
Abstract
The cytotoxic and antitumour activities of five organotin complexes (1–5) with o-hydroxy-benzoic or p-hydroxy-benzoic acids were evaluated in a series of in vitro and in vivo experiments. All complexes exhibited strong cytotoxic activity against all cancer cells lines, whereas complexes 1, 2 and 4 induced apoptosis at significantly lower doses than complexes 3 and 5. Human cancer cells treated with increasing concentrations of complexes 1, 2 and 4 gradually lost their ability to form colonies. Only complexes 1 and 2 inhibited colony formation efficiency in rat leiomyosarcoma cells. Histopathology of liver and kidney showed mild damage and lung oedema after a single injection of 10 mg/kg body wt of complex 1 to Wistar rats. At higher doses (100 mg/kg body wt) brain stem oedema was observed. Daily administration of tumour-bearing Wistar rats (leiomyosarcoma) with 1 mg/kg body wt of complex 1 until death reduced the mean tumour growth rate by more than 3× fold and prolonged mean survival time by 120%. These findings indicate that the organotin complexes with ortho- or para-hydroxy-benzoic acids possess potent cytotoxic and antitumour activity and they could be used as potential chemotherapeutic agents.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cisplatin is one of the most potent anticancer agent used alone or in combination with chemotherapy agents to treat various human tumours (Miller et al. 2010). However, the serious side effects (mainly nephrotoxicity) exerted by cisplatin (Miller et al. 2010) in conjunction with the increased tumour resistance (Koberle et al. 2010) lead to the development of platinum and non-platinum metal complexes with similar anticancer activity (Garbutcheon-Singh et al. 2011). Especial interest was invested on organotin derivatives characterised by potent cytotoxic and anticancer activity (Arjmand et al. 2014; Amir et al. 2014; Muhammad and Guo 2014). Organotin derivatives can cause direct DNA damage (Liu et al. 2006) acting through different mechanisms than cisplatin, e.g., the rise of cytosolic Ca2+ which leads the cells to apoptosis (Aw et al. 1990) or the inhibition of essential macromolecules synthesis and the mitochondrial metabolism (Alama et al. 2009). Moreover, the cytotoxicity exerted by some organotin complexes is correlated with the inhibition of peroxidation of linoleic acid induced by lipoxygenase (Abdellah et al. 2009).
Organotin derivatives show potent cytotoxic properties that can be even 200 times higher of cisplatin (Verginadis et al. 2011). Ttriphenyltin(VI) complexes of non-steroidal anti-inflammatory drugs were found to be highly cytotoxic against the cancer cell lines MCF-7 (breast), T-24 (urinary bladder) and A549 (lung) (Dokorou et al. 2011), while triphenyltin 2-(triphenyltinmercapto) nicotinate exhibited an IC50 as low as 5.5 nM against leiomyosarcoma (LMS) cells (Verginadis et al. 2011). Moreover, organotin(IV) derivatives of L-cysteine show cytotoxic activity against six human cancer cells lines: leukaemia (HL60), liver (Bel7402), stomach (BGC823), cervix epithelial (HeLa), nasopharyngeal (KB) and lung (PG) (Chasapis et al. 2004). Hence, these compounds exert cytotoxic activity against cisplatin-sensitive and cisplatin-resistant cell lines. In vivo experiments in tumour bearing animals (LMS) with organotin metal complexes prolonged the mean survival time (MST) of the animals (Verginadis et al. 2011; Metsios et al. 2012) and inhibited tumour growth (Verginadis et al. 2011) with minor side effects (mainly in the kidney) (Verginadis et al. 2011; Metsios et al. 2012) or liver damage (Metsios et al. 2012).
The aim of this study was to examine the cytotoxic effects of organotin compounds with ortho- and para-hydroxybenzoic acid on malignant cell lines and to evaluate their toxic effects as well as their antitumour properties in a series of in vitro and in vivo experiments.
Materials and methods
Cell lines and culture conditions
Human cell lines MCF-7 (breast adenocarcinoma - ATCC®, HTB-22™), HeLa (cervical cancer—ATCC®, CCL-2™), U2-OS (human osteosarcoma—ATCC®, HTB-96™) and MRC-5 (normal foetal lung fibroblasts - ATCC®, CCL-171™) as well as cells (LMS) isolated from tumours histological identified as LMS in Wistar rats (Avdikos et al. 2007) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, 1% penicillin/streptomycine and 0.5% L-glutamine (1% L-glutamine for MRC-5 cells). Cells were maintained in culture dishes at 37 °C in a 5% CO2 and 95% air atmosphere incubator.
Synthesis of organotin compounds 1–5
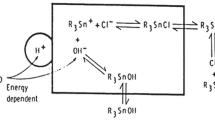
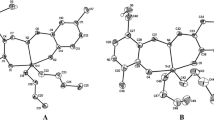
The Organotin(IV) complexes 1–5 were synthesised according to the method described earlier (Abdellah et al. 2009). Thus, a suspension of the ligand (2-hydroxybenzoic acid or 4-hydroxybenzoic acid) in distilled water was treated with an equimolar amount of KOH, and a clear solution was immediately formed. A methanolic solution of di-organotin(IV) or tri-organotin(IV) chloride was then added to the above solution. The white precipitation formed is filtered off, washed with water and it dried in vacuo over silica gel. The reaction route for the preparation is shown in Fig. 1. The analytical and spectroscopic data (m.p., elemental analysis, FT-IR, 1H-NMR and ESI-MS) of 1–5 are identical to those reported in reference Abdellah et al. (2009) (Table 1). The molecular diagram of complexes 1 and 2 is presented in Fig. 2.

Reaction route for the preparation of 1–5 (Abdellah et al. 2009)

Molecular diagram with atomic numbering of a [Me2Sn(o-BZA)2] (1) and b [(n-Bu)2Sn(o-BZA)2] (2)
Cell proliferation assay
Cell viability was evaluated using MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) assay. Cancer cells (LMS, HeLa, MCF-7 and U2-OS) and normal cells (MRC-5) were cultured at a density of 3 × 103 and 5 × 103, respectively, in 96-well plates for 24 h. There were then washed with PBS and a solution containing different concentrations of complexes 1–5 or cisplatin (diluted in DMSO) was added. After incubation for another 48 h, 50 μl of MTT were added in each well from a stock solution (5 mg/ml) and the cells were incubated for another 4 h at 37 °C. The formed formazan crystals were solubilized with 50 μl of DMSO under gentle shaking and absorbance was determined at 570 nm (subtract background absorbance measured at 690 nm) using a microplate spectrophotometer (Multiskan Spectrum, Therno Fisher Scientific, Waltham, USA). Data were expressed as a percentage of control measured in the absence of complexes (DMSO control). All experiments were performed in triplicate.
Colony formation efficiency assay
LMS, HeLa, MCF-7 and U2-OS cells were seeded into six-well plates at a density of 6 × 104 cells per well and incubated for 24 h. Cells were then washed with PBS, treated with media containing various concentrations of complexes 1–5 in DMSO and incubated for 48 h. LMS cells were also treated with cisplatin (0.5, 1 and 2.5 μΜ). Supernatants and cells were collected and counted using Neubauer cytometer and 5 × 102 cells were seeded onto six-well plates. After 7 days, dishes were fixed with methanol–acetic acid 3:1 at −20 °C for 10 min, dried and stained with Giemsa dye 1:10 for 30 min. The dishes were then washed, dried and the colonies were counted.
Quantification of apoptosis
LMS cells were seeded into six-well plates at a density of 6 × 104 cells per well and incubated for 24 h before the experiment. Cells were washed with PBS, treated with media containing various concentrations of complexes 1–5 diluted in DMSO and cisplatin (IC50 value) and incubated for 48 h. Supernatants and cells were collected, centrifuged and cell pellets were suspended in calcium buffer 1× at a rate 105 cells/100 μl. Cells were stained with 5 μl of Annexin V-fluorescein isothiocynate and 4 μl of propidium iodide in a dark room for 15 min. DNA content was determined on a FACScan flow cytometer (Partec ML, Partec GmbH, Germany).
DNA fragmentation assay
DNA fragmentation was estimated as described by Verginadis et al. (Verginadis et al. 2011). Briefly, LMS cells were treated with complexes 1–5 for 48 h. Supernatants and cells were collected and incubated with lysis buffer. Fragmented DNA was digested with RNAse and Proteinase K and precipitated with isopropanol. Finally, DNA fragments were fractionated by 1.2% agarose gel and were visualised by ethidium bromide under ultraviolet light.
Animals and treatment
Female Wistar rats, aged 3–4 months and weighting 242 ± 13 g, were reared at controlled room temperature (20 ± 2 °C), with periodical lighting (12 h light/12 h dark). Standard Wistar rat diet and water ad libitum were used in all the experiments. Experiments on animals were handled with human care in accordance with the European Union directive for the care and the use of laboratory animals (EEC Directive 2010/63/EU) and according to the permission number 20EEP02.
Acute toxicity
Forty female Wistar rats were divided into four groups (ten animals per group) and injected intra-peritoneally (i.p.) with a single dose of 1, 10 and 100 mg/kg body wt of complex 1 in order to achieve a logarithmic increase of the dose. Control group was injected with a similar volume of the solvent (1 ml of PBS/DMSO (7:1)). All animals were starved for 3–4 h before treatment. Twenty minutes after the administration, the rats were allowed to access freely food and water. The animals were observed for their behaviour twice a day. Body weight (body wt) was recorded every 2 days and used as an index of toxicity. The percentage change in body wt was calculated as: Body wt (%) = [(mean body wt on day D − mean body wt on day 1)/mean body wt on day 1] × 100, where D = day of the experiment.
The animals survived were sacrificed by ether on day 15. The internal organs were removed en bloc and fixed in 10% buffered formalin. Tissue sections of formalin-fixed, paraffin-embedded tumours were stained with haematoxylin–eosin. The sections were examined under light microscope.
Subacute toxicity
Subacute toxicity was studied in 30 female Wistar rats that were divided into three groups (10 animals per group), and received i.p. daily dosages of solvent (0.5 ml), 0.5 and 1 mg/kg body wt of complex 1 for 4 weeks. Animals were monitored for their behaviour once per day. Body weight was recorded every 2 days and was used as an index of toxicity, while the percentage change in body wt was calculated as previously. All animals were sacrificed by ether at the end of experiment. Immediate autopsy, fixation of the organs in 10% buffered formalin and a histopathological examination were performed as described previously (Verginadis et al. 2011).
Antitumour activity in vivo
Twenty-nine female Wistar rats were inoculated with 3 × 106 LMS cells/1 ml Hank’s balanced salt solution on the upper scapula after anaesthetisation (mixture of anaesthesia was composed of ketamine 10% (Imalgene), midazolame 40% (Roche) and saline solution 0.9, 50% to a total volume of 0.5 ml per animal). After the appearance of a palpable tumour mass (smaller than 1.5 cm diameter), the inoculated animals were divided into two groups: the control group (CG, consisted of 15 animals) and the treatment group (TG, consisted of 14 animals). All animals in TG Group were treated daily till death with 1 mg/kg body wt of complex 1 (diluted in 0.5 ml PBS/DMSO 7:1), while the animals of the CG received daily 0.5 ml PBS/DMSO(7:1). The animals were observed for their behaviour once a day. After the animals’ death, an autopsy, fixation of the organs in 10% formaldehyde and a histological examination were performed. For both groups, the MST of the animals, tumour weight inhibition (TWI%), mean tumour weight and mean tumour growth rate (MTGR) were calculated. The tumour growth rate for each animal was calculated by the following equation: Tumour growth rate (g/d) = tumour weight (g)/survival time (days), while TWI% was calculated as: TWI% = [(mean control tumour weight-mean treatment tumour weight)/ mean control tumour weight] × 100.
Statistical analysis
The data are expressed as mean ± S.D., and the difference between two groups was evaluated using Student’s t-test and two-way analysis of variance (SPSS version 17.0, Statistical Package for the social Sciences software, SPSS, Chicago, USA). A probability level of p < 0.05 was used to establish significance.
Results
Cytotoxicity
Complexes 1–5 were tested for their in vitro cytotoxic activity against all cell lines with the MTT assay. The IC50 values for cell growth proliferation after 48 h of incubation were calculated. Cisplatin was used as the reference treatment. Moreover, the survival of MRC-5 cells, at the IC50 values of the complexes against the LMS cells, was also calculated.
All complexes showed potent cytotoxic activity against the three human cells lines ranging from 108 to 724 nM for the MCF-7 cells, 21 to 295 nM for the HeLa cells and 97 to 820 nM for the U2-OS cells. The IC50 value of complex 2 was 304 and 211 times lower than the corresponding value of cisplatin for the MCF-7 and U2-OS cells, respectively while the IC50 value of complex 4 was 264 times lower for the HeLa cells. Among the tested compounds, complexes 3 and 5 presented the weaker cytotoxic activity (Table 2).
Furthermore, the cytotoxic activities of complexes 1–5 and cisplatin was studied in LMS cells. Complex 1 presented the lower IC50 (31 nM) followed by complex 2 (43 nM) and 4 (45 nM). The IC50 values for complexes 3 and 5 were higher than the other 3 complexes but still extremely lower compared to cisplatin. Trypan blue assay was previously performed by Abdellah et al. (2009) in order to determine the cell membrane integrity. The complexes have high activity against LMS cells while complex 4 was the most active one. Except from complex 5 the rest of the complexes exhibited no inhibitory activity on growth proliferation of MRC-5 cells (IC50 concentration of complexes against LMS cells) (Table 3).
Colony formation efficiency after treatment
The cells were tested for their ability to form colonies after drug treatment compared to untreated cells. Complexes 1 and 4 presented a dose-dependent inhibition of MCF-7, HeLa and U2-OS cells colony formation. All human cancer cell lines treated with dosages higher than 20 nM of complex 2 lost their ability to form colonies (Fig. 3)

Formation of colonies for a MCF-7, b HeLa and c U2-OS cells after treatment with the complexes 1, 2 and 4. Different letters indicate the significant differences (p < 0.05) among the current and the exact previous (lower) dose
After treatment with complexes 1 and 2, LMS cells gradually lost their ability to form colonies. Complex 2 at the highest dose (100 nM) completely abolished the formation of colonies while LMS cells resisted treatment with complex 4 and were able to form colonies even at the highest concentration of the complex (Fig. 4a). Complexes 3 and 5 failed to inhibit colony formation at concentrations lower than 100 nM (results not shown). On the other hand, cisplatin also exerted a dose-dependent inhibition of colony formation of LMS cells but at significantly higher doses than complexes 1, 2 and 4 (Fig. 4b).

Formation of colonies for LMS cells after treatment with a the complexes 1, 2 and 4 and b cisplatin. Different letters indicate the significant differences (p < 0.05) among the current and the exact previous (lower) dose
Flow cytometry and DNA fragmentation
Flow cytometry assay was used to quantify apoptotic or necrotic death in LMS cells treated with compounds 1–5 or the IC50 of cisplatin (5.5 μΜ). Compared to untreated LMS cells that showed a total of 6.2 ± 1.3 and 1.7 ± 0.5% apoptotic and necrotic cell death, respectively, the cells treated with complex 1 at 50 nM showed 22.4 ± 1.8% apoptosis and 2.1 ± 0.2% necrosis. At higher concentrations (100 nM), the cells showed 21.1 ± 2.8% apoptosis and 1.6 ± 0.6% necrosis. As for complexes 2 and 4, cells showed 33.8 ± 2.3 and 13.7 ± 2.6 apoptosis and 1.8 ± 0.1 and 2.2 ± 1.4% necrosis after treatment with 50 nM of the complexes, respectively and 38.7 ± 2.2 and 28.9 ± 1.6% apoptosis and 7.5 ± 0.9 and 2.8 ± 1.2% necrosis after treatment with 100 nM. Complexes 3 and 5 induced apoptosis of LMS cells at higher concentrations (>300 nM) (results not shown). Apoptosis induced by cisplatin (IC50 value) was estimated at 35.0 ± 3.0% and necrosis at 3.3 ± 1.6% (Fig. 5a, b).

Quantification of apoptotic and necrotic cells after treatment with 50 and 100 nM of complex 1, 2 and 4 and cisplatin (CP, 5.5 μΜ) for 48 h. *Statistically significant from untreated cells, p < 0.05. Data are presented as mean ± SD
The characteristic laddering pattern observed at the electrophoresis gel after treatment of LMS cells with 150 and 100 nM of complexes 1 and 2 and 4, respectively, indicated that cells underwent apoptosis triggered by the DNA damage and fragmentation (Fig. 5c).
Acute toxicity
Ninety percent of the animals at the highest dosage group (100 mg/kg body wt) died within an hour after the i.p. administration of complex 1, while 40% of the animals that received 10 mg/kg body wt died within 24 h. None of the animals from the other two groups (control and 1 mg/kg body wt) died during the experiment (Table 4).
All animals that received 1 mg/kg body wt of complex 1 did not show any signs of toxicity. The absence of toxicity was verified by the histologic examination. Animals injected with 10 mg/kg body wt of complex 1 showed reduced mobility and difficulty in breathing during the first 24 h. Animals presented intent seizures right after the administration of the complex 1 at the highest dose (100 mg/kg body wt). The main organs targeted were liver, kidneys and lungs. The diseased animals showed cerebral oedema as well (Fig. 6).

Acute toxicity report. Healthy samples from the control group: a liver, b lung, c kidney and d brain stem and representative histological images from animals that received 100 mg/kg body wt of complex 1: e liver, f lung, g kidney and h brain stem. Tissue sections were stained with Hematoxylin–eosin. Liver and lung sections were magnified by ×200 while kidney and brain stem sections by ×400
Subacute toxicity
None of the animals presented any signs of toxicity. Mild kidney damage was only observed at animals treated daily with the highest dose (1 mg/kg body wt of complex 1) (results not shown).
Antitumour activity
Daily administration of tumour bearing Wistar rats with 1 mg/kg with complex 1 until death prolonged the MST (120% increase) and reduced the MTGR by more than 3× fold and with minimal side effects (Table 5). Furthermore tumour weight inhibition was calculated as TWI% = 60.2%. Long terms survival was noted for LTS = 1/15. This animal belonged in TG group and was excluded from the above calculations.
It is worth mentioning that total tumour regression was observed in one animal treated with complex 1. The histopathological examination showed an abscess cavity at the site of the inoculation. This animal was the only one with an observed lung metastasis, which was resistant to treatment.
Discussion
Ever since cisplatin was introduced in cancer therapy, many non-conventional platinum compounds and non-platinum metal complexes were designed and tested. Among them, organotin derivatives have shown potent cytotoxic and antitumour properties (Arjmand et al. 2014).
Verginadis et al. showed that a triorganotin complex with 2-mercapto-nicotinic acid inhibits LMS cell proliferation with the corresponding IC50 value of the complex being significantly lower than that of cisplatin (41 nM versus 25 μM) (Verginadis et al. 2011). Di-n-butyltin(IV) compounds related to salicylic acid were found to be 4–10×-fold more effective than cisplatin against A204, MCF-7 and T24 human tumour cells (Ross et al. 1991). This is in accordance with our results where all complexes exerted higher cytotoxic activity than cisplatin against MCF-7, HeLa, U2-OS and LMS cells.
Flow cytometry revealed that complexes 1, 2 and 4 caused LMS cell death through apoptosis and inhibited human cancer cells and LMS cell’s ability to form colonies. Moreover, electrophoresis of the LMS cell’s DNA showed that apoptosis was triggered by the DNA damage and fragmentation after incubation with complexes 1, 2 and 4. Such DNA damage is shown for several organotin derivatives for instance tributiltin (Liu et al. 2006). The inability of complexes 3 and 5 to induce apoptosis at similar, low doses as complexes 1, 2, and 4 a well as to inhibit colony formation could be possibly attributed to an alternative apoptotic mechanism and specifically to the rise of intracellular Ca2+ that would lead the cell to apoptosis (Nakatsu et al. 2007; Reader et al. 1999; Viviani et al. 1995) but would also allow the cells to develop after the complex was removed from the culture media. This is only a hypothesis that would need additional experiments to be verified although the relative lower cytotoxic activity of complexes 3 and 5 compared to 1, 2, and 4 suggest that they should be excluded from further research. Apoptosis seems to be the main type of cell death caused by many organotin derivatives even though the path that leads to apoptosis may differ between different complexes (Nakatsu et al. 2007; Grondin et al. 2007; Tada-Oikawa et al. 2008). Diorganotin(IV) and triorganotin(IV) complexes of meso-tetra(4-sulfonatophenyl) porphine induce apoptosis in A375 human melanoma cells via both death receptor-mediated and mitochondrial apoptotic pathways (Amir et al. 2014), while triphenyl tin benzimidazolethiol induces mitochondrial-mediated apoptosis in human cervical cancer cells (Hoti et al. 2003). On the other hand, trimethyl tin seems to cause both apoptotic and necrotic death in human hepatoma G2 cells (Cai et al. 2009).
Organotin derivatives are known to cause neurotoxicity and bile duct damage (Boyer 1989; Winship 1988). Complex 1 caused the death of 90% of the animals after a single dose of 100 mg/kg within an hour of the i.p. administration. Since the main pathological findings (brain stem and lung oedema and mild liver and kidney damage) were not capable to cause the animal’s death, a possible cause was metabolic acidosis (Matsuno-Yagi and Hatefi 1993). This is a very likely explanation considering that all animals presented Kussmaul type of breath before death. Similar findings were reported after i.p. administration of triphenyltin 2-(triphenyltinmercapto) nicotinate with kidney damage being more severe reaching total tubular necrosis (Verginadis et al. 2011). A simple i.p. administration of the complex at lower doses (10 mg/kg BW) showed toxic effects and caused death to the 40% of the animals within 24 h. Repeated administration of the complex at very low doses (1 mg/kg/day) for 30 days caused only slight kidney damage. The same dosage was used to study the antitumour effects of the complex.
Tumour-bearing animals treated with complex 1 showed prolongation of survival time and tumour growth inhibition. Other organotin derivatives have higher TWI% values (Verginadis et al. 2011), while oral administration of triethyltin(IV)-lupinylsulfide hydrochloride showed as high as 90% tumour volume inhibition (Muhammad and Guo 2014). Not all organotin complexes are active in vivo. Organotin complexes with salicylic acid derivatives are inactive against L1210 leukaemia in mice (Gielen et al. 1993), while triphenyltin 5-sulfosalicylate and triphenyltin 5-aminosalicylate offer no protection against murine colon carcinoma colon 26 (Gielen 1995). Both treated and untreated animals showed slight liver damage and minor lung oedema, which could be attributed to the tumour, but only treated animals showed kidney damage, as expected by the subacute toxicity results. It is noteworthy to mention that one of the treated animals showed total tumour regression at the site of the inoculation. Unfortunately, this animal was the only one presenting lung metastasis. The lung metastasis presented to this animal was resistant to treatment and showed no signs of necrosis during the histological evaluation.
Conclusions
In this study, we evaluated the cytotoxic and antitumour properties of five organotin complexes (1–5) with o-hydroxy-benzoic or p-hydroxy-benzoic acids. Amongst them, complex 1 presented the highest cytotoxic activity against LMS cells (IC50 = 31 ± 1.6 nM), complex 2 against MCF-7 and U2-OS cells (IC50 = 108 ± 2.6 nM and 97 ± 2.1 nM, respectively) and complex 4 against HeLa cells (IC50 = 21 ± 1.1 nM). The induction of apoptosis was identified as the mechanism of LMS cell death. Complexes 1 and 2 showed a higher inhibitory effect on LMS colony formation efficiency compared to complex 4. The following acute toxicity study revealed a mildly toxic effect of complex 1 at a dose of 10 mg/kg body weight in the liver, kidney and lungs of adult Wistar rats. Tumour-bearing Wistar rats administrated daily with 1 mg/kg body weight of complex 1 lived longer than the control rats (45.1 versus 20.7 days) probably due to the deceleration of tumour growth rate (1.6 versus 5.2 g/day) caused by the complex. In short, our results demonstrated that complex 1 exerted potent cytotoxic activity, tumour growth retardation and prolongation of MST in tumour-bearing (LMS) Wistar rats. Thus, it could be used as a potential candidate for cancer treatment at primary site of the tumour. Further investigation is needed to confirm the antitumour properties of the other organotin complexes.
References
Abdellah MA, Hadjikakou SK, Hadjiliadis N, Kubicki M, Bakas T, Kourkoumelis N, Simos YV, Karkabounas S, Barsan MM, Butler IS (2009) Synthesis, characterization, and biological studies of organotin(IV) derivatives with o- or p-hydroxybenzoic acids. Bioinorg Chem Appl 2009:542979
Alama A, Tasso B, Novelli F, Sparatore F (2009) Organometallic compounds in oncology: implications of novel organotins as antitumor agents. Drug Discov Today 14:500–508
Amir MK, Khan S, Rehman Z, Shah A, Butler IS (2014) Anticancer activity of organotin(IV) carboxylates. Inorg Chim Acta 423:14–25
Arjmand F, Parveen S, Tabassum S, Pettinari C (2014) Organo-tin antitumor compounds: Their present status in drug development and future perspectives. Inorg Chim Acta 423:26–37
Avdikos A, Karkabounas S, Metsios A, Kostoula O, Havelas K, Binolis J, Verginadis I, Hatziaivazis G, Simos I, Evangelou A (2007) Anticancer effects on leiomyosarcoma-bearing Wistar rats after electromagnetic radiation of resonantradiofrequencies. Hell J Nucl Med 10:95–101
Aw TY, Nicotera P, Manzo L, Orrenius S (1990) Tributiltin stimulates apoptosis in rats thymocytes. Arch Biochem Biophys 283:46–50
Boyer IJ (1989) Toxicity of dibutyltin, tributyltin and other organotin compounds to humans and to experimental animals. Toxicology 55:253–298
Cai J, Wang M, Li B, Wang C, Chen Y, Zuo Z (2009) Apoptotic and necrotic action mechanisms of trimethyltin in human hepatoma G2 (HepG2) cells. Chem Res Toxicol 22:1582–1587
Chasapis CT, Hadjikakou SK, Garoufis A, Hadjiliads N, Bakas T, Kubicki M, Ming Y (2004) Organotin(IV) derivatives of L-cysteine and their in vitro anti-tumor properties. Bioinorg Chem Appl 2004:43–54
Dokorou V, Primikiri A, Kovala-Demertzi D (2011) The triphenyltin(VI) complexes of NSAIDs and derivatives. Synthesis, crystal structure and antiproliferative activity. Potent anticancer agents. J Inorg Biochem 105:195–201
Garbutcheon-Singh KB, Grant MP, Harper BW, Krause-Heuer AM, Manohar M, Orkey N, Aldrich-Wright JR (2011) Transition metal based anticancer drugs. Curr Top Med Chem 11:521–542
Gielen M (1995) Tin-based antitumour drugs: new developments. Met Based Drugs 2:99–103
Gielen M, de Vos D, Meriem A, Boualam M, el Khloufi A, Willem R (1993) The acute toxicity and in vivo antitumour activity against L1210 leukemia of triphenyltin 3,5-diisopropylsalicylate, bis (di-n-butyl-(s)-2-pyrrolidone-5-carboxylato)tin oxide and di-n-butyltin bis(3-amino-4-methylbenzoate). Vivo 7:171–174
Grondin M, Marion M, Denizeau F, Averill-Bates DA (2007) Tributyltin induces apoptotic signaling in hepatocytes through pathways involving the endoplasmic reticulum and mitochondria. Toxicol Appl Pharmacol 222:57–68
Höti N, Ma J, Tabassum S, Wang Y, Wu M (2003) Triphenyl tin benzimidazolethiol, a novel antitumor agent, induces mitochondrial-mediated apoptosis in human cervical cancer cells via suppression of HPV-18 encoded E6. J Biochem 134:521–528
Köberle B, Tomicic MT, Usanova S, Kaina B (2010) Cisplatin resistance: preclinical findings and clinical implications. Biochim Biophys Acta 1806:172–182
Liu HG, Wang Y, Lian L, Xu LH (2006) Tributiltin induces DNA damage as well as oxidative damage in rats. Environ Toxicol 21:166–171
Matsuno-Yagi A, Hatefi Y (1993) Studies on the mechanism of oxidative phosphorylation. ATP synthesis by submitochondrial particles inhibited at F0 by venturicidin and organotin compounds. J Biol Chem 268:6168–6173
Metsios A, Verginadis I, Simos Y, Batistatou A, Peschos D, Ragos V, Vezyraki P, Evangelou A, Karkabounas S (2012) Cytotoxic and anticancer effects of the triorganotin compound [(C6H5)3Sn(cmbzt)]: an in vitro, ex vivo and in vivo study. Eur J Pharm Sci 47:490–496
Miller RO, Tadagavadi RK, Ramesh G, Reeves WB (2010) Mechanisms of cisplatin nephrotoxicity. Toxins (Basel) 2(11):2490–2518
Muhammad N, Guo Z (2014) Metal-based anticancer chemotherapeutic agents. Curr Opin Chem Biol 19:144–153
Nakatsu Y, Kotake Y, Ohta S (2007) Concentration dependence of the mechanisms of tributyltin-induced apoptosis. Toxicol Sci 97:438–447
Reader S, Moutardier V, Denizeau F (1999) Tributyltin triggers apoptosis in trout hepatocytes: the role of Ca2+, protein kinase C and proteases. Biochim Biophys Acta 1448:473–485
Ross M, Gielen M, Lelieveld P, De Vos D (1991) Cytotoxic activity of di-n-butyltin(IV)(X-A-B-Y) compounds related to salicylic acid against human tumour cells. Anticancer Res 11:1089–1091
Tada-Oikawa S, Kato T, Kuribayashi K, Nishino K, Murata M, Kawanishi S (2008) Critical role of hydrogen peroxide in the differential susceptibility of Th1 and Th2 cells to tributyltin-induced apoptosis. Biochem Pharmacol 75:552–561
Verginadis II, Karkabounas S, Simos Y, Kontargiris E, Hadjikakou SK, Batistatou A, Evangelou A, Charalabopoulos K (2011) Anticancer and cytotoxic effects of a triorganotin compound with 2-mercapto-nicotinic acid in malignant cell lines and tumor bearing Wistar rats. Eur J Pharm Sci 42:253–261
Viviani B, Rossi AD, Chow SC, Nicotera P (1995) Organotin compounds induce calcium overload and apoptosis in PC12 cells. Neurotoxicology 16:19–25
Winship KA (1988) Toxicity of tin and its compounds. Advers Drug React Acute Poison Rev 7:19–38
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Agiorgiti, M.S., Evangelou, A., Vezyraki, P. et al. Cytotoxic effect, antitumour activity and toxicity of organotin derivatives with ortho- or para-hydroxy-benzoic acids. Med Chem Res 27, 1122–1130 (2018). https://doi.org/10.1007/s00044-018-2135-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-018-2135-7