
Abstract
The discovery of three new natural products (1, 4, and 5), one semi-synthetic derivative (3) along with two known compounds (2 and 6) were isolated from an endophytic fungus Cryptosporiopsis sp. The structural elucidations of 1–6 were authenticated by one-dimensional and two-dimensional nuclear magnetic resonance, mass spectroscopy, and X-ray diffraction analysis. Herein, we intervention of diazomethane as tool that help in the crystallization and isolation of inseparable mixtures of compounds 1 and 2. Compounds (1–6) were screened for cytotoxic activity against six cancer cell lines in which the 4-epi-ethisolide (2) exhibited moderate activity with IC50 values 11 µM in HL-60, whereas the compound 3 lost its cytotoxic potentiality, but it displayed moderate antimicrobial activity. The result illustrates that the methylene moiety in 2 plays significant role in cytotoxic potential.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant endophytic fungi live in the intercellular spaces of plant tissues for all or part of their life cycle without causing apparent pathogenic symptoms. Endophytes are a poorly investigated group of microorganisms and have proved to be an important source of bioactive and chemically novel metabolites with an enormous metabolic diversity (Strobel and Daisy 2003; Nalli et al. 2017). These organisms have the ability to produce a plethora of substances with unique structures and interesting biological activities to modern medicine, agriculture, and industry. Recently, increasing the number of reports on their identification and on the production of secondary metabolites witnessed the enormous potential of endophytes as rich and reliable natural resource (Strobel 2003). The genus Cryptosporiopsis, with over 15 species, has attracted considerable attention as a source of numerous compounds with antimicrobial and anticancer activities (Debbab et al. 2012; Kharwar et al. 2011; Kumar et al. 2013). Cryptosporiopsis species are anamorphs of ascomycetes in the genera Pezicula and Neofabraea Dermataceae and have been isolate as endophytes of several plants (Sigler et al. 2005; Zilla et al. 2013). With the aim of discovering novel bioactive metabolites from endophytic fungi, we investigated the chemical constituents of the endophytic fungal strain Cryptosporiopsis sp. H2-1 (NFCCI 2856), an endophyte associated with Clidemia hirta found in Hawaii. Based on the morphological characteristics and on sequence analysis of the internal transcribed spacer (ITS) region described in our previous report, the endophyte was placed under the genus Cryptosporiopsis.
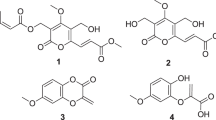
Herein we report the isolation, characterization of three new (1, 4, and 5), two known compounds 4-epi-Ethisolide (2), Orsellinic acid (6) from H2-1 (Fig. 1). In addition, we developed an alternate approach for the separation of 1 and 2 by using as a chemical tool, which induced crystallization and afforded compound 3 as a natural derivative that in turn helped us to elucidate the structure of 2. The structural elucidation of (1–6) were established on the basis of 1D and 2D nuclear magnetic resonance (NMR) spectra analysis and the relative configuration of 3 was finally determined via single crystal X-ray diffraction by using the anomalous dispersion of Mo Kα radiation. The antimicrobial and tumor cell growth inhibitory activity of these compounds (1–6) is also discussed.

Structures of compounds (1–6) isolated from H2-1
Results and discussion
Re-evaluation of the ITS region of H2-1 by re-sequencing and obtaining a longer nucleotide sequence (545 bases as against 499 bases in the earlier study), displayed a 98% sequence similarity with Cryptosporiopsis californiae. Thus, this isolate can be regarded as a member of Cryptosporiopsis californiae. The DNA sequence was updated in the GenBank database. For the isolation of bioactive metabolites from H2-1, 5 L of fermentation broth was extracted with ethyl acetate that yielded 4.0 g of crude extract. Flash column chromatography of the crude extract on silica gel and further purification with high performance liquid chromatography (HPLC) led to the isolation of three new (1, 4, and 5), one semi synthetic derived (3) and two known (2 and 6) compounds. The known molecules were identified by comparison of spectroscopic data with those reported in the literature as 4-epi-Ethisolide (2) (Krohn et al. 1994) and Orsellinic acid (6) (Jayaprakasha and Rao 2000).
Structure elucidation of the new compounds 1, 3, 4, and 5
Compounds 1 and 2 was obtained as a mixture by flash column chromatography initially it seems to be the single by thin layer chroamtography (TLC) and HPLC using PDA detector due to similar polarity of these compounds, but the NMR spectral results do not corroborate with the mass spectral data. 1H & 13C NMR clearly indicated that terminal methylene moiety was present in one of the dihydrofuran nucleus which was treated with diazomethane to form a pyrazoline (3) as a crystalline solid and 1 as colorless viscous. Furthermore the compounds 1 and 2 was purified by preparative HPLC equipped with ELSD detector at (rt 14.2) and (rt 15.5) by using C18 column (5 µ, 10 × 250 mm) (Fig. 2). Furthermore, we establish the relative configuration of 2 by X-ray diffraction and NOESY correlation along with the cytotoxic potential.

Comparison of HPLC chromatogram of 1 and 2 in (PDA and ELSD detector)
The new compound 1 was obtained as colorless viscous oil with \(\left[ {\rm{\alpha }} \right]_D^{25} - 65.8\) (c 0.1, CHCl3). The IR absorptions at 1777, 1622 cm−1 indicate the presence of two carbonyl functionalities. The molecular formula was determined to be C9H12O4 based on a High-resolution electrospray ionisation mass spectrometry (HRESIMS) (at m/z 183.0171 [M − H]− analysis and NMR data, indicating 4 degrees of unsaturation. The 1H and 13C NMR data (Table 1) of 1 resembled those of 4-epi-Ethiosolide (2), except for the disappearance of exocyclic double bond signals in 4-epi-Ethiosolide (2) and the presence of a one methine and one methyl signals in 1. These characteristics implied that the exocyclic double bond in 4-epi-Ethiosolide (2) was reduced in 1. The above inference was confirmed by the detailed analysis of 1D and 2D NMR data of 1. The 1H NMR, 13C NMR, and HSQC spectra indicated the presence of two ester carbonyls (C-2 and C-6), two oxygenated aliphatic methines (C-4 and C-6a), two aliphatic methines (C-3 and C-3a) one methylene (C-1′′), two methyls (C-1′ and C-2′′), explaining 2 out of four degrees of unsaturation.
Therefore, compound 1 should be a bicyclic (ring A and B; Fig. 1) structure, to satisfy the remaining two degrees of unsaturation. Interpretation of the 1H–1H COSY spectrum showed the presence of two substructures (C-2′′ ↔ C-1′′ ↔ C-4 ↔ C-3a ↔ C-3 ↔ C-1′, and C-3a ↔ C-6a) as shown by bold faced lines in (Fig. 3). Additionally, the Heteronuclear Multiple Bond Correlation (HMBC) analysis completed all of the carbon-carbon bonds in 1 (Fig. 3). The presence of ring-A was indicated by the HMBC correlations of H-6a/C-4, of H-3a/C-4. The position of ethyl group (C-2′′ ↔ C-1′′), located at C-4, was determined on the basis HMBC correlations from H-1′′ and H-2′′ to C-4, and H-3a to C-1′′. Further, the key HMBC correlations of H-3a/C-3 and C-2, of H-3/C-2 and C-3a confirmed the presence of ring-B. The mark difference between compounds 2 and 1, the exocylic double bond was reduced to methyl group, was confirmed by the HMBC correlations from H-1′ to C-3, C-2, and C-3a, and from H-3 and H-3a to C-1′ (Fig. 3). Even though no HMBC correlation was found between H-4/C-6, and H-6a/C-2, ester linkages were given in order to satisfy ring residues and mass of the compound 1. The relative configurations of 1 were determined on the basis of the NOESY experiments as being compatible with computer modeling (Fig. 4). The NOE correlations of H-4/H-3, and H2-1′′/H-3a/H3-1′/H-6a suggested that H-4 and H-3 were α-oriented and that H2-1′′, H-3a, H3-1′, and H-6a were β-oriented, as depicted by molecular modeling analysis (Fig. 4). On the basis above observed NOESY correlations, the relative configurations at stereocenters C-6a, C-3a, and C-4 of 1 were concluded to be identical to those of 2. Therefore, the structure of 1 was authenticated as (3 S,3aR,4 R,6aR)-4-ethyl-3-methyldihydrofuro[3,4-b]furan-2,6(3 H,4 H)-dione.

Selected 1H-1H COSY ( ), HMBC (
), HMBC ( ) correlation for compounds 1, 3, 4, and 5
) correlation for compounds 1, 3, 4, and 5

Key NOESY correlations of 1 and 4
Compound 3a colorless crystalline solid in acetone was obtained after reacting with diazomethane of 2. The ORTEP view of 3 with displacement ellipsoids drawn at 40% H atoms are shown as small spheres of arbitrary radii (Fig. 5).

Single-crystal X-ray structure of 3
The HRESIMS exhibited the prominent ion peak at m/z 225.1217 [M + H]+ established the molecular formula C10H12N2O4 which was further authenticated by 1H and 13C NMR spectral data analysis. The IR spectrum of 3 exhibited absorption bands at 1758 and 1735 cm−1 accounting for the presence of bis-lactone groups. An examination of the 1H and 13C NMR data (Table 1) showed the structure of 3 to be similar to that of 2, except the signal for terminal methylene group have been disappeared. In contrast, an extra pyrazoline ring could be located at C-3. Further analysis of the 1H NMR spectrum of 3 displayed resonances for one aza-methylene [δ H 4.68 m, H1-3′′; 4.89 m, H1-3′′] and one methylene [δ H 1.92 m, 2.26 m]. The 13C NMR data and HSQC spectrum denoted the presence of 10 resonances out of these three belongs to sp 3 methylenes, of which one was aza-methylene [δ C 79.63,(C-3′)] and one aliphatic methylene [δ C 29.18 (C-4′)] of pyrazoline ring respectively. The 1H−1H COSY data of 3 revealed the presence of one isolated spin system: H2-11 ↔ H2-12 indicating the presence of pyrazoline ring which was further confirmed on the basis of the HMBC correlations from H2-3′ to C-3, from H2-4′ to C-2. Thus, the structure of 3 was determined as depicted in (Fig. 1). Structure of compound 2 was traced back from the structure of 3 in which terminal methylene was converted into pyrazoline ring by 1–3 cycloaddition reaction with diazomethane.
Compound (4) was obtained as a brownish powder. Its molecular formula was established from 13C and HRESIMS data (m/z 183.0671 [M − H]−) as C9H12O4, indicating four degrees of unsaturation. The IR spectrum indicated the presence of hydroxyl (3451 cm−1) and two carbonyl (1778, 1745 cm−1) functionalities. The 1H NMR spectrum (Table 1) exhibited one n-propyl group (δ H 1.60, m and 2.12, m; 1.46, m and 0.96, t, J = 7.3 Hz), one oxygenated methane (δ H 5.14, d, J = 7.6), and one singlet methyl (δ H 2.25, s). 13C and Distortionless Enhancement by Polarization Transfer (DEPT) spectra displayed nine carbon resonances (Table 1) attributable to four quaternary carbons [including two carbonyls (δ C 172.7, 166.7) and two olifinic ones (δ C 146.8, 139.9)], one oxygenated methane (δ C 81.2), one methyl (δ C 11.5), and three other resonances assignable to one n-propyl group. These data suggested that 4 is a derivative of α,β-Dimethyl-(Z)-4-hydroxypent-2-enedioic anhydride (Wiley and Ellert 1957). The HMBC correlations from oxygenated methane H-3 (δ H 5.14) to C-4 (δ C 146.8), C-5 (δ C 139.9) and C-1′ (δ C 34.7) located the hydroxyl group at C-3 (δ C 81.3) (Fig. 3). The correlations of Me-1′′ with C-4 (δ C 146.8), C-6 (δ C 166.7) and C-1′ (δ C 34.7) in HMBC spectrum confirmed that methyl is located at C-5. Similarly, the location of n-propyl group at C-4 was determined by HMBC correlation from H2-1′ and H2-2′ to C-4, and H-3 to C-1′. Furthermore, in the NOESY spectral analysis, the strong NOESY correlation between H1-3 and H2-1′ were revealed that H1-3 was spatially close to the n-propyl group and was assigned α-orientation, as depicted by molecular modeling analysis (Fig. 5). The successive 2D spectral experiments accomplished the assignments of all proton and carbon signals of 4, and thus its chemical structure was established as (S)-3-hydroxy-5-methyl-4-propyl-2H-pyran-2,6(3 H)-dione.
New metabolite (5) was isolated as white, amorphous powder, and its molecular formula was determined as C11H16O3 from the HRESIMS and 13C NMR data, indicating 4 degrees of unsaturation. The IR spectrum displayed absorption bands at 1693 and 1658 cm−1, indicative of the presence of two carbonyl functionalities. A close inspection of the 1H and 13C NMR spectra of 5 by DEPT and HSQC experiments revealed the presences of two carbonyl (C-2 and C-12), two sp 2 methine (C-3 and C-4) and seven sp 3 methylene carbons (C-5, C-6, C-7, C-8, C-9, C-10, and C-11). The 1H–1H COSY analysis of 5 demonstrated the presence of one contiguous methylene unit from H-3 to H2-11 via alkene protons H-3 (δ H 5.78; δ C 122.5) and H-4 (δ H 6.91; δ C 151.2), and placed between two carbonyl carbons C-2 (δ C 170.1) and C-12 (δ C 177.7). The above inference was further confirmed by HMBC correlations of H-3/C-2, C-4, and C-5, of H-4/C-2 and C-6, of H-10 and H-11/C-12 (Fig. 3). The E-configuration were assigned for ∆3,4 alkene unit on the basis of large coupling constant (J = 15.6 Hz) between H-3 and H-4. Finally, the anhydride bond between C-2 and C-12 was assigned to satisfy ring residues and mass of the compound 5. Thus, on the basis of above spectral evidences, the structure of new molecule 5 assigned as (E)-oxacyclododec-3-ene-2,12-dione.
Compounds (1–6) were evaluated for antimicrobial activities against the Gram negative bacterium, Escherichia coli, Gram positive bacteria, Staphylococcus aureus and Bacillus cereus, and the fungal pathogen, Candida albicans. All the molecules were inactive at the concentration of 100 µg/ml except 3. The compound 3 was moderate active against E. coli, B. cereus and C. albicans with IC50 values of 90, 65, and 70 µg/ml, respectively. Furthermore all isolates (1–6) were screened for cytotoxic activity against six human cancer cell lines including human myeloid leukemia (HL-60), colon cancer (COLO 205), breast cancer (MDA-MB-231), Prostate cancer (PC-3), Pancreatic cancer (MIA PaCa-2), and lung cancer (A549) using the MTT method (Alley et al. 1988). Interestingly, compound 2, which having unusual allylic double bond at C-3, showed moderate cytotoxic activity (IC50 11 µM in HL-60), while other isolates exhibited less cytotoxicity (IC50 > 100 µM). The cytotoxic results of all the compounds are summarized in (Table 2).
Furthermore, the effect of 2 on cell cycle phase distribution in Human leukemia HL-60 cells was examined at 10, 20, and 30 µM concentration for 24 h duration (Fig. 6). There is concentration dependent increase in SubG1 DNA fraction (DNA < 2 N) in 2 treated HL-60 cells. At 10 µM concentration, 17% cells showed SubG1 population which increased to 41 and 69% at 20 and 30 µM concentrations respectively, whereas control cells showed 4% of apoptotic SubG1 population.

Cell cycle analysis in human leukemia HL-60 cells treated with compound 2 for 24 h. HL-60 cells were treated with 2 at 10, 20, and 30 µM concentrations for 24 h and stained with propidium iodide (PI) to determine cell cycle phase distribution. Hypo diploid population (Sub G0, < 2n DNA) considered here as apoptotic population. Data are representative of one of the two similar experiments
We also investigated the effect of 2 on mitochondrial membrane potential (MMP) loss in human leukemia HL-60 cells. Mitochondrial potential loss were analyzed by using dihydrorodamine (cationic fluorescence dye) which reduced in mitochondria of cancer cells. Finally, fluorescence was measured using a flow cytometer. The untreated control cells showed about 5% MMP loss, while HL-60 cells treated with 2 exhibited MMP loss in concentration dependent manner (Fig. 7). At 10 µM concentration MMP loss was 34% while at 20 and 30 µM concentrations MMP loss were 65 and 86%, respectively.

Compound 2 triggers mitochondrial potential loss in HL-60 cells at 10, 20, and 30 µM concentration for 24 h and stained with Rhodamine 123. Loss in mitochondrial potential was indicated by decrease in dye fluorescence (M1 in the figure). Data are representative of one of the two similar experiments. The untreated control cells showed about 5% MMP loss, while HL-60 cells treated with 2 exhibited MMP loss in concentration dependent manner (Fig. 7). At 10 µM concentration MMP loss was 34% while at 20 and 30 µM concentrations MMP loss were 65 and 86%, respectively
The apoptosis induction by 2 in HL-60 cells was checked through Hoechst staining. Apoptotic bodies’ formation increases in concentration dependent manner from 10 to 30 µM concentration of 2. As evident by blebbing and apoptotic bodies (Indicated by arrows in Fig. 8), observed in cells treated with 2, showed altered cellular morphology including cell wall deformation, shrinkage of cell size and nuclear condensation. Further, the nuclei of untreated cells were round and healthy.

Hoechst staining and Phase contrast microscopy of 2 treated HL-60 cells. Cells were incubated with 2 at 10, 20, and 30 µM concentration for 24 h. Cells were stained with Hoechst 33258 dye and visualized by microscopy. Condensed nuclei and apoptotic bodies were indicated by arrows. Untreated control show healthy round nuclei, while the treated cells show scattered apoptotic bodies, which increases in concentration dependent manner. Phase contrast microscopy of treated cells showed blebbing and cell shrinkage. Untreated control cells are healthy and round
As shown in (Fig. 9), compound 2 caused the activation of both Procaspase-9 and Procaspase-8 in concentration dependent manner. Hence, two induced apoptosis involve both extrinsic and intrinsic pathway. Sequential activation of different caspase signaling takes place during apoptosis which merge on caspase-3. Compound 2 triggers caspase-3 cleavage in HL-60 cells. PARP-1 cleavage seen at 10 µM concentration in HL-60 cells which increases in dose dependent manner.

Compound 2 triggers apoptotic signaling cascade in human leukemia HL-60 cells. Cells were treated with 2 at indicated concentrations for 24 h. Lystaes were prepared after 24 h and equal amount of proteins (50–70 µg) were applied for SDS-PAGE. Western blot analyzes of indicated proteins were performed in whole cell lysate as described in material and methods. β Actin used as internal loading control
We next assessed the effect of 2 on Bcl-2/Bax ratio which determines the cell susceptibility to apoptosis (Korsmeyer 1999, Oltvai et al. 1993). Compound 2 decrease the expression of both proteins and the overall Bcl-2/Bax ratio decrease in concentration dependent manner (Fig. 10). Furthermore, expression of Bcl-xL also decreased in cells treated with compound 2.

Influence of 2 on Bcl-2/ Bax ratio in HL-60 cells. Cells were treated with 2 for 24 h and whole cell protein lysates were prepared. Proteins were separated by SDS-PAGE and subjected to western blot analysis of Bax and Bcl-2. Relative density of each b and was measured as arbituary units by Quantity One Software
Experimental
General
Melting points of synthesized compounds were recorded on electrical melting point apparatus (Buchi, D-545) and are uncorrected. High resolution mass spectra were obtained on Agilent 6540 (Q-TOF) high resolution mass spectrometer, in the electrospray (ESIMS) mode. 1H NMR spectra were recorded (Brucker Avance) at 400 and 500 MHz and 13C NMR at 125 MHz in CDCl3 and CD3OD, chemical shifts values are reported in δ (ppm) units and coupling constants values in hertz. Tetramethylsilane was used as internal standard. Optical rotation was measured on a Perkin Elmer 341 polarimeter in a 1 dm cell at 25 °C. Infrared spectra were recorded with a PerkinElmer spectrum 65 Fourier transform infrared spectrometer and wavelengths (λ) are given in cm−1. Column chromatography was performed using silica gel (100−200 mesh; Merck) and Sephadex LH-20. Semipreparative HPLC was performed on an Agilent HPLC with a RP-18 column (250 × 4.6 mm, 5 μm; Agilent), a photodiode array detector and auto injector function (Agilent 1260 series).
Extraction and isolation
The endophytic fungus was isolated from the tissue of the C. hirta and processed as described previously (Zilla et al. 2013). The culture was identified and a voucher specimen (NFCCI 2856) has been deposited to the National Fungal Culture Collection of India. The endophytic fungal strain Cryptosporiopsis californiae was cultivated in 5 L of PD broth (HiMedia, India) in an incubator shaker (New Brunswick) at 25 °C and 200 rpm for 15 days. The whole fermentation broth was filtered through a cheese cloth to separate the supernatant from the mycelia. Further, filtrate was homogenized with ethyl acetate (3 L) and extracted thrice. The crude extract was passed through the Na2SO4 and concentrated under reduced pressure by using Rotavapour (Buchi) which afforded a dark brownish, oily residue (4.0 g). The whole EtOAc soluble part was chromatographed on silica gel using gradient elution from 100% (v/v) hexane to 100% EtOAc (v/v), to give fractions (Fr. 1–29). Fr. 5–7 (hexane/EtOAc, 95:5 v/v elution 0.248 g) provided mixture of 1 and 2, further purified by semipreparative reversed-phase HPLC using (C-18 column, 5 µ, 10 × 250 mm, isocratic (MeCN−H2O, 20:80, 30 min, 0.8 mL/min, UV detection at 254 nm) yielded compound 1 and 2. Purification of Fr 9–14 (hexane/EtOAc, 90:10 v/v elutions 0.720 g) by preparative TLC gives 4 (21.8 mg). Compound 5 was isolated from Fr 18–24 (hexane/EtOAc, 60:40 v/v elution 92.4 mg) by using sephadex (LH-20) column and methanol was used as an eluting agent at a flow rate 1.5 mL/min. Preperative HPLC (MeCN−H2O, 40:60, 1.5 mL/min) of Last Fr. 26–29 afforded 6 (18.6 mg).
4-Ethyl-3-methyldihydrofuro[3,4-b]furan-2,6(3H,6aH)-dione (1)
(C9H12O4) colorless gummy liquid; \(\left[ {\rm{\alpha }} \right]_D^{25} - 65.8\) (c 1.0, CHCl3); 1D and 2D NMR data, see Table 1; HRESIMS m/z 183.0171 [M − H]− (calcd for C9H12O4, 184.0736).
4-Ethyl-3a,4,4′,5′-tetrahydro-2H-spiro[furo[3,4-b]furan-3,3′-pyrazole]-2,6(6aH)-dione (3)
(C10H12N2O4) white needles (MeOH); \(\left[ {\rm{\alpha }} \right]_D^{25} - 63.0\) (c 1.4, CHCl3); 1D and 2D NMR data, see Table 1; HRESIMS m/z 225.1217 [M + H]+ (calcd for C10H12N2O4, 224.0797).
3-Hydroxy-4-methyl-5-propyl-2H-pyran-2,6(3H)-dione (4)
(C9H12O4) brown amorphous solid; mp 198−200 °C; \(\left[ {\rm{\alpha }} \right]_D^{25} - 104.0\) (c 1.5, CHCl3); 1D and 2D NMR data, see Table 1; HRESIMS m/z 183.0671 [M − H]− (calcd for C9H12O4, 184.0736).
(E)-oxacyclododec-3-ene-2,12-dione (5)
(C11H16O3) white amorphous solid; mp 190−192 °C; \(\left[ {\rm{\alpha }} \right]_D^{25} - 133\) (c 1.4, CHCl3); 1D and 2D NMR data, see Table 1; HRESIMS m/z 197.0143 [M + H]+ (calcd for C11H16O3, 196.1099).
X-ray crystallographic analysis of compound 3
C10H12N2O4, M = 224.22, crystallizes in orthorhombic system, space group P 212121, cell parameters: a = 5.3538(5), b = 10.7853(9), c = 18.0366(19) Å and Z = 4, D calcd = 1.182 Mg/m3, μ (Mo Kα) = 0.078 mm−1, F(000) = 688, and T = 293(2) K. A crystal of dimensions 0.30 × 0.20 × 0.20 mm3 was collected on X’calibur, computer-controlled single crystal x-ray diffractometerhaving CCD Camera, (φ−ω scans), Mo Kα radiation (λ = 0.71073 Å). The structure was refined with fullmatrix least-squares calculations on F2 using SHELXL-97 (Sheldrick 2008). A total of 2925 reflections, collected in the θ range 3.78–25.98°, yielded 1215 unique reflections (Rint = 0.0336). The final stage converged to R1 = 0.0411 (wR2 = 0.0865) for 869 observed reflections [with I > 2σ(I)] and goodness-of-fit = 1.009. Crystallographic data for the structure of 3 have been deposited with the Cambridge Crystallographic Data Center as (CCDC 1015922). A copy of the data can be obtained free of charge on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: + 44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Chemicals used, cell culture and treatment
Propidium iodide (PI), Dihydrorhodamine 123 (DHR123), DNase-free RNase,3-(4,5,-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Eukaryotic proteinase inhibitor cocktail, Streptomycin, Kanamycin were purchased from M/s Sigma chemicals Co., St. Louis. Mouse anti-human antibodies to Bax (#SC20067), PARP-1(#SC8007), Bcl-2 (#SC7382), Casapase-9 (#SC56077), Procaspase-3(#SC7272), Bcl-xL (SC#8392), actin (#SC-8432) goat anti-mouse IgG-HRP (#SC2031) were from M/s Santa Cruz, USA. Mouse anti Caspase-8 (#AM46) were from M/s BD, Pharmingin. Electrophoresis reagents and protein markers were from M/s Bio-Rad, USA while Hyper film and ECL reagents from M/s Amersham Biosciences (GE Healthcare), UK.
Human myeloid leukemia cell line (HL-60), colon cancer cell line (COLO 205), breast cancer cell line (MDA-MB-231), prostate cancer cell line (PC-3), pancreatic cancer cell line (MIA PaCa-2) and lung cancer cell line (A549) were purchased from ECACC, England. Cells were grown in RPMI/DMEM/MEM growth medium containing 10% FCS, 100 µg kanamycin and streptomycin per ml media. Cells were grown in CO2 incubator (Thermocon Electron Corporation, Houston, TX) having 95% humidity and 37 °C temperature. Cells were treated with compounds dissolved in DMSO while the untreated control cultures received only the DMSO with concentration less than 0.2%.
Cytotoxicity (MTT) assay
Human myeloid leukemia cell line HL-60 were seeded (16000 cells/well/100 µL of media) into 96-well plates. After 4 h, cells were treated with compounds and placed in incubator for 48 h. MTT dye was added to the plates 4 h prior to experiment termination at concentration of 2.5 mg/mL, dissolved in media. MTT formazon crystals formed were dissolved in DMSO and absorbance at 570 nm was recorded. Cell survival concentration was calculated as (A570 of treated cells/A570 of control cells) × 100% and from there IC50 values were determined.
Cell cycle analysis
Most of cytotoxic drugs act by damaging DNA of cancer cells. HL-60 cells (0.5 × 106/2 mL medium) were seeded in 24 well culture plate. After 4 h, cells were treated at 10, 20, and 30 µM concentration of 2 for 24 h. Cells were collected at 400 × g, washed with PBS twice and fixed overnight in 70% alcohol at 40 °C. Next day cells were collected at 400 × g, washed with PBS twice, suspended in PBS and subjected to RNase digestion (200 µg/mL) at 37 °C for 60 min. Finally, cells were incubated with PI (10 µg/mL) for 30 min and analyzed immediately on flow cytometer (FACS Aria BD Biosciences, San Jose, CA). The fluorescence intensity of sub-G1 cell fraction (DNA < 2n) represent the apoptotic cell population.
Mitochondrial membrane potential loss study
Mitochondria play an important role in activating apoptotic induction in cancer cells. Damaged mitochondria release a variety of proteins that activates the procaspase cascade inside the cells and triggers apoptosis. Cells (0.5 × 106/2 mL medium/ 24 well plate) were treated with 2 at 10, 20 and 30 µM concentrations for 24 h. Dihydrorhodamine-123 (Rh123) was added 40 min prior to experiment termination. Cells were collected at 400 × g, washed with PBS twice and changes in mitochondrial trans membrane potential due to mitochondrial damage were analyzed on flow cytometer.
Hoechst staining and phase contrast microscopy
Cells (0.5 × 106/2 mL medium/ 24-well plate) were treated with 2for 24 h time period at 10, 20, and 30 µM concentrations. Cells were collected at 400 × g, washed twice with PBS and fixed in 500 µL of fixing solution (cold acetic acid: methanol (1 + 3), v/v) overnight at 4 °C. Cells were collected at 400 × g, washed with fixing solution and dispensed in 70 µL of fixing solution. Cells were spreaded on a clean cold slide and dried overnight at room temperature. Next day cells were stained with Hoechst 33258 (5 µg/mL in 0.01 M citric acid and 0.45 M disodium phosphate containing 0.05% Tween 20) for 40 min at room temperature. Slides were washed with water followed by in PBS. While wet, 40 µL of mounting fluid (PBS: glycerol, 1:1) were poured over the slide, covered with glass cover slip and sealed with nailpolished. Cells were observed under microscope at 20× (Olympus IX70) for any nuclear morphological changes due to addition of compounds in human leukemia HL-60 cells. For phase contrast microscopy, treated and control cells were photographed using microscope.
Conclusions
In summary herein, we report three new natural products (1, 4, and 5), two known (2 and 6) and intervention of diazomethane led to separation afforded compound 3 from an endophytic fungus Cryptosporiopsis sp. along with moderate cytotoxicity of 2 and antimicrobial activity of 3. In account of that we developed a chemical engineering tool which helps in the crystallization and isolation of inseparable mixtures which is difficult to isolate from the column chromatography and HPLC. This discovery can extensively be used as an alternative source to generate the natural product-like libraries through chemically engineered extracts. Herein, we treated the crude extract with excess amount of diazomethane which illustrate the structural diversity in HPLC chromatogram (See the Fig. S32) further research study will be achieved in near future. The approach relies on the power of numbers, that is, in the chemical alteration of a sizable fraction of the starting complex mixture.
References
Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR (1988) Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res 48:589–601
Debbab A, Aly A, Proksch P (2012) Endophytes and associated marine derived fungi ecological and chemical perspectives. Fungal Divers 57:45–83
Jayaprakasha GK, Rao LJ (2000) Phenolic constituents from the lichen Parmotrema stuppeum (Nyl.) Hale and their antioxidant activity. Zeitschrift fur Naturforschung C 55:1018–1022
Kharwar RN, Mishra A, Gond SK, Stierle A, Stierle D (2011) Anticancer compounds derived from fungal endophytes: their importance and future challenges. Nat Prod Rep 28:1208–1228
Krohn K, Ludewig K, Aust HJ, Draeger S, Schulz B (1994) Biologically active metabolites from fungi. 3. sporothriolide, discosiolide, and 4-epi-ethisolide-new furofurandiones from Sporothrix sp., Discosia sp., and Pezicula livida. J Antibiot 47:113–118
Korsmeyer SJ (1999) BCL-2 gene family and the regulation of programmed cell death. Cancer Res 59:1693–1700
Kumar M, Qadri M, Sharma PR, Kumar A, Andotra SS, Kaur T, Kapoor K, Gupta VK, Kant R, Hamid A, Johri S, Taneja SC, Vishwakarma RA, Riyaz-Ul-Hassan S, Shah BA (2013) Tubulin inhibitors from an endophytic fungus isolated from Cedrus deodara. J Nat Prod 76:194–199
Nalli Y, Arora P, Wadhwa B, Fayaz FA, Vishwakarma RA, Gupta VK, Hassan SY, Ali A (2017) Diapolic acid A–B from an endophytic fungus, Diaporthe terebinthifolii depicting antimicrobial and cytotoxic activity. J Antibiot 70:212–215
Oltvai ZN, Milliman CL, Korsmeyer SJ (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74:609–619
Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr A 64:112–122
Sigler L, Allan T, Lim SR, Berch S, Berbee M (2005) Two new Cryptosporiopsis species from roots of ericaceous hosts in western North America. Stud Mycol 53:53–62
Strobel G, Daisy B (2003) Bioprospecting for microbial endophytes and their natural products. Microbiol Mol Biol Rev 67:491–502
Strobel GA (2003) Endophytes as sources of bioactive products. Microbes Infect 5:535–544
Wiley R, Ellert H (1957) Notes-2-pyrones. XXIV. Derivatives of α,β-dimethylglutaconic anhydride. J Org Chem 22:330–331
Zilla MK, Qadri M, Pathania AS, Strobel GA, Nalli Y, Kumar S, Guru SK, Bhushan S, Singh SK, Vishwakarma RA, Riyaz-Ul-Hassan S, Ali A (2013) Bioactive metabolites from an endophytic Cryptosporiopsis sp. inhabiting Clidemia hirta. Phytochemistry 95:291–297
Acknowledgements
The authors are grateful to the CSIR-New Delhi for providing financial support (project no. P81101) for the work of SK. He also acknowledges the AcSIR for their enrolment in Ph. D program. We acknowledge to D. Singh for running and processing NMR experiments. We kindly acknowledge Department of Science & Technology for single crystal X-ray diffractometer as a National Facility under Project No. SR/S2/CMP-47/2003. The manuscript bears the institutional publication no. IIIM/1777/2015.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Sunil Kumar and Yedukondalu Nalli contributed equally to this work.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Kumar, S., Nalli, Y., Qadri, M. et al. Isolation of three new metabolites and intervention of diazomethane led to separation of compound 1 & 2 from an endophytic fungus, Cryptosporiopsis sp. depicting cytotoxic activity. Med Chem Res 26, 2900–2908 (2017). https://doi.org/10.1007/s00044-017-1989-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-1989-4