
Abstract
Rhizoma Coptidis is an alkaloid-rich herbal drug used in Chinese herbal medicine. Currently, conventional methods for screening and isolating alkaloids are labor-intensive and time-consuming. In the present study, ultrafiltration liquid chromatography with photodiode array detection coupled to electrospray ionization tandem mass spectrometry (ultrafiltration liquid chromatography-photodiode array detector-electrospary ionization mass spectrometry (LC-PDA-ESI/MS)) were applied to screen and identify α-glucosidase inhibitors in R. Coptidis. High-speed countercurrent chromatography and reverse-phase medium-pressure liquid chromatography were applied to separate and isolate the active constituents. As a result, five major compounds in R. Coptidis were screened and identified as α-glucosidase inhibitors by ultrafiltration LC-PDA-ESI/MS. Five ligands, jatrorrhizine, epiberberine, coptisine, palmatine, and berberine, were isolated by reverse-phase medium-pressure liquid chromatography and high-speed countercurrent chromatography. The purities of the five compounds as determined by high-performance liquid chromatography were 76.20, 75.13, 82.24, 93.78, and 92.01%, respectively. The results indicate that systematic isolation of bioactive components in R. Coptidis guided by ultrafiltration LC-PDA-ESI/MS is a feasible and efficient technique that could be extended to separation of other enzyme inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In recent years, traditional Chinese medicine has gained attention because of its advantages compared to conventional medicinal practices. According to statistical data from the World Health Organization, approximately 171 million patients with diabetes mellitus (DM) were reported in 2000, with a predicted increase to 366 million by 2030 (Wild et al. 2004). Prevention and treatment of DM has become a worldwide health problem. α-Glucosidase inhibitors are widely used to treat type 2 DM. These compounds inhibit α-glucosidase, preventing the release of α-glucose from food carbohydrates, and decrease postprandial blood glucose and insulin levels (Derosa and Maffioli 2012). The adverse effects of α-glucosidase inhibitors include diarrhea, flatulence, and abdominal discomfort. Therefore, in order to ameliorate adverse effects, it is essential to discover α-glucosidase inhibitors from low toxicity natural sources.
R. Coptidis, also known as “Huanglian,” is the dry rhizome of Coptis chinensis Franch, Coptis deltoidea C. Y. Cheng et Hsiao, or Coptis teeta Wall (family Ranunculaceae). R. Coptidis is one of the 50 fundamental herbs in Chinese herbalism, commonly used to treat a variety of diseases, including headache, fever, constipation, and diarrhea (Ren et al. 2007). In addition, R. Coptidis extracts and decoctions have demonstrated significant antimicrobial activity against a variety of organisms including bacteria, viruses, fungi, protozoans, and helminths.
Alkaloids are the main bioactive chemical constituents of R. Coptidis. Our preliminary in vitro screening assay revealed the presence of α-glucosidase inhibitors in R. Coptidis. However, it is still unclear which compounds in the extract are active. The present study investigates the potent inhibition of α-glucosidase activity in the extract of R. Coptidis, and illustrates the use of this extract for prevention and treatment of DM.
Screening and identification of active compounds from crude extracts are important research tasks, yet the systematical elucidation of the pharmacological role of natural products is challenging. For this reason, increasing attention is directed to researching new methods for isolating active components.
Bioassay-guided fractionation, purification, and structure identification are conventional procedures for screening and identifying compounds in plants. Conventional bioassay-guided fractionation has been used to select reference drugs. However, it causes loss of activity owing to dilution effects or decomposition during the isolation process, is labor-intensive and inefficient. Therefore, it is necessary to find rapid and effective strategies to screen and identify the active compounds in R. Coptidis.
Ultrafiltration LC-MS has been widely used for screening and analysis of biologically active compounds from botanical extracts because of its online screening ability, high speed, ease of operation, and high reliability. Ultrafiltration facilitates the separation of ligand–receptor complexes from unbound compounds, whereas LC-MS identifies the ligands. Moreover, low sample consumption, the lack of need for immobilization, and reuse of enzymes are the most important advantages of ultrafiltration LC-MS for high-throughput screening and identification of active compounds (Delavenne et al. 2014).
Isolating and purifying alkaloids is challenging because of the complexity of the compounds involved. In recent decades, several methods have been reported for the separation of alkaloids from R. Coptidis extracts. These methods include colorimetry (Qian and Yang 2014), capillary electrophoresis (Chen et al. 2008), microwave-assisted extraction (Zou et al. 2010), micellar electrokinetic chromatography (Chang and Sun 2006), and high-performance liquid chromatography (HPLC) with different detection systems, such as mass spectrometry (Luo et al. 2005; Yan et al. 2012). Recently, High-speed countercurrent chromatography (HSCCC), a support-free liquid–liquid partition chromatography, has been used successfully as an efficient method in the isolation and purification of alkaloids. Since 1987, analytical HSCCC is used to separate a variety of natural products, including alkaloids, anthraquinones, flavonoids, and saponins (Tong and Yan 2007; Xie et al. 2014). HSCCC has the advantage of sample recovery without employing a solid phase, preventing the irreversible absorption of analytes. It is especially suitable for preparative isolation of natural products in industry, compared with conventional liquid–solid separation methods (Zhang et al. 2010). In this study, using HSCCC as the separation system created the problem of excessive exposure to toxic organic solvents, including ethyl acetate, methanol, n-heptane, and in particular, chloroform and methylene chloride. Therefore, it is vital to develop a new system that can lower the environmental pollution caused by toxic solvents.
Reverse-phase medium-pressure liquid chromatography (RP-MPLC) is an effective, simple, and economical separation method with short separation times and a column packed with relatively large particles to reduce operating pressure and material costs. The MPLC system with an ultraviolet (UV) detector and an automatic fraction collector can be adjusted and expanded with a large loading amount according to different purification tasks, and the eluting reagent can be collected according to peak and retention times, or the volume of the fractions during the elution process.
In our previous research, an LC-MS assay for screening and characterizing active compounds from natural products was developed. Here, HSCCC and RP-MPLC were employed to isolate α-glucosidase inhibitors from R. Coptidis, guided by ultrafiltration-based alkaloid screening. Five active compounds were obtained. The structures of the five α-glucosidase inhibitors from R. Coptidis are shown in Fig. 1. This is the first report on using HSCCC and RP-MPLC guided by ultrafiltration experiments for the screening and isolation of α-glucosidase inhibitors from R. Coptidis.

Chemical structures of target compounds isolated in this study. (1) Jatrorrhizine; (2)Epiberberine; (3) Coptisine; (4) Palmatine; (5) Berberine
Experimental
Apparatus
HSCCC separation was carried out on a DE Spectrum HSCCC (Dynamic Extractions, Slough, UK). The multilayer coil separation column was prepared by winding a 28 m × 2.6 mm inner diameter (I.D.). A polytetrafluoroethene tube was directly fixed onto one of the holders, forming multiple coiled layers to give a total capacity of 28 mL (for the analytical separation column) and 125 mL (for the semi-preparative separation column). The β value changed from 0.33 at the internal terminal to 0.58 at the external terminal (R = 8 cm, β = r/R, where R is the distance between the central axis of the centrifuge and the holder axis, and r is the distance from the coil to the holder shaft). The rotation speed was adjusted to a range of 0–1600 r.p.m.; 1400 r.p.m. was used in the present study. The HSCCC system was equipped with a solvent delivery module of BT 8100 (Biotronic, Maintal, Germany), an integrator of D-2500 (Merck Hitachi, Darmstadt, Germany), and an injection valve with a sample loop of 10 mL. RP-MPLC separation was performed in an Isolera one MPLC system (Biotage, Switzerland). ESI-MS was performed on an LCQ FLEET ion-trap mass spectrometer (Thermo Finnigan, San Jose, USA). HPLC was carried out on a Waters 2695 coupled with a Waters 2998 PDA (Milford, USA). NMR spectral acquisition was carried out with a Bruker AV 500 spectrometer operated at 500 MHz for 1H, and at 125 MHz for 13C (Bruker BioSpin, Rheinstetten, Germany).
Reagents and materials
R. Coptidis was purchased from Hebei Kangpai Medicinal Store (Hebei, China) and was identified by Professor Chunming Liu (Changchun Normal University, Changchun, China). α-Glucosidase (E.C.3.2.1.20) from Saccharomyces cerevisiae was obtained from Sigma Aldrich (St Louis, MO, USA) and ammonium acetate from Fluka (Bueke, Switzerland). The ultrafiltration chamber used was Microcon YM-10 (Millipore, Bedford, MA) with a molecular weight cutoff of 10,000 Da. The solvents for HPLC analysis were of LC grade and purchased from Fisher Chemicals (Pittsburgh, PA, USA). The other solvents utilized for preparation of the crude extract of R. Coptidis and fractionation, ultrafiltration, and isolation were all of analytical grade and were purchased from Beijing Chemical Engineering Company (Beijing, China). All aqueous solutions were prepared with pure water produced by a Milli-Q water (18.2 MU) system (Millipore, Bedford, MA, USA).
Preparation of the R. Coptidis extract
R. Coptidis (5.0007 g) was extracted with 50 mL of ethanol/water (70:30, v/v) for 12 h, three times. After filtration, the combined extracts were concentrated under reduced pressure and re-dissolved in 20 mL methanol (crude extract concentration 200 mg mL−1). The extract was stored at 4 °C before ultrafiltration liquid chromatography-photodiode array detector-electrospary ionization mass spectrometry (LC-PDA-ESI/MS).
R. Coptidis (198.001 g) was successively extracted with 600 mL of ethanol/water (70:30, v/v) for 12 h, four times. The mixture was filtered and the extraction was repeated twice. Next, the combined filtrate was dried at 40 °C, and re-dissolved in 300 mL of water. The solution was defatted twice. The lower phase was extracted successively five times using 300 mL of n-butanol each time. Finally, 13.714 mg of crude extract was obtained. The extract was stored at −20 °C before HSCCC and RP-MPLC separation.
Determination of α-glucosidase inhibitory activity
The inhibition assay of α-glucosidase was performed according to the modified literature method (Li et al. 2009).
Reaction mixture containing 40 μL of the tested compound in phosphate buffer and 40 μL of 10 mM phosphate buffer (pH 6.80) containing 0.5 U mL−1 α-glucosidase, was added to each well and incubated at 37 °C for 5 min, followed by the addition of 20 μL of 0.5 mM p-nitrophenyl α-d-glucopyranoside (Biochem) to the mixture of treatment-terminated wells. After the plate was incubated at 37 °C for 30 min, 100 μL of 0.1 mol L−1 sodium carbonate solution was added to each well to stop the reaction. Absorbance was immediately recorded at 405 nm with a Tecan GENiOS multifunctional microplate reader (Mannedorf, Switzerland). Control contained the same reaction mixture with equal volume of phosphate buffer substituted in place of the solution of the tested compound. Acarbose (Bayer) dissolved in water was used as a positive control. The inhibition (%) was calculated via [(A a−A b)/A a] × 100%, where A a and A b are the absorbance of the control and sample tested, respectively. The IC50 values of samples (the concentration of a sample that is required for 50% inhibition of yeast α-glucosidase) were calculated with nonlinear regression analysis and expressed as the mean of three repeated experiments.
Screening by ultrafiltration LC-PDA-ESI/MS procedure
Ultrafiltration LC-PDA-ESI/MS screening based on MS includes the following three steps: incubation, ultrafiltration, and analysis. A total of 20 μL of α-glucosidase (E.C.3.2.1.20) (0.2 U, dissolved in 10 mM ammonium acetate buffer, pH 6.8) and 5 μL of R. Coptidis extract (200 mg mL−1, dissolved in methanol) was added to 105 μL of 10 mM ammonium acetate buffer (pH 6.8). The mixture was incubated at 37 °C for 40 min and ultrafiltrated using a Sigma 1–14 centrifuge (Sigma, Germany) with ultramembrane filter (Microcon YM-10, Millipore, Bedford, MA) at 10,000 × g for 10 min at room temperature. The filter was washed three times by centrifugation with 100 μL aliquots of ammonium acetate buffer (pH 6.8) to separate the unbound low-mass molecules. The bound compounds were released by adding 100 μL of methanol solution (50:50, v/v) (pH 3.3), followed by centrifugation at 10,000 × g for 10 min, which was repeated three times. A control experiment without the enzyme was carried out before each screening. All of the binding assays were performed in duplicate and analyzed in triplicate. The ultrafiltrates were injected directly into the LC-PDA-ESI/MS system for analysis.
LC-PDA-ESI/MS was performed on a Thermo Scientific LCQ Fleet ion trap mass spectrometer equipped with electrospray ionization (ESI) source (Thermo, USA). The analysis was carried out with an Agilent extend-C18 column (250 × 4.6 mm I.D., 5 μm) and a C18 guard column. The mobile phase consisted of methanol (A) and 0.2 % aqueous acetic acid (B) with the gradient elution conditions as follows: 22–24% A at 0–2 min, 24–27% A at 2–25 min, 27–30% A at 25–30 min, 30–32% A at 30–35 min, and 32–40% A at 35–60 min; the chromatogram was acquired at 225 nm. ESI-MS experiments in positive mode were used for data collection. The following settings were applied to the instrument: sheath gas flow rate, 50 bar; auxiliary gas flow rate, 10 bar; electrospray voltage of the ion source, 4.5 kV; capillary voltage, 35 V; capillary temperature, 350 °C. The full scan of ions ranged from 100 to 1500 Da. In-source collision induced dissociation (ISCID) experiment was performed to obtain detailed structural information.
HSCCC partition coefficient (K) value examination
There were 14 sets of solvent systems that were chosen for performing K evaluation: ethyl acetate/n-butanol/acetonitrile/water (4.5:0.5:0.5:4.5, 4.5:0.5:3.0:4.5, 4.5:0.5:2.5:4.5, 4.5:0.5:1.5:4.5, v/v/v/v), ethyl acetate/n-butanol/ethanol/water (3.0:2.0:0.5:6.0, 3.0:1.7:0.5:6.0, 3.0:1.6:0.5:7.0, 3.0:1.5:0.5:6.0, 3.0:0.5:1.5:2.5, 3.0:1.0:1.0:6.0, 3.0:1.2:0.7:6.0, v/v/v/v), and n-butanol/methanol/water (4.5:1:4.5, 4.5:3:4.5, 4.5:1.5:4.5, v/v/v). Approximately 3.0 mL of each phase was delivered into a 10 mL test tube, and approximately 20 mg of the extract from R. Coptidis was added. The test tube was capped, shaken vigorously for 1 min, and left until complete separation of phases occurred. After two clear layers were formed, 1.0 mL of each phase was dissolved in 1.0 mL of methanol and quantitatively analyzed to determine the K values of the compounds at 225 nm. Two-phase solvent systems with different ratios were prepared and equilibrated in a separation funnel at room temperature. The K value was calculated in the peak area of the target compound in the upper phase divided by that in the lower phase (K = A s/A m).
Separation by HSCCC
Separation assays were performed to isolate potential α-glucosidase inhibitors from R. Coptidis. In this study, two stages of HSCCC separation were employed to separate target compounds. In the first stage, jatrorrhizine, epiberberine, and coptisine were separated. The multilayer coil was filled with ethyl acetate/n-butanol/ethanol/water (3.0:2.0:0.5:6.0, v/v/v/v) as the stationary upper phase. The lower phase was pumped into the column in a tail to head direction at a flow rate of 2.0 mL min−1 and the coiled column was rotated at 1400 r.p.m. After the two phases had established the hydrodynamic equilibrium in the rotating column, 1.0 mL of a sample solution (a mixture of 1.0 mL of each phase containing 23.26 mg of extract of R. Coptidis) was injected into the injection valve for the analytical isolation. The eluted compounds were monitored at 225 nm and collected into test tubes with a fraction collector.
In the second stage, palmatine and berberine were separated. The multilayer coil was filled with the ethyl acetate/n-butanol/ethanol/water (3.0:1.0:1.0:6.0, v/v/v/v) as the stationary upper phase. Sample solution (2 mL, a mixture of 1.0 mL of each phase containing 23.26 mg of extract of R. Coptidis for the semi-preparative isolation, and 1.0 mL for the analytical isolation) was injected into the column. The rotating column was stopped at 250 min for the semi-preparative isolation. The other parameters for HSCCC were the same as in the first stage. The fractions were collected according to the elution profile and evaporated in a rotary evaporator. The residues were stored in a refrigerator prior to HPLC and ESI-MSn analyses. After the separation was completed, the retention rate of the stationary phase was computed as the volume of the stationary phase collected from the column to the total column capacity.
RP-MPLC separation procedure
RP-MPLC separation was carried out in an Isolera one MPLC system (Biotage Switzerland), with a Biotage® SNAP Cartridge KP-C18-HS column (12 g). The binary mobile phase consisted of methanol (solvent A) and 0.2% aqueous acetic acid (solvent B). A binary gradient elution program with a flow rate of 8 mL/min was used for the separation. The system was run with a gradient program: 76% B, 5 min; 76–73% B, 10 min; 73–70% B, 15 min; 70–60% B, 18 min; 60% B, 20 min. The column was first equilibrated with 15 mL 80% B and the crude extract (10 mg), which was dissolved in 2 mL of 50% methanol, was then injected into the column. Peaks of interest were monitored at 225 nm by a UV detector. The fractions (10 mL) were collected into test tubes with a fraction collector, and further analyzed by HPLC. All fractions of the same compound determined by HPLC were combined.
HPLC analysis for K value and purity examination
To analyze the K values of potential α-glucosidase inhibitors and monitor the peak fractions from HSCCC and RP-MPLC, a Waters 2695 Series HPLC system equipped with a SunFireTM C18 (250 × 4.6 mm I.D., 5 μm, Waters, MA, USA) column, a Waters 2998 DAD, a quaternary pump, degasser, and a thermostatic auto sampler were used. The gradient elution was performed as follows: 22–24% A at 0–2 min, 24–27% A at 2–25 min, 27–30% A at 25–30 min, 30–32% A at 30–35 min, 32–40% A at 35–60 min; flow rate: 0.8 mL min−1; detection wavelength: 225 nm; column temperature 30 °C.
Results and discussion
Ultrafiltration LC-ESI-MS α-glucosidase assay
The ultrafiltration LC-PDA-ESI/MS method is used for screening bioactive compounds from complex mixtures, particularly for natural products with minimal sample preparation. When the extract of R. Coptidis is incubated with α-glucosidase, active ligands bind to α-glucosidase, and unbound small molecules can be separated from alkaloid-α-glucosidase complexes or α-glucosidase using an ultrafiltration membrane. In this manner, the ultrafiltration chamber functions as a solution-phase extraction device, retaining alkaloids tightly bound to the receptor, while washing away unbound compounds. The alkaloid–receptor complex can be disrupted by adding organic solvents or changing the pH value, and the released alkaloids can be identified by LC-PDA-ESI/MS analysis.
The trapped ligands from R. Coptidis were analyzed by HPLC after incubation with α-glucosidase and ultrafiltration affinity purification. Compared with samples incubated with denatured enzyme, the peaks of the compounds incubated with α-glucosidase showed higher intensities after being specifically bound to α-glucosidase. The LC-DAD profiles of ligands (potential α-glucosidase inhibitors) in R. Coptidis are shown in Fig. 2. Five peaks eluting at 12.3, 15.2, 18.7, 21.0, and 24.9 min, respectively, showed binding affinity to α-glucosidase.

HPLC chromatograms obtained by ultrafiltration chromatography of R. Coptidis extract (a); Screening procedure for binding to α-glucosidase. The solid line represents the experiment with α-glucosidase (b), and the dashed line represents the experiment with denatured α-glucosidase (c). (1) Jatrorrhizine; (2) Epiberberine; (3) Coptisine; (4) Palmatine; (5) Berberine
The MSn data of the chemical compounds from R. Coptidis were acquired in positive ion mode. Six peaks appeared in the HPLC–MS total ion chromatograms. Compounds related to peaks 1–6 in Fig. 2 exhibited intense protonated molecular ions [M+H]+ at m/z 338, 336, 320, 352.8, 336.2, and 327.3, confirming the molecular weights of peaks 1–6 of Fig. 2 to be 337, 335, 319, 351.8, 335.2, and 326.3, respectively (Table 1). The ISCID experiments also further validated that peaks 1–5 of Fig. 2 were indeed jatrorrhizine, epiberberine, coptisine, palmatine, and berberine respectively, as reported in literature (Li et al. 2010); the compound related to peak 6 has not been identified in this study.
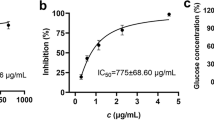
The activities of the compounds that inhibit α-glucosidase were estimated by in vitro assays to further examine the α-glucosidase inhibitors from R. Coptidis. Palmatine and berberine display remarkable inhibitory activity towards α-glucosidase, compared to acarbose (an inhibition rate of 50.47% at 1.0 mg/mL). Inhibitory activity order of the compounds is palmatine > berberine > coptisine > jatrorrhizine > epiberberine (Table 2). The data obtained demonstrate that the extract considerably inhibits α-glucosidase, indicating the usefulness of screening and isolating active compounds of the R. Coptidis extract.
Selection of two-phase solvent systems of HSCCC
The two-phase solvent system plays an important role in successful separation of target compounds in HSCCC. Partition coefficient (K) is the most important parameter in solvent system selection. It should be close to 1 to get an efficient separation and a suitable run time. If the K value is much smaller than 1, the solutes will be eluted close to each other near the solvent front, which may result in loss of peak resolution; if it is much greater than 1, the solutes will be eluted in excessively broad peaks, leading to extended elution time (Shi et al. 2007). Moreover, the settling time of the two-phase solvent system should be less than 30 s, and a satisfactory retention of the stationary phase should be attained. For the separation of alkaloids such as berberine and jatrorrhizine, the chloroform/methanol/dilute hydrochloric acid solvent system has been successfully used in HSCCC. However, this solvent system is toxic to humans and the environment because it contains chloroform and methanol. Therefore, we chose low-toxic solvents such as ethyl acetate and n-butanol for our experiment.
In order to get an optimal range of K values (Table 3), efficient separation, suitable separation time, and valid retention of stationary phase, three solvent systems (ethyl acetate/n-butanol/acetonitrile/water, ethyl acetate/n-butanol/ethanol/water, and n-butanol/methanol/water) at different volume ratios were tested according to the chemical properties and polarities of alkaloids. The K values of compounds 1, 2, and 3 were evaluated, as the ratio of the two K values (α = K 1/K 2, where K 1 > K 2) should be greater than 1.5 for semi-preparative multilayer separation column according to rule 5 of the “HSCCC Golden rules” (Ito 2005). The K value of coptisine should not be close to that of jatrorrhizine and epiberberine, and the K value of palmatine should not be close to that of berberine and coptisine.
The K value data indicates that when n-butanol/methanol/water systems (4.5:1:4.5, 4.5:1.5:4.5, 4.5:3:4.5, v/v/v) were used as the two-phase solvent system, the compounds dissolved readily in the upper phase but scarcely in the lower phase. This proves that the solvent systems composed of ethyl acetate/n-butanol/acetonitrile/water or n-butanol/methanol/water could not be used to elute the compounds. When n-butanol/methanol/water (4.5:1:4.5, 4.5:1.5:4.5 and 4.5:3:4.5, v/v/v), ethyl acetate/n-butanol/acetonitrile/water (4.5:0.5:0.5:4.5, 4.5:0.5:1.5:4.5, 4.5:0.5:2.5:4.5, 4.5:0.5:3.0:4.5, v/v/v/v) systems were used, the compounds jatrorrhizine, epiberberine, and coptisine dissolved readily in the upper phase but scarcely in the lower phase. Therefore, the solvent systems mentioned above were not considered further. When the solvent systems of ethyl acetate/n-butanol/ethanol/water (3.0:2.0:0.5:6.0, 3.0:1.7:0.5:6.0, 3.0:1.6:0.5:7.0, 3.0:1.5:0.5:6.0 and 3.0:1.2:0.7:6.0, v/v/v/v) were used as the two-phase solvent system, all compounds dissolved in the upper and lower phase. The K values of compounds jatrorrhizine, epiberberine, and coptisine changed from 0.49 to 2.82, but the K values of palmatine and berberine increased from 3.36 to 12.41. When the solvent systems of ethyl acetate/n-butanol/ethanol/water (3.0:1.0:1.0:6.0 and 3.0:0.5:1.5:2.5, v/v/v/v) were used as the two-phase solvent system, the K values of palmatine and berberine changed from 1.61 to 3.17, but the K values of jatrorrhizine, epiberberine, and coptisine increased from 0.18 to 0.94, and jatrorrhizine could not be dissolved in the upper phase. Therefore, the solvent systems of ethyl acetate/n-butanol/ethanol/water (3.0:2.0:0.5:6.0, 3.0:1.7:0.5:6.0, 3.0:1.6:0.5:7.0, 3.0:1.5:0.5:6.0, and 3.0:1.2:0.7:6.0, v/v/v/v) were chosen as candidate solvent systems for the separation of jatrorrhizine, epiberberine, and coptisine, and the solvent systems of ethyl acetate/n-butanol/ethanol/water (3.0:1.0:1.0:6.0 and 3.0:0.5:1.5:2.5 v/v/v/v) were chosen as candidate solvent systems for the separation of palmatine and berberine. When the solvent system of ethyl acetate/n-butanol/ethanol/water (3.0:1.5:0.5:6.0, v/v/v/v) was used, the K ratios were K 2/K 1 = 1.46 and K 3/K 2 = 1.62, but the settling time of the two-phase solvent system was unacceptably high. After a series of experiments, the solvent system of ethyl acetate/n-butanol/ethanol/water (3.0:2.0:0.5:6.0, v/v/v/v) was chosen for the separation of jatrorrhizine, epiberberine, and coptisine. With the same method, the solvent system of ethyl acetate/n-butanol/ethanol/water (3.0:1.0:1.0:6.0, v/v/v/v) was chosen for the separation of palmatine and berberine. Due to the large K values, palmatine and berberine could be separated satisfactorily with this system according to the chromatogram, with the K value ratio (K 5/K 4) = 1.28.
HSCCC separation of potential α-glucosidase inhibitors from R. Coptidis
HSCCC was chosen for the procedure. HSCCC is a continuous liquid–liquid partition chromatography based on partitioning of compounds between two immiscible liquid phases without support matrix. At the first stage, the solvent system of ethyl acetate/n-butanol/ethanol/water (3.0:2.0:0.5:6.0, v/v/v/v) was used for the separation of jatrorrhizine, epiberberine, and coptisine, and tested on the analytical separation column. The results (Fig. 3a and b) indicate that when the solvent system mentioned above was used, the target compounds were not well separated. Therefore, jatrorrhizine, epiberberine, and coptisine were separated by medium-pressure liquid chromatography.

HSCCC chromatogram of R. Coptidis. (a) Ethyl acetate/n-butanol/ethanol/water at a volume ratio of 3.0:1.5:0.5:6.0, on the analytical column (28 mL; flow rate of the mobile phase: 1.0 mL/min). (b) Ethyl acetate/n-butanol/ethanol/water at a volume ratio of 3.0:2.0:0.5:6.0, on the analytical column. (c) Ethyl acetate/n-butanol/ethanol/water at a volume ratio of 3.0:1.0:1.0:6.0, on the analytical column. (d) Ethyl acetate/n-butanol/ethanol/water at a volume ratio of 3.0:1.0:1.0:6.0, on the semi-preparative column (125 mL; flow rate of the mobile phase: 2.0 mL/min). (1) Jatrorrhizine; (2) Epiberberine; (3) Coptisine; (4) Palmatine; (5) Berberine
At the second stage, results indicated that HSCCC separation was successfully achieved if the solvent system of ethyl acetate/n-butanol/ethanol/water (3.0:1.0:1.0:6.0, v/v/v/v) was used (Fig. 3c and d). Furthermore, three flow rates were selected to get the best semi-preparative isolation. At first, we chose a flow rate of 1.5 mL/min, which required 240 min to complete elution. When the flow rate was changed to 2.0 mL/min, the elution time was reduced to 120 min. As expected, the target compounds were well separated. Although the elution time was further reduced with the flow rate of 2.5 mL/min, some compounds were not well isolated. A higher flow rate could reduce separation time, but separation and isolation of target compounds would be negatively affected. Decreasing flow rate would have improved the retention of the stationary phase and resolution. However, decreasing flow rate would extend the total separation time. Eventually, the flow rate of 2.0 mL/min was chosen for the semi-preparative isolation. Ultimately, a flow rate of 2.0 mL/min, a revolution speed of 1400 r.p.m., and a temperature of 30 °C were used in the experiment, with the retention rate of the stationary phase in the coil being approximately 71.5%. Figure 3c and d show HSCCC diagrams of the extract of R. Coptidis on an analytical column (containing 14.25 mg of the extract) and a semi-preparative column (containing 67.31 mg of the extract) eluted with ethyl acetate/n-butanol/ethanol/water (3.0:1.0:1.0:6.0, v/v/v/v) solvent system. HPLC–DAD was used to analyze the potential α-glucosidase inhibitors from the extract of R. Coptidis. Under the optimal separation conditions, 1.2 and 0.5 mg of palmatine and berberine were obtained, respectively, with >90.0% purity. Figure 4 shows the HPLC chromatograms of each fraction.

HPLC chromatograms of palmatine (a) and berberine (b) isolated by HSCCC, and the crude extract of R. Coptidis
RP-MPLC separation and compounds analysis
MPLC is an effective, fast, and economical separation method that is simple to operate. The theory of RP-MPLC separation is similar with RP-HPLC; therefore, we selected the separation conditions according to HPLC. Methanol (solvent A) and 0.2 % acetic acid (solvent B) were used as a binary mobile phase in RP-MPLC. As the flow rate of the mobile phase affects separation, different flow rates were explored before 8.0 mL/min was chosen for the experiment. Finally, a satisfactory separation of the three compounds in the extract of R. Coptidis was achieved. Figure 5 shows the RP-MPLC separation of a sample containing 40 mg of extract of R. Coptidis resolved in methanol and water (1:1, v/v).

RP-MPLC chromatograms of the crude extract of R. Coptidis
The analytical RP-MPLC separation produced three major peaks in one run. The fractions of R. Coptidis extract corresponding to each peak isolated by MPLC were analyzed by HPLC, and the results are shown in Fig. 6. The three compounds in RP-MPLC separation were identified by a comparison of congruent retention time with that of the extract of R. Coptidis. Finally, 0.6, 0.4, and 0.4 mg of fractions 1 (jatrorrhizine), 2 (epiberberine), and 3 (coptisine) were obtained, with 76.20, 75.13, and 82.24% purity, respectively.

HPLC profiles of compounds 1–3 in RP-MPLC separation (a–c) and the crude extract of R. Coptidis. (1) Jatrorrhizine; (2) Epiberberine; (3) Coptisine
Identification of α-glucosidase inhibitors by ESI-MSn
Based on UV spectra together with ESI-MS data and HPLC retention time of authentic standards, RP-MPLC and HSCCC peak fractions were identified. MS and NMR spectroscopy were used for structure identification.
RP-MPLC Peak 1: ESI-MS (m/z): 338 [M + H]+. 1H NMR (400 MHz, CD3OD); δ 9.74 (1H, s, H–8), 8.78 (1H, s, H–13), 8.10 (1H, d, J = 8.8 Hz, H–11), 7.99 (1H, d, J = 8.8 Hz, H–12), 7.66 (1H, s, H–1), 6.86 (1H, s, H–4), 4.90 (2H, t, J = 6.4 Hz, H–6), 4.19 (3H, s, 9–OCH3), 4.10 (3H, s, 10–OCH3), 4.02 (3H, s, 2–OCH3), 3.20 (2H, t, J = 6.4 Hz, H–5); 13C NMR (100 MHz, CD3OD): δ 151.9 (C–9), 151.8 (C–2), 149.7 (C–3), 146.2 (C–8), 145.7 (C–10), 140.3 (C–13a), 135.5 (C–4a), 130.3 (C–12a), 128.1(C–12), 124.4(C–11), 123.3 (C–13b), 121.0 (C–13), 119.5 (C–8a), 116.0 (C–4),110.1 (C–1), 62.6 (9–OCH3), 57.7 (10–OCH3), 57.4 (2–OCH3), 57.0 (C–6), 27.7 (C–5). The data above indicates that the compound is jatrorrhizine (Hussain et al. 1989).
RP-MPLC Peak 2: ESI-MS (m/z): 336 [M + H]+. 1H NMR (400 MHz, CD3OD); δ 9.73 (1H, s, H–8), 8.86 (1H, s, H–13), 7.88 (2H, overlapped, H–11, 12), 7.66 (1H, s, H–1), 7.05 (1H, s, H–4), 6.46 (2H, s, –OCH2O–), 4.89 (2H, t. J = 6.4 Hz, H–6), 3.99 (3H,s, 2–OCH3), 3.94 (3H, s, 3–OCH3), 3.28 (2H, t, J = 6.4 Hz, H–5); 13C NMR (100 MHz, CD3OD): δ 153.9 (C–3), 153.3 (C–2), 151.0 (C–10), 149.2 (C–9), 145.8 (C–8), 145.3 (C–13a), 139.2 (C–12a), 134.6 (C–4a), 130.0 (C–12), 123.1 (C–11), 122.3 (C–13), 120.6 (C–13b), 113.7 (C–8a), 112.3 (C–4), 109.9 (C–l), 106.2 (–OCH2O–), 57.4 (C–6), 57.0 (2–OCH3), 56.7 (3–OCH3), 27.8 (C–5). The data above indicates that the compound is epiberberine (Jung et al. 2008).
RP-MPLC Peak 3: ESI-MS (m/z): 320 [M + H]+. 1H NMR (400 MHz, CD3OD): δ 9.74 (1H, s, H–8), 8.74 (1H, s, H–13), 7.88 (1H, d, J = 8.4 Hz, H–11), 7.85 (1H, d, J = 8.4 Hz, H–12), 7.66 (1H, s, H–1), 6.96 (1H, s, H–4), 6.46 (2H, s, –OCH2O–), 6.10 (2H, s, –OCH2O–), 4.88 (2H, t, J = 6.4 Hz, H–6), 3.24 (2H, t, J = 6.4 Hz, H–5); 13C NMR (100 MHz, CD3OD): δ 152.2 (C–3), 150.0 (C–2). 149.3 (C–10), 145.8 (C–8). 145.4 (C–9), 139.0 (C–13a), 134.5 (C–12a), 131.8 (C–4a), 123.2 (C–1), 122.5 (C–12). 122.3 (C–11), 122.0 (C–13), 113.7 (C–13b), 109.4 (C–8a), 106.5 (C–4), 106.2 (–OCH2O–), 103.7 (–OCH2O–), 57.3 (C–6), 28.2 (C–5). The data above indicates that the compound is coptisine (Wang et al. 2004).
HSCCC Peak 4: ESI-MS (m/z): 352 [M + H]+, 337 [M–CH3+H]+, 308 [M–C2 H4 O+H]+, 322 [M–C2H6+H]+, 307 [M–C2 H6 –CH3+H]+. 1H NMR (500 MHz, DMSO-d6): δ 9.06 (1H, s, H–13), 8.21 (1H, d, J = 9.2 Hz, H–11), 8.04 (1H, d, J = 9.1 Hz, H–12), 7.72 (1H, s, H–1), 7.10 (1H, s, H–4), 4.98 (2H, t, J = 6 Hz, H–6), 4.10 (12H, s, 9–OCH3), 4.07 (12H, s, 10–OCH3), 3.92 (12H, s, 2–OCH3), 3.86 (12H, s, 3-OCH3), 3.24 p.p.m. (2H, t, J = 6 Hz, H–5). 13C NMR (125 MHz, DMSO–d6): δ 150.6 (C–10), 150.2 (C–3), 148.0 (C–2), 145.8 (C–8), 144.1 (C–9), 138.0 (C–13a), 133.4 (C–12a), 131.0 (C–4a), 127.2 (C–11), 123.8 (C–12), 121.8 (C–8a), 120.8 (C–13b), 120.5 (C–13), 108.8 (C–4), 105.9 (C–1), 62.3 (10–OCH3), 57.4 (9–OCH3), 56.6 (–OCH2O–), 55.4 (C–6), 26.7 p.p.m. (C–5). The data above indicates that the compound is palmatine (Lee et al. 2010).
HSCCC Peak 5: ESI-MS (m/z): 336 [M + H]+, 321 [M–CH3+H]+, 320 [M–CH3]+, 306 [M–CH3–CH3+H]+. 1HNMR (500 MHz, DMSO-d6): δ 9.87 (1H, s, H–8), 8.93 (1H, s, H–13), 8.21 (1H, d, J = 9.1 Hz, H–11), 8.01 (1H, d, J = 9.1 Hz, H–12), 7.81 (1H, s, H–1), 7.10 (1H, s, H–4), 6.17 (2H, s, –OCH2O–), 4.93 (2H, t, J = 6.0 Hz, H–6), 4.10 (3H, s, 9–OCH3), 4.05 (3H, s, 9–OCH3), 3.21 p.p.m. (2H, t, J = 6.0 Hz, H–5). 13C NMR (125 MHz, DMSO-d6): δ 150.6 (C–10), 150.2 (C–3), 148.0 (C–2), 145.8 (C–8), 144.1 (C–9), 138.0 (C–13a), 133.4 (C–12a), 131.0 (C–4a), 127.2 (C–11), 123.8 (C–12), 121.8 (C–8a), 120.8 (C–13b), 120.5 (C–13), 108.8 (C–4), 105.9 (C–1), 102.4 (–OCH2O–), 62.3 (10–OCH3), 57.4 (9–OCH3), 55.4 p.p.m. (C–6), 26.7 (C–5). The data above indicates that the compound is berberine (Li et al. 2010).
Conclusions
In this study, five α-glucosidase inhibitors, jatrorrhizine, epiberberine, coptisine, palmatine, and berberine, were successively isolated from R. Coptidis by HSCCC and RP-MPLC under the guidance of ultrafiltration-based alkaloid screening. The UF-LC-PDA-ESI/MS method is a valuable tool for effectively screening and identifying active compounds in complex mixtures. Moreover, HSCCC and RP-MPLC proved effective, simple, fast, and economical separation methods. Therefore, the method described in this research is useful for systematic screening and isolation of enzyme inhibitors from R. Coptidis and other crude extracts.
References
Chang LC, Sun SW (2006) Micellar electrokinetic chromatography for separation of a mixture of coptis alkaloids, scute flavonoids, and rhubarb anthraquinones and bianthrones. J Pharm Biomed Anal 40:62–67
Chen JH, Zhao HQ, Wang XR, Lee FS, Yang HH, Zheng L (2008) Analysis of major alkaloids in Rhizoma coptidis by capillary electrophoresis-electrospray-time of flight mass spectrometry with different background electrolytes. Electrophoresis 29:2135–2147
Delavenne X, Montbel A, Hodin S, Zufferey P, Basset T (2014) Quantification of total and unbound tranexamic acid in human plasma by ultrafiltration liquid chromatography/tandem mass spectrometry: Application to pharmacokinetic analysis. J Pharm Biomed Anal 91:32–36
Derosa G, Maffioli P (2012) α-Glucosidase inhibitors and their use in clinical practice. Arch Med Sci 8:899–906
Hussain RA, Kim J, Beecher CW, Kinghorn AD (1989) Unambiguous carbon-13 NMR assignments of some biologically active protoberberine alkaloids. Heterocycles 29:2257–2260
Ito Y (2005) Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J Chromatogr A 1065:145–168
Jung HA, Yoon NY, Bae HJ, Min BS, Choi JS (2008) Inhibitory activities of the alkaloids from Coptidis Rhizoma against aldose reductase. Arch Pharm Res 31:1405–1412
Lee WC, Kim JK, Kang JW, Oh WY, Jung JY, Kim YS, Jung HA, Choi JS, Lee SM (2010) Palmatine attenuates D-galactosamine/ lipopolysaccharide-induced fulminant hepatic failure in mice. Food Chem Toxicol 48:222–228
Li HL, Song FR, Xing JP, Tsao R, Liu ZQ, Liu SY (2009) Screening and structural characterization of alpha-glucosidase inhibitors from hawthorn leaf flavonoids extract by ultrafiltration lc-dad-ms(n) and sori-cid fticr ms. J Am Soc Mass Spectrom 20:1496–1503
Li YB, Zhang TJ, Zhang XL, Xu HY, Liu CX (2010) Chemical fingerprint analysis of Phellodendri Amurensis Cortex by ultra performance LC/Q-TOF-MS methods combined with chemometrics. J Sep Sci 33:3347–3353
Luo XB, Chen B, Yao SZ (2005) Simultaneous analysis of protoberberine, indolequinoline and quinolone alkaloids in coptis-evodia herb couple and the chinese herbal preparations by high-performance liquid chromatography-electrospray mass spectrometry. Talanta 66:103–110
Qian P, Yang XW (2014) Five new alkaloids from Coptidis Rhizoma-Euodiae Fructus couple and their cytotoxic activities against gastrointestinal cancer cells. Fitoterapia 93:74–80
Ren LL, Xue XY, Zhang FF, Xu Q, Liang XM (2007) High performance liquid chromatography-mass spectrometry analysis of protoberberine alkaloids in medicine herbs. J Sep Sci 30:833–842
Shi SY, Huang KL, Zhang YP, Zhao Y, Du QZ (2007) Purification and identification of antiviral components from Laggera pterodonta by high-speed counter-current chromatography. J Chromatogr B 859(1):119–124
Tong SQ, Yan JZ (2007) Large-scale separation of hydroxyanthraquinones from rheum palmatum L. by ph-zone-refining counter-current chromatography. J Chromatogr A 1176:163–168
Wang DW, Liu ZQ, Guo MQ, Liu SY (2004) Structural elucidation and identification of alkaloids in Rhizoma Coptidis by electrospray ionization tandem mass spectrometry. J Mass Spectrom 39:1356–1365
Wild S, Roglic G, Green A, Sicree R, King H (2004) Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 27:1047–1053
Xie Z, Sun Y, Lam S, Zhao M, Liang Z, Yu X, Yang D, Xu X (2014) Extraction and isolation of flavonoid glycosides from Flos Sophorae Immaturus using ultrasonic-assisted extraction followed by high-speed countercurrent chromatography. J Sep Sci 37:957–965
Yan R, Mu Q, Wang Y, Liu Y, Di X (2012) Relative determination approach to the metabolites of protoberberine alkaloids in Rat urine by liquid chromatography Tandem mass spectrometry for the comparative studies on Rhizome coptidis and Zuojinwan preparation. Iran J Pharm Res 11:1265–1273
Zhang YC, Liu CM, Zhang ZK, Wang J, Wu GM, Li SN (2010) Comprehensive separation and identification of chemical constituents from Apocynum venetum leaves by high-performance counter-current chromatography and high performance liquid chromatography coupled with mass spectrometry. J Chromatogr B 878:3149–3155
Zou JG, Liu F, Xiao-Long XU, Liu YY, Peng HL, Wang HP (2010) Optimization of microwave-assisted extraction of total alkaloids from semen strychni. Food Sci 31:116–119
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31170326, 31370374), the team project of Jilin Provincial Science and Technology Department (No. 20130413043GH), the Academic Innovation Foundation of Changchun Normal University (cscxy2015003), the project of Jilin Provincial Education Department ([2013] 253), and the Natural Science Foundation of Changchun Normal University (No. [2013] 001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Tang, Y., Li, S., Li, S. et al. Screening and isolating potential α-glucosidase inhibitors from Rhizoma Coptidis by ultrafiltration LC-PDA-ESI/MS combined with high-speed countercurrent chromatography and reverse-phase medium-pressure liquid chromatography. Med Chem Res 26, 3384–3394 (2017). https://doi.org/10.1007/s00044-017-2031-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-2031-6