, dd, J = 1.5 Hz, J = 9.31 Hz, H-3), 7.03 (1H, dd, J = 1.5 Hz, J = 8.1 Hz, H-11), 7.14 (1H, dd, J = 4.5 Hz, J = 8.1 Hz, H-10), 7.50 (1H, s, H-5), 7.51 (1H, dd, J = 2.7 Hz, J = 9.3 Hz, H-4), 7.68 (1H, dd, J = 1.5 Hz, J = 4.5 Hz H-9), 9.50 (1H, s, 3J(1H–119/117Sn = 41.5 Hz, H-7); 13C NMR (75.57 MHz, DMSO-d6) δ: 168.7 (C-2), 163.9 (C-7), 154.9 (C-12), 144.5 (C-8), 138.9 (C-5), 136.2 (C-6), 134.7 (C-9), 132.9 (C-4), 125.6 (C-10), 125.0 (C-11), 121.5 (C-3), 117.0 (C-1), 34.1(C-tert-Bu), 31.5(CH3-tert-Bu), 27.1 (C-γ, 3J(119Sn–13C) = 37.0 Hz), 26.3 (C-β, 2J(119Sn–13C) = 95.2 Hz), 24.5 (C-α, 1J(119Sn–13C) = 684.7 Hz, θ = 143.2°), 13.9 (C-δ); 119Sn NMR (112.07 MHz, CDCl3)δ: −193, 119Sn NMR (112.07 MHz, DMSO-d6)δ −231; MS:(FAB+) [m/z] (%): [M+ + 1, 503] (35), [M+-2Bu, 389] (40), [M+-C5H3NO, 296] (10); HR-MS (FAB+) m/z: 503.1721 (Calc. for C24H35N2O2Sn), observed: 503.1726.
10,10-Di-butyl-5-fluoro-1,6-diaza-10-stanna-9,11-dioxatetracycle[8.7.0.01,80.12,17] heptadeca-1,3,5,7,13,15-heptaene (3d)
Compound 3d was synthetized from 0.1125 g (0.803 mmol) of 5-fluorosalicylaldehyde, 0.088 g (0.803 mmol) of 2-amino-3-hydroxypyridine, and 0.2 g (0.8027 mmol) of dibutyltin oxide, affording 0.335 g (90%) of a dark red solid: m.p. 74–75 °C (dec); molar conductance, ΛM (1 × 10−3 M, methanol): 13.4 μS cm−1 (non-electrolyte); UV–Vis [methanol, λmáx/nm (ε M−1 cm−1)]: 206 (18,921), 252 (12,272), 285 (8263) π → π* (Aromatic), 386 (4498) π → π* (C=N), 470 (11,099) n → π* (C=N); IR (KBr) cm−1: 1593 ν(C=N), 1229 ν(C–O), 619 ν(Sn–C), 531 ν(Sn–O), 492 ν(Sn–N), 1111 ν(C–F); 1H NMR (300.52 MHz, DMSO-d6) δ: 0.75 (6H, t, J = 7.2 Hz, H-δ), 1.15–1.27 (4H, sext, J = 7.21 Hz, H-γ), 1.33–1.51 (8H, m, H-α, H-β), 6.69 (1H, dd, J = 4.8 Hz, J = 9.3 Hz, H-3), 7.06 (1H, d, J = 8.1 Hz, H-11), 7.17 (1H, dd, J = 4.5 Hz, J = 8.1 Hz, H-10), 7.31 (1H, td, J = 3.3 Hz, J = 8.4 Hz H-4), 7.51 (1H, dd, J = 3.0 Hz, J = 9.0 Hz, H-6), 7.70 (1H, dd, J = 1.2 Hz, J = 4.2 Hz, H-9), 9.47 (1H, s, 3J(1H–119/117Sn) = 37.3 Hz, H-7); 13C NMR (75.57 MHz, DMSO-d6)δ: 167.1 (C-2), 162.7 (C-7), 155.2 (C-12), 153.2 (d, JC–F = 231.2 Hz, C-4), 144.3 (C-8), 135.0 (C-9), 126.0 (C-10), 125.8 (d, 1JC–F = 24.2 Hz, C-3), 125.4 (C-11), 123.6 (d, 2JC–F = 7.6 Hz, C-3), 120.2 (d, 1JC–F = 22.7 Hz, C-6), 117.1 (d, 2JC–F = 8.3 Hz, C-1), 27.2 (C-γ, 3J(119/117Sn–13C) = 43.8/37.0 Hz), 26.2 (C-β, 2J(119/117Sn–13C) = 102.8/96.7 Hz) 26.2 (C-γ), 25.2 (C-α, 1J(119Sn–13C) = 624.2 Hz, θ = 137.2°), 13.9 (C-δ); 119Sn NMR (112.07 MHz, CDCl3)δ: −192; 119Sn NMR (112.07 MHz, DMSO-d6)δ: −246; MS: (FAB+) [m/z] (%): [M+ + 1, 465] (90), [M+-2Bu, 366] (45), [M+-C5H3NO, 273] (20); HR-MS (FAB+) m/z: 465.0910 (Calc. for C20H25N2O2SnF), observed: 465.0907.
10,10-Di-butyl-5-chloro-1,6-diaza-10-stanna-9,11-dioxatetracycle [8.7.0.01,80.12,17] heptadeca-1,3,5,7,13,15-hexaene (3e)
Compound 3e was prepared from 0.1258 g (0.803 mmol) of 5-chlorosalicylaldehyde, 0.088 g (0.803 mmol) of 2-amino-3-hydroxypyridine, and 0.2 g (0.803 mmol) of dibutyltin oxide, affording 0.335 g (87%) dark red solid; m.p. 45–46 °C (dec); molar conductance, ΛM (1 × 10−3 M, methanol): 3.1 µS cm−1 (non-electrolyte); UV–Vis [methanol, λmáx/nm (ε M−1 cm−1)]: 207 (25,033), 229 (24,691) π → π* (Aromatic), 387 (5726) π → π* (C=N), 469 (4032) n → π* (C=N); IR (KBr) cm−1: 1605 ν(C=N), 1229 ν(C–O), 1110 ν(C–Cl), 695, 566 ν(Sn–C), 527 ν(Sn–O), 475 ν(Sn–N); 1H NMR (300.52 MHz, DMSO-d6)δ: 0.74 (6H, t, J = 7.2 Hz, H-δ), 1.21-1.25 (4H, sext, J = 7.2 Hz, H-γ), 1.35–1.50 (8H, m, H-α, H-β), 6.68 (1H, d, J = 9.0 Hz, H-3), 7.07 (1H, d, J = 8.1 Hz, H-11), 7.17 (1H, dd, J = 4.2 Hz, J = 8.1 Hz, H-10), 7.38 (1H, dd, J = 2.7 Hz, J = 9.0 Hz, H-4), 7.72 (1H, d, J = 2.7 Hz, H-6), 7.71 (1H, dd, J = 3.3 Hz, J = 6.0 Hz, H-9), 9.47 (1H, s, 3J(1H–119/117Sn) = 35.2 Hz, H-7); 13C NMR (75.57 MHz, DMSO-d6)δ: 169.1 (C-2), 162.5 (C-7), 155.3 (C-12), 144.3 (C-8), 137.0 (C-4), 135.4 (C-6), 135.0 (C-9), 126.0 (C-10), 125.5 (C-11), 124.3 (C-3), 119.3 (C-1), 119.0 (C-5), 27.2 (C-γ, 3J(119Sn–13C) = 40.8 Hz), 26.2 (C-β, 2J(119Sn–13C) = 105.8 Hz), 25.7 (C-α, 1J(119/117Sn–13C) = 731.5/697.5 Hz. θ = 147.9°), 13.9 (C-δ); 119Sn NMR (112.07 MHz, CDCl3)δ: −192; 119Sn NMR (112.07 MHz, DMSO-d6)δ: −254; MS: (FAB+) [m/z] (%): [M+ + 1, 481] (90), [M+-2Bu, 366] (45), [M+-C5H3NO, 273] (20); HR-MS (FAB+) m/z: 481.0705 (Calc. for C20H25N2O2SnCl), observed: 481.0703.
10,10-Di-butyl-5-bromo-1,6-diaza-10-stanna-9,11-dioxatetracycle [8.7.0.01,80.12,17] heptadeca-1,3,5,7,13,15-hexaene (3f)
Compound 3f was prepared from 0.161 g (0.803 mmol) of 5-bromosalicylaldehyde, 0.088 g (0.803 mmol) of 2-amino-3-hydroxypyridine, and 0.2 g (0.803 mmol), affording 0.343 g (82%) dark red solid; m.p. 47–49 °C (dec.); molar conductance, ΛM (1 × 10–3 M, methanol): 2.8 µS cm−1 (non-electrolyte); UV–Vis [methanol, λmáx/nm (ε M−1 cm−1)]: 206 (17,112), 229 (19,019), 256 (16,575) π → π* (Arom), 385 (4302) π → π* (C=N), 469 (9974) n → π* (C=N); IR (KBr) cm−1: 1600 ν(C=N), 1230 ν(C–OArom.), 1076 ν(C–Br), 678, 565 ν(Sn–C), 526 ν(Sn–O), 443 ν(Sn–N); 1H NMR (300.52 MHz, DMSO-d6)δ: 0.73 (6H, t, J = 7.2 Hz, H-δ), 1.14–1.26 (4H, sext, J = 7.2 Hz, H-γ), 1.33–1.51 (8H, m, H-α, H-β), 6.63 (1H, d, J = 9.0 Hz, H-3), 7.07 (1H, dd, J = 1.2 Hz, J = 8.1 Hz, H-11), 7.17 (1H, dd, J = 4.2 Hz, J = 8.1 Hz, H-10), 7.46 (1H, dd, J = 2.4 Hz, J = 9.0 Hz, H-3), 7.70 (1H, dd, J = 1.5 Hz, J = 4.5 Hz, H-9), 7.83 (1H, d, J = 2.7 Hz, H-6), 9.46 (1H, s, 3J(1H–119/117Sn) = 35.2 Hz, H-7); 13C NMR (75.57 MHz, DMSO-d6)δ: 169.4 (C-2), 162.4 (C-7), 155.3 (C-12), 144.3 (C-8), 139.6 (C-4), 138.5 (C-6), 135.1 (C-9), 126.0 (C-10), 125.5 (C-11), 124.7 (C-3), 119.8 (C-1), 106.4 (C-5), 27.2 (C-γ, 3J(119Sn–13C) = 39.3 Hz), 26.2 (C-β, 2J(119Sn–13C) = 107.3 Hz), 25.7 (C-α, 1J(119/119Sn–13C) = 734.5/700.5 Hz, θ = 148.2°), 14.0 (C-δ); 119Sn NMR (112.07 MHz, CDCl3)δ: −192; 119Sn NMR (112.07 MHz, DMSO-d6)δ: −255; MS: (FAB+) [m/z] (%): [M+ + 1, 525] (100), [M+-2Bu, 411] (95), [M+-C5H3NO, 318] (25); HR-MS (FAB+) m/z: 525.0200 (Calc. for C20H26N2O2SnBr), observed: 525.0197.
10,10-Di-n-butyl-5-iodo-1,6-diaza-10-stanna-9,11-dioxatetracycle[8.7.0.01,80.12,17] heptadeca-1,3,5,7,13,15-hexaene (3g)
Compound 3g was prepared from 0.199 g (0.803 mmol) of 5-Iodosalicylaldehyde, 0.088 g (0.803 mmol) of 2-amino-3-hydroxypyridine, and 0.2 g (0.803 mmol) of dibutyltin oxide, affording 0.406 g (89%) dark red solid, m.p. dec 60–62 °C; molar conductance, ΛM (1 × 10−3 M, methanol): µS cm−1 (non-electrolyte); UV–Vis [methanol, λmáx/nm (ε M–1 cm−1)]: 236 (24740) π → π* (Arom), 389 (5329) π → π* (C=N), 471 (12028) n → π* (C=N); IR (KBr) cm−1: 1592 ν(C=N), 1236 ν(C–O), 1077 ν(C–I), 670, 599, ν(Sn–C), 526 ν(Sn–O), 449 ν(Sn–N); 1H NMR (300.52 MHz, DMSO-d6) δ: 0.73 (6H, t, J = 7.5 Hz, H-δ), 1.14–1.60 (4H, sext, J = 7.2 Hz, H-γ), 1.33–1.50 (8H, m, H-α, H-β), 6.51 (1H, d, J = 9.0 Hz, H-3), 7.07 (1H, dd, J = 1.5 Hz, J = 8.1 Hz, H-11), 7.17 (1H, dd, J = 4.5 Hz, J = 8.1 Hz, H-10), 7.58 (1H, dd, J = 2.4 Hz, J = 9.0 Hz, H-4), 7.70 (1H, dd, J = 1.2 Hz, J = 4.2 Hz, H-9), 7.94 (1H, d, J = 2.4 Hz, H-6), 9.44 (1H, s, 3J(1H–119/117Sn) = 35.5 Hz, H-7); 13C NMR (75.57 MHz, DMSO-d6) δ: 169.8 (C-2), 162.4 (C-7), 155.3 (C-12), 145.0 (C-4), 144.7 (C-6), 144.3 (C-8), 135.0 (C-9), 126.0 (C-10), 125.5 (C-11), 125.1 (C-3), 120.9 (C-1), 76.4(C-5), 27.2 (C-γ, 3J(119Sn–13C) = 37.8 Hz), 26.2 (C-β, 2J(119Sn–13C) = 105.8 Hz), 25.6 (C-α, 1J(119/117Sn–13C) = 730.0/697.5 Hz, θ = 147.7°), 14.0 (C-δ); 119Sn NMR (112.07 MHz, CDCl3) δ: −192; 119Sn NMR (112.07 MHz, DMSO-d6) δ: −253; MS: (FAB+) [m/z] (%): [M+ + 1, 573] (23), [M+-2Bu, 459] (15), [M+-C5H3NO, 366] (2); HR-MS (FAB+) m/z: 573.0061 (Calc. for C20H26N2O2SnI), observed: 573.0064.
10,10-Di-n-butyl-5-nitro-10-stanna-1,6-diaza-9,11-dioxatetracycle[8.7.0.01,80.12,17] heptadeca- 1,3,5,7,13,15-hexaene (3h)
Compound 3h was synthetized from 0.134 g (0.803 mmol) of 5-nitrosalicylaldehyde, 0.088 g (0.803 mmol) of 2-amino-3-hydroxypyridine, and 0.2 g (0.803 mmol) dibutyltin oxide, affording 0.306 g (80%) of a light red solid; m.p. dec 98–100 °C; molar conductance, ΛM (1 × 10−3 M, methanol): 2.8 S cm−1 (non-electrolyte); UV–Vis [methanol, λmáx/nm (ε M–1 cm−1)]: 206 (23,224), 238 (22,197), 278 (25,375) π → π* (Arom.), 348 (18,384) π → π* (C=N), 450 (19,606) n → π* (C=N); IR (KBr) cm−1: 1598 ν(C=N), 1228 ν(C–OArom.),1382 νsym(NO2), 1542 νas(NO2), 687, 600, ν(Sn–C), 542 ν(Sn–O), 466 ν(Sn–N); 1H NMR (300.52 MHz, DMSO-d6) δ: 0.72 (6H, t, J = 9.0 Hz, H-δ), 1.15–1.40 (8H, m, H-α, H-β), 1.41 (4H, s, H-γ, 6.69 (1H, d, J = 9.6 Hz, H-3), 7.12 (1H, dd, J = 1.2 Hz, J = 8.1 Hz, H-11), 7.21 (1H, dd, J = 4.5 Hz, J = 8.4 Hz, H-10), 7.75 (1H, dd, J = 1.5 Hz, J = 4.5 Hz, H-9), 8.14 (1H, dd, J = 3.0 Hz, J = 9.3 Hz H-4), 8.68 (1H, d, J = 2.7 Hz, H-6), 9.61 (1H, s, 3J(1H–119/117Sn) = 24.0 Hz, H-7); 13C NMR (75.57 MHz, DMSO-d6) δ: 175.1 (C-2), 162.5 (C-7), 155.4 (C-12), 144.4 (C-8), 136.4 (C-5), 135.5 (C-9), 135.3 (C-6), 131.0 (C-4), 126.4 (C-10), 124.3 (C-3), 126.0 (C-11), 117.4 (C-1), 27.8 (C-α, 1J(119Sn–13C) = 706.6 Hz, θ = 145.4°), 27.3 (C-γ, 3J(119Sn–13C) = 41.7 Hz), 26.1 (C-β, 2J(119Sn–13C) = 122.4 Hz), 13.9 (C-δ);119Sn NMR (112.07 MHz, CDCl3) δ: −189; 119Sn NMR (112.07 MHz, DMSO-d6)δ: −293; MS: (FAB+) [m/z] (%):[M+ + 1, 492] (40), [M+-2Bu, 378] (25), [M+-C5H3NO, 285] (5); MS-HR (FAB+) m/z: 492.0945 (Calc. for C20H26N3O4Sn), Observed: 492.0953.
Cytotoxicity assay
The cytotoxicity of the compounds was assessed by sulforhodamine B assay and cis-platin was used as a positive control, as described previously [30] on cancer cell lines: HCT-15 (colorectal), U-251 (glioblastoma), MCF-7 (breast), MDA-MB231 (breast), SKLU-1 (non-small cell lung), and K-562 (chronic myelogenous leukemia).
Stability
The complexes showed solubility in polar protic (methanol, ethanol) and polar aprotic solvents (dimethyl sulfoxide, dichloromethane) and chloroform. To evaluate their cytotoxicity of the prepared complexes and their stability under biological medium, solutions of the complexes 3a–3h in DMSO–phosphate buffered saline (1:9 v/v) were prepared and their UV–Vis spectra recorded subsequently, 72 h after, the absorption bands remain without change, additionally any new peaks showed up. The UV–Vis spectroscopy allowed us to corroborate the stability of organotin(IV) complexes under physiological conditions.
In vivo toxicity assay (Artemia Salina)
The brine shrimp lethality test (Artemia Salina) was used to evaluate the toxicity of all complexes as previously described [31]. Brine shrimp cysts (Artemia Salina) were incubated with nonnatural salt-water (Instant Ocean). Fifty milligrams of cysts were sprayed and hatched at 20–30 °C. After 2 days, nauplii were taken and a sample at 20 mM in DMSO was prepared for testing. Aliquots were taken and diluted from this stock solution, with deionized water to rich the required concentrations. 0.1 mL of seawater containing ten larvae and 0.1 mL solution to examine were added to every single well of 96-well microplates. The experiments were assayed three times. Additionally, a solution of DMSO was tested as the negative control. Dead larvae, were registered 24 h later in a Nikon inverted microscope (4×). To kill the shrimp 0.1 mL of ethanol was added. The LC50 value was calculated using the Reed–Muench method [32].
Results and discussion
Synthesis
The complexes 3a–h were obtained via a multicomponent reaction as depicted in Scheme 1. The corresponding 5-R-salicylaldehyde, 2-amino-3-hydroxypyridine and dibutyltin oxide were refluxed in a mixture toluene:methanol (80:20) as solvent. The reaction proceeds efficiently giving products in yields of 80–90%. All complexes are soluble in conventional organic solvents. The molar conductance in MeOH solution indicates that complexes 3a–h are in the range 2.3–65.8 Λ (ohm−1 cm2 mol–1), demonstrating the non-electrolytic nature of these complexes.

Synthesis of oganotin(IV) compounds via multicomponent reaction.
The compound 3d has been described previously. It was prepared by from the Schiff base ligand with dibutyltin dichloride and Et3N with methanol as solvent. Its antibacterial activity was assayed against Gram-positive (Bacillus subtilis and Staphylococcus aureus) and Gram-negative (Escherichia coli and Pseudomonas aeruginosa) bacteria and exhibit a remarkable activity associated to the presence of bromine in the structure [33]. However, neither the reaction yield nor the physical properties were reported. Additionally, a series dimethyltin(IV) complexes from 2-amino-3-hydroxypyridine have been also described in the literature, and the in vitro antibacterial (S. aureus) and antifungal (Aspergillus spp) activities evaluated showing high activity. For example, methyltin(IV) complexes were isolated by a two-step reaction using dimethyltin(IV) dichloride and the corresponding Schiff base ligand (prepared from 5-R-salicylaldehyde (R=Me, NO2) or 3,5-dichloro and 3,5-diiodo salicylaldehyde) and trimethylamine. However, the yields of the reactions were not reported either for the synthesis of the ligands or the complexes [34].
Electronic absorption spectra
The solution spectra of 3a–3h in MeOH showed a similar band pattern. The absorption bands in the 200–470 nm region were assigned to the π–π* electronic transitions of the aromatic ring 206–288 nm (εmax = 16232–27331 M−1 cm−1); the second band was observed in the range of 348–380 nm (εmax = 5329–18384 M−1 cm−1) due to the π–π* transition of azomethine (C=N); and the band at 450–471 nm (εmax = 120028–19606 M−1 cm−1) to a n–π* (C=N) transition.
NMR
The 1H and 13C spectra of the complexes (see Supplementary Material) were assigned by using one and two-dimensional NMR experiments (DEPT, COSY, HSQC, and HMBC) in CDCl3. The multiplicity patterns and the 1H NMR integration were consistent with the structures represented in Scheme 1. Evidence of the coordination of tin was provided by the presence of characteristic signals of the butyl groups attached to the tin atom in the aliphatic region, a triple signal for methyl group in the range of 0.72–0.76 ppm, and multiple signals for methylenes from 1.15 to 1.53 ppm. All complexes showed the well-defined signals for the aromatic and pyridine rings. Additionally, azomethine (C=N) was observed as a single signal in the range from 9.44 to 9.61 ppm, which shows the expected satellite signals attributable to spin-spin coupling 3J(119/117Sn−1H) among the tin nucleus and the azomethine proton, with values from 35 to 47 Hz (Table 1) indicating a configuration E and corroborating of the Sn–N bond formation [35, 36].
The 13C NMR spectra displayed the expected signals for all complexes. The methyl and methylene groups of the butyl fragment bonded to the tin were detected in the aliphatic region from 13.0 to 27.8. Additionally, for all complexes it was also possible to calculate the coupling constants values 1J(13C–119/117Sn), 2J(13C–119/117Sn), and 3J(13C–119/117Sn) from the satellite signals from butyl groups (Table 1). This permitted us to confirm the 13C NMR assignation and calculate the C–Sn–C bond angle in solution, using the values of the coupling constant 1J(13C–119Sn) and the Holecek equation [37], which were found in the range of 135–148° (Table 1). The signals for the aromatic and pyridine rings were clearly identified. The azomethine carbon C-7 was observed at 162.4–163.9 ppm.
A sharp single signal in the range of −189 to −194 ppm was detected in the 119Sn NMR for all compounds revealing the presence of discrete species (see Supplementary Material), the spectroscopic evidence suggests a pentacoordinate environment in a non-coordinated solvent (CDCl3) [37]. However, when the experiments were conducted in DMSO-d6, chemical shifts in the region from −231 to −293 ppm were found (Table 2), the change in the chemical shift to lower frequencies observed corresponds to hexacoordinated species [37], and indicates an increase in the coordination number associated to the coordination of the DMSO to Sn (IV).
X-ray crystallography
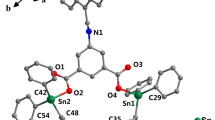
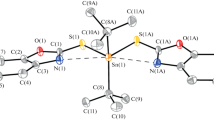
Crystals of compound 3a were obtained by slow evaporation from methanol and the X-ray structure analysis showed the dinuclear molecular structure represented in Fig. 1, and it reveals the formation of a dimeric assembly in solid state through an intermolecular O(2)…Sn(1) bridge forming a Sn2O2 four-membered ring. The bond distance Sn(1)…O(2)#1 is 2.715 (2) Å. This value is shorter than the sum van der Waals radii (3.69 Å). In the dinuclear unit, the two ligands act as tridentates and the SnBu2 fragment is coordinated by the two oxygen atoms from the deprotonated phenols O(1) and O(2) and the iminic nitrogen. The ligand chelates the tin atom and forms two rings: a six and a five-membered ring. The torsion angle of C(12)–O(2)–Sn(1)–C(13) is −178.8, which evidences the coplanarity of the five-membered ring and the pyridine ring. The Sn atom is hexacoordinated due to the intermolecular bridge through the Sn(1) and the oxygen O(2). The oxygens atoms bridging between the two Sn atoms form a Sn2O2 planar ring, which is formed with the oxygen atom from the five-membered ring. The Sn–O(2) bond length of 2.148 (2) Å is slightly shorter than Sn–O(1), 2.170(2) Å. Both are comparable with those informed in the literature for tin (IV) compounds [38] and shorter than those described for the analogous complexes whose organic substituents bonded to the tin atom are methyls [39, 40]. Additionally, both bonds are slightly longer than the sum of the covalent radii Sn–O (2.05 Å), evidencing a strong interaction between the two atoms. The Sn–N bond distance is 2.217(2) Å. The value of the C–Sn–C bond angle is 147.4 (1)°, and this indicates that it is non-linear and shorter than the O–Sn–O bond angle 157.4 (1)°. A comparison of the C–Sn–C bond angle value with that obtained in solution (135°) from the NMR coupling constants reveals that this angle is shorter in solution than in solid state. The data collection, refinement parameters, and selected bond lengths and angles are displayed in Tables 3, 4.

Crystal structure of complex 3a; all hydrogen atoms were omitted for clarity. Thermal ellipsoids at 50% level probability
Cytotoxic activity (cancer cell lines)
The cytotoxicity of complexes 3a–3h was assessed by using the sulforhodamine B microculture assay to estimate cell growth on a group of human cancer cell lines U-251 (glioblastoma), K-562 (chronic myelogenous leukemia), HCT-15 (human colorectal cancer), MCF-7 (human breast cancer), SKLU-1 (non‐small‐cell lung cancer), and MDA-MB-231 (human breast cancer), Table 5. The synthetized complexes have structural similarities such the pyridine and phenolate moiety, the principal structural difference is the substitution on the aromatic ring in the five position. The substituents, H, CH3, F, Cl, Br, and I were selected as representative examples to analyze their biological response in terms of isosteric and bioisoteric replacement. The biosisosters are substituents or groups with similar physical or chemical properties, and are used to diminish the toxicity, modify or increase the biological activity with minor changes in the chemical structure, such as: hydrogen-fluorine replacement, that has and influence in the pharmacological properties, associated to formation of interactions with biological receptors due to the remarkable electronegativity differences, while the steric and lipophilic properties are preserved. The replacement chlorine-methyl that has been used in series of biologically active compounds because of its similarity in size and lipophilicity [41]. The cytotoxic data indicated that all tested complexes were significantly more cytotoxic than cis-platin, which was used as a positive control. Although the organotin (IV) complexes exhibited varying levels of cytotoxicity, in general, the complexes 3a–3h were found to be highly cytotoxic to K-562 with lower cytotoxicity toward HCT-15 and MCF-7, indicating that organotin (IV) complexes possess selective cytotoxicity profiles against distinct cell lines. As summarized in Table 5, complexes 3a–3h displayed potent antiproliferative activity against K-562 with IC50 values around 0.20–0.42 μM where he complex 3f (IC50 = 0.20) emerges as the most cytotoxic meanwhile, the nitro derivative (3h) was the most cytotoxic toward U-251 (IC50 = 0.25), HCT-15 (IC50 = 0.83), and MDA-MB-231 (CI50 = 0.27) cells.
Regarding cytotoxic activity against SKLU-I cells, the tert-butyl substituent (3c) exhibited a potent activity as well as 3e and 3h. On the other hand, for antiproliferative activity toward MCF-7, the iodine derivative (3g) showed a high toxicity (IC50 = 0.23).
On the basis of the IC50 values, the comparative analysis of the two breast cancer cell lines MCF-7 and MDA-MB-231 indicates that the organotin complexes 3a–3h were less active toward MCF-7 cells.
In general terms, the isosteric exchange of hydrogen by fluorine produced only a slight increase in the cytotoxicity for U-251, HCT-15, and MDA-MB 251 cell lines. The contrary was observed for pyridoxal organotin (IV) complexes [28]. However, a substantial increase in the cytotoxicity was evident for MCF-7 due to the bioisosteric replacement of methyl by iodine.
Brine shrimp lethality (Aremia Salina)
The toxicity analysis is normally conducted in mice. However, the present tendency is to use different approaches to reduce animal assays. The brine shrimp (Artemia Salina) lethality test is an appropriated preliminary in vivo toxicity analysis susceptible to several of chemical substances. It is well known that the accessibility of the eggs, the simplicity of incubation and the fast growing of the nauplii, as well as the simplicity of preserving Artemia population provide a practical and efficient bioassay for carrying out toxicological evaluations. Furthermore, Artemia Salina test is used frequently to evaluate toxicity as an approach to human toxicity and as evidence of antitumoral activity [42]. This allows us to compare the results with the cancer cell line inhibition experiment.
The toxicity of organotin(IV) complexes 3a–3h, was assessed using the brine shrimp lethality assay. According to the results shown in Table 6, the complexes screened for lethality in Artemia Salina showed toxicity. The complex 3e LC50 (9.77 μM) displayed strong toxicity. In contrast, the complexes 3h, 3d, and 3c were less toxic LC50 (39.81, 26.92, and 22.39 μM). The data also showed that the isosterical replacement of hydrogen by fluorine decreased the toxicity in Artemia Salina. Meanwhile, in cancer cell lines, the replacement had a slight effect on the cytotoxic response. In contrast, the bioisosteric replacement of methyl by Iodine showed a slight increase in the toxicity toward Artemia Salina.
The comparison between LC50 and IC50 mean values indicates that the complexes 3a–3h did not show relation in the grade of toxicity between the two assays. 3a–3h exhibited strong cytotoxicity in cancer cell lines (IC50 0.20–1.31 μM) and low toxicity in Artemia Salina (LC50 9.70–39.81 μM).
Conclusions
In summary, herein we inform the synthesis of organotin(IV) complexes, 3a–3h, prepared by a multicomponent reaction. All isolated complexes were examined by their in vitro cytotoxic activity toward cancer cell lines: HCT‐15 (human colorectal cancer), U‐251 (glioblastoma), K‐562 (chronic myelogenous leukemia), SKLU‐1 (non‐small‐cell lung cancer), MCF‐7, and MDA-MB-231 (human breast cancer). In general, the synthesized organotin complexes exhibited a similar cytotoxicity profile. However, they were more active toward K-562 than HCT-15. A comparative analysis between the two breast cancer cell lines tested indicated that the complexes 3a–3h are more active toward MDA-MB231 than MCF-7. The isosteric exchange of hydrogen by fluorine had a minor impact on the antiproliferative activity of U-251, HCT-15, and MDA-MB231 cell lines, and the bioisosterical exchange of methyl by iodine resulted in a substantial increase in the cytotoxic activity against MCF-7. The isosteric replacement is an alternative that should be explored in the design of organotin complexes as cytotoxic agents.
References
Abdel Aziz AA, Seda SH. Synthesis, structural features and biochemical activity assessment of N,N′-bis-(2-mercaptophenylimine)-2,5-thiophenedicarboxaldehyde Schiff base and its Co(II), Ni(II), Cu(II) and Zn(II) complexes. Appl Organomet Chem. 2017;31:e3879.
Abu-Dief AM, Mohamed IMA. A review on versatile applications of transition metal complexes incorporating Schiff bases. Beni-Suef Univ J Basic Appl Sci. 2015;4:119–33.
Al Zoubi W, Al-Hamdani AAS, Kaseem M. Synthesis and antioxidant activities of Schiff bases and their complexes: a review. Appl Organomet Chem. 2016;30:810–7.
Al Zoubi W, Ko YG. Schiff base complexes and their versatile applications as catalysts in oxidation of organic compounds: part I. Appl Organomet Chem. 2017;31:e3574.
Asadi M, Asadi Z, Sadi SB, Zarei L, Baigi FM, Amirghofran Z. Synthesis, characterization and the interaction of some new water-soluble metal Schiff base complexes with human serum albumin. Spectrochim Acta A Mol Biomol Spectrosc. 2014;122:118–29.
Qin W, Long S, Panunzio M, Biondi S. Schiff bases: a short survey on an evergreen chemistry tool. Molecules. 2013;18:12264–89.
da Silva CM, da Silva DL, Modolo LV, Alves RB, de Resende MA, Martins CVB, et al. Schiff bases: A short review of their antimicrobial activities. J Adv Res. 2011;2:1–8.
Ganguly R, Sreenivasulu B, Vittal JJ. Amino acid-containing reduced Schiff bases as the building blocks for metallasupramolecular structures. Coord Chem Rev. 2008;252:1027–50.
Liu X, Hamon J-R. Recent developments in penta-, hexa- and heptadentate Schiff base ligands and their metal complexes. Coord Chem Rev. 2019;389:94–118.
Gasser G, Ott I, Metzler-Nolte N. Organometallic anticancer compounds. J Med Chem. 2011;54:3–25.
Haas KL, Franz KJ. Application of metal coordination chemistry to explore and manipulate cell biology. Chem Rev. 2009;109:4921–60.
Hambley TW. Developing new metal-based therapeutics: challenges and opportunities. Dalton Trans. 2007;43:4929–37.
Ndagi U, Mhlongo N, Soliman ME. Metal complexes in cancer therapy—an update from drug design perspective. Drug Des Dev Ther. 2017;11:599–616.
Zaki M, Arjmand F, Tabassum S. Current and future potential of metallo drugs: revisiting DNA-binding of metal containing molecules and their diverse mechanism of action. Inorg Chim Acta. 2016;444:1–22.
Tzubery A, Tshuva EY. Cytotoxic titanium(IV) complexes of salalen-based ligands. Eur J Inorg Chem. 2017;12:1695–705.
Dasari S, Bernard Tchounwou P. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78.
Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107:1387–407.
Oberoi HS, Nukolova NV, Kabanov AV, Bronich TK. Nanocarriers for delivery of platinum anticancer drugs. Adv Drug Deliv Rev. 2013;65:1667–85.
Mjos KD, Orvig C. Metallodrugs in medicinal inorganic chemistry. Chem Rev. 2014;114:4540–63.
Adeyemi JO, Onwudiwe DC. Organotin(IV) dithiocarbamate complexes: chemistry and biological activity. Molecules. 2018;23:2571.
Antonenko TA, Shpakovsky DB, Vorobyov MA, Gracheva YA, Kharitonashvili EV, Dubova LG, et al. Antioxidative vs. cytotoxic activities of organotin complexes bearing 2,6-di-tert-butylphenol moieties. Appl Organomet Chem. 2018;32:e4381.
Devi J, Yadav J, Singh N. Synthesis, characterisation, in vitro antimicrobial, antioxidant and anti-inflammatory activities of diorganotin(IV) complexes derived from salicylaldehyde Schiff bases. Res Chem Intermed. 2019;45:3943–68.
Hu L, Wang H, Xia T, Fang B, Shen Y, Zhang Q, et al. Two-photon-active organotin(IV) complexes for antibacterial function and superresolution bacteria imaging. Inorg Chem. 2018;57:6340–8.
Iqbal H, Ali S, Shahzadi S. Antituberculosis study of organotin(IV) complexes: a review. Cogent Chem. 2015;1:1.
Dubé D, Brideau C, Deschênes D, Fortin R, Friesen RW, Gordon R, et al. 2-Heterosubstituted-3-(4-methylsulfonyl)phenyl-5-trifluoromethyl pyridines as selective and orally active cyclooxygenase-2 inhibitors. Bioorg Med Chem Lett. 1999;9:1715–20.
Galván-Hidalgo JM, Chans GM, Ramírez-Apan T, Nieto-Camacho A, Hernández-Ortega S, Gómez E. Tin(IV) Schiff base complexes derived from pyridoxal: synthesis, spectroscopic properties and cytotoxicity. Appl Organomet Chem. 2017;31:e3704.
Galvan-Hidalgo JM, Gomez E, Ramirez-Apan T, Nieto-Camacho A, Hernandez-Ortega S. Synthesis and cytotoxic activity of dibutyltin complexes derived from pyridoxamine and salicylaldehydes. Med Chem Res. 2015;24:3621–31.
Galván-Hidalgo JM, Ramírez-Apan T, Nieto-Camacho A, Hernández-Ortega S, Gómez E. Schiff base Sn(IV) complexes as cytotoxic agents: synthesis, structure, isosteric and bioisosteric replacement. J Organomet Chem. 2017;848:332–43.
Sheldrick GM. SHELXS-2014 and SHELXL-2014. Germany: University of Gottingen; 2014.
Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst. 1991;83:757–66.
Rocha-Del Castillo E, Gómez-García O, Andrade-Pavón D, Villa-Tanaca L, Ramírez-Apan T, Nieto-Camacho A, et al. Dibutyltin(IV) complexes derived from L-DOPA: synthesis, molecular docking, cytotoxic and antifungal activity. Chem Pharm Bull. 2018;66:1104–13.
Reed LJ, Muench H. A simple method of estimating fifty per cent endpoints. Am J Epidemiol. 1938;27:493–7.
Sedaghat T, Habibi R, Motamedi H, Khavasi HR. Synthesis, structural characterization and antibacterial activity of diorganotin(IV) complexes with ONO tridentate Schiff bases containing pyridine ring. Chin Chem Lett. 2012;23:1355–8.
Öztaş NA, Yenişehirli G, Ancın N, Öztaş SG, Özcan Y, İde S. Synthesis, characterization, biological activities of dimethyltin(IV) complexes of Schiff bases with ONO-type donors. Spectrochim Acta A Mol Biomol Spectrosc. 2009;72:929–35.
Kawakami K, Tanaka T. Remarkable configurational changes of some dimethyltin complexes derived from ONO tridentate schiff bases in strong donor solvents. J Organomet Chem. 1973;49:409–15.
Matsubayashi G-e, Tanaka T, Nishigaki S, Nakatsu K. Nuclear magnetic resonance studies of some N′-substituted pyridine-2-carbaldimine adducts of n-butyltrichlorotin(IV) and the molecular structure of n-butyltrichloro(N′-phenylpyridine-2-carbaldimine-NN′)-tin(IV). J Chem Soc Dalton Trans. 1979;3:501–5.
Holeček J, Nádvorník M, Handlíř K, Lyčka A. 13C and 119Sn NMR spectra of Di-n-butyltin(IV) compounds. J Organomet Chem. 1986;315:299–308.
Roy M, Roy S, Devi NM, Singh CB, Singh KS. Synthesis, structural characterization and antimicrobial activities of diorganotin(IV) complexes with azo-imino carboxylic acid ligand: crystal structure and topological study of a doubly phenoxide-bridged dimeric dimethyltin(IV) complex appended with free carboxylic acid groups. J Mol Struct. 2016;1119:64–70.
Basu Baul TS, Chaurasiya A, Lyčka A, Rojas-León I, Höpfl H. Molecular aggregations of bicyclodioxazastannone produced from multicomponent reactions involving functionalized 2-hydroxybenzaldehydes, α- or β-amino acids and a dimethyltin precursor. J Organomet Chem. 2019;898:120859.
Öztaş SG, Şahin E, Ancın N, İde S, Tüzün M. Structural and spectral studies of N-(3-hydroxypyridine-2-yl)salicylideneimine and N-(3-hydroxypyridine-2-yl)-5-bromosalicylideneimine and their dimethyltin(IV) complexes. Z Kristallogr Cryst Mater. 2003;218:492.
Patani GA, LaVoie EJ. Bioisosterism: a rational approach in drug design. Chem Rev. 1996;96:3147–76.
de las M, Oliva M, Gallucci N, Zygadlo JA, Demo MS. Cytotoxic activity of argentinean essential oils on Artemia salina. Pharm Biol. 2007;45:259–62.
Acknowledgements
The authors thank PAPIIT IN204417 and IN206020 for financial assistance and the technical support for the determination of the IR and mass spectrometry to María del Carmen García, María del Rocío Patiño, and Javier Pérez.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Galván-Hidalgo, J.M., Roldán-Marchán, D.M., González-Hernández, A. et al. Organotin (IV) complexes from Schiff bases ligands based on 2-amino-3-hydroxypyridine: synthesis, characterization, and cytotoxicity. Med Chem Res 29, 2146–2156 (2020). https://doi.org/10.1007/s00044-020-02630-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-020-02630-4