
Abstract
Inflammation is mediated by a variety of soluble factors, including a group of secreted polypeptides known as cytokines. The anti-inflammatory cytokines are a series of immune regulatory molecules that control the pro inflammatory cytokine response. Cytokines act in concert with specific cytokine inhibitors and soluble cytokine receptors to regulate the human immune response. The aim of the present study is to probe the anti-inflammatory potential of the crude extract and the bioactive metabolite isolated from marine bacteria Streptomyces sp. on key inflammatory mediators like tumor necrosis factor-α and interleukin-6. Here, we isolated ten known pyrazine-1,4-dione substituted cyclic dipeptide by semi-preparative HPLC and studied their anti-inflammatory activities against tumor necrosis factor-α and interleukin-6. Compound 3, 4, 5, 7 and 8 showed good inhibition of the both the cytokines in lipopolysaccharide-stimulated macrophages. The study reveal that compound 7 was to be specific inhibitor for tumor necrosis factor-α which efficiently inhibited tumor necrosis factor-α release in a dose-dependent manner and decreased lipopolysaccharide induced tumor necrosis factor-α production in human peripheral blood mononuclear cells in both the in vitro and in vivo experiments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microbial natural products are an important source of both existing and new drugs (Solanki et al. 2008). With marine species comprising approximately half of the total global biodiversity, the sea offers an enormous resource for novel compounds and it has been classified as the largest remaining reservoir of natural molecules to be evaluated for drug activity (De Vries and Beart 1995). To date, most attention has been applied to marine ecosystems, especially deep sea environments, as a rich source of novel microbes and hence natural products. So far several distinct classes of biomolecules were isolated from sea microbes prove to be having the potent immunomodulatory and antitumor activities (Remsing et al. 2003). Among the producers of commercially important metabolites, actinomycetes have proven to be a prolific source with a surprisingly small group of taxa accounting for the vast majority of compounds. Actinomycetes are responsible for the production of about half of the discovered secondary metabolites, notably antibiotics, antitumor agents, immunosuppressive agents and enzymes (Wu et al. 2007; Williams et al. 2007; Sujatha et al. 2005). Secondary metabolites produced by actinomycetes possess a wide range of biological activities and the vast majority of these compounds are derived from the single genus Streptomyces which are responsible for about two-thirds antibiotics, the most widely recognized secondary metabolites, including many antimicrobials such as erythromycin, streptomycin and tetracycline with unusual structures and potent biological activities (Lam 2006). That is one of the reasons the isolation of unusual environments for bioactive natural products is of great interest.
Inflammation, the response of tissue to injury, is characterized in the acute phase by increased blood flow and vascular permeability along with the accumulation of fluid, leukocytes, and inflammatory mediators such as cytokines (Yuan et al. 2006). The human immune response is regulated by a highly complex and intricate network of control elements. Under physiological conditions, these cytokine inhibitors serve as immunemodulatory elements that limit the potentially injurious effects of sustained or excess inflammatory reactions (Akyol et al. 2012). The release of pro-inflammatory cytokines is an important mechanism by which the immune cells regulate the inflammatory responses and contribute to various inflammatory and autoimmune disorders (Gayathri et al. 2007). Tumor necrosis factor-α (TNF-α) and Interleukin-6 (IL-6) are two of the most important cytokines. These two cytokines act as pro-inflammatory mediators of a variety of autoimmune diseases such as pain and joint destruction in RA (Smolen and Steiner 2003).
TNF-α is a pro-inflammatory cytokine produced mainly by macrophages and T cells that play a key role in many physiological immune processes, but when produced in excess it can cause severe damage to the host. Over-expression of TNF-α is correlated with many pathological conditions like Crohn’s disease, ulcerative colitis (Papa Gobbi et al. 2016), diabetes (Hotamisligil et al. 1995), multiple sclerosis (Selmaj et al. 1991), stroke (Lovering and Zhang 2005) and cirrhosis (Such et al. 2001).
IL-6 is a multifunctional cytokine synthesized and released by a variety of cells, including macrophages, in response to infection, tissue damage, or immunological challenge, playing an important role in the regulation of inflammation, immune responses, the acute-phase reaction, and haematopoiesis. IL-6 acts both at local and systemic levels affecting the functionality of several cell types, including not only immune system cells, but also hepatocytes, skeletal muscle cells and adipocytes. As a pro-inflammatory cytokine, IL-6 is present in high levels in both the synovium and serum of RA patients, and in the serum and ascitic fluid (AF) of cirrhotics (Ruiz-Alcaraz et al. 2011) inducing many different cellular and physiological responses during these chronic inflammation processes.
Nowadays, a big effort has been made in terms of TNF-α inhibition, but still no small molecule has been approved so far as reliable inhibitors of this cytokine. So far, TNF-α inhibitor drugs in clinics are proteins having undesirable side effects. IL-6 inhibitors have proved to be potentially useful for treatment of Alzheimer’s disease, psychiatric disorders, cancer, diabetes, and depression (Rosler et al. 2001); (Jahromi et al. 2000) converting the designing of IL-6 inhibitory agents into a major hope in the area of anti-inflammatory drug development.
All the compounds were analyzed for their ability to decrease lipopolysaccharide (LPS) induced TNF-α and IL-6 production in human peripheral blood mononuclear cells (PBMCs). Co-incubation of 1–10 with LPS at 5 h showed a considerable inhibition of TNF-α and IL-6 at 10 µM, when compared to LPS-induced control cells and rolipram standard.
Result and discussion
The 5 litres fermentation batch of marine-derived bacteria Streptomyces sp. were extracted three times with EtOAc. In the preliminary screening, the resulting extract was tested for their anti-inflammatory activities, both in vitro and in vivo experiments, against key inflammatory mediators TNF-α and IL-6, and the results showed that at 1 µg/mL concentration, the extract inhibited TNF-α and IL-6 release in a dose-dependent manner and decreased LPS induced TNF-α and IL-6 production in human PBMCs. We analyzed extract by preparative HPLC and developed a method through which maximum number of compounds were resolved (Fig. 1).

HPLC chromatogram: flow rate 0.7 mL/min; solvents water (A) acetonitrile (B); column Eclipse XDB-C18, 5 µm, 9.4 × 250 mm; column temp. 30; injection vol. 35 µL
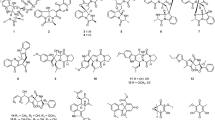
Consequently, ten known pyrazine-1,4-dione substituted cyclic dipeptide class of secondary metabolites were separated with >95 % purity (1–10) (Fig. 2). The structures of the isolated compounds were characterized as cyclo(L-Tyr-L-Pro) (1) (Rudi et al. 1994), cyclo(L-Pro-L-Val) (2) (Takaya et al. 2007), cyclo(L-4-OH-Pro-L-Leu) (3) (Cronan et al. 1998), cyclo(L-lle-D-Pro) (4), cyclo(D-trans-4-OH-Pro-D-phe) (5) (Fdhila et al. 2003), cyclo(L-lle-L-Pro) (6) (Takaya et al. 2007), cyclo(L-Leu-L-Pro) (7) (Takaya et al. 2007); (Jayatilake et al., 1996), cyclo(D-Pro-L-Phe (8) (Debitus et al. 1998), cyclo(D-6-Hyp-L-Phe) (9) (Park et al. 2006), cyclo(D-Pro-D-Phe) (10) (Fdhila et al. 2003) by analysis of HRESIMS, NMR (1D and 2D spectroscopy), and their comparison with those of the literature data (Fig. 2). All the compounds (1–10) were screened for their ability to decrease lipopolysaccharide induced TNF-α and IL-6 production in human peripheral blood mononuclear cells (PBMCs) (Gayathri et al. 2007; Lin and Lin 2011; Ammon 2010).

Structures of all the compounds isolated from Streptomyces sp
LPS, a component of the gram-negative bacterial cell wall, is one of the major causative agents of gram-negative sepsis. LPS is a potent inducer of monocytes and macrophages, which are key mediators of the innate immune response. Stimulation of cells with LPS leads to a cascade of intracellular signalling events that ultimately result in production and secretion of cytokines and other inflammatory mediators that constitute the pro-inflammatory response. The potential anti-inflammatory properties of compounds were evaluated in PBMC because this macrophage model produces high concentrations of IL-6 and TNF-α in culture upon activation with LPS, and reduced the need for additional animals required to obtain and use primary cultures. In the in vitro experiments incubation of compounds with LPS at 5 h showed a considerable inhibition of TNF-α and IL-6 over three concentrations (10, 5 and 1 μM), when compared to LPS-induced control cells and rolipram standard (Table 1). The compounds 3, 4, 5, 7 and 8 against TNF-α, and 5 and 8 against both IL-6 (Table 1) showed maximum inhibitory effects, having % inhibition >40 at 10 μM concentrations. The compounds 3, 4, and 7 were exhibited poor % inhibition against IL-6, whereas 1, 2, 6, 9 and 10 was not detected (nd) against both TNF-α and IL-6. In the in vitro experiments, the compounds 7 (IC50 9.4 µM) against TNF-α, and 5 (IC50 8.3 µM) against IL-6 were proven the most potent among all (Table 1). For all active compounds from above in vitro analysis, in vivo evaluation was performed in mice to determine their ability to replicate in animal models (Table 2). At a dose of 20 mg/kg concentration, compounds 4 (44.8 %), and 7 (63.4 %) against TNF-α, and 4 (41.4 %) and 7 (57.1 %) against IL-6 were proven the most potent among all. In summary, the compound 7 found to be specific inhibitor for TNF-α, in both in vitro and in vivo experiments, which efficiently inhibits TNF-α and IL-6 release in a dose-dependent manner and decreased LPS induced TNF-α and IL-6 production in human PBMCs. The cytokines IL-6 and TNF-α were evaluated because these pro-inflammatory cytokines play a key role in the inflammatory response and can be easily quantified in supernatant and serum samples. Levels of IL-6 and TNF-α were not detectable in untreated cells and animals that served as control in vitro and in vivo respectively. However, cell culture data did not always accurately predict the results in an animal model. Thus, the compounds under observation are studied in vivo. This was supported by the findings that there is considerable variation in both in vitro and in vivo results. Discrepancies between cell culture data and animal data are not entirely surprising considering only one cell type exists in the culture studies, whereas multiple cell types capable of producing pro-inflammatory cytokines are present in a whole animal system. Additionally, other inflammatory mediators may influence the cytokine response.
Material and methods
General experimental procedures
High resolution mass spectra were obtained on Agilent 6540 (Q-TOF) high resolution mass spectrometer, in the electro spray ionisation (ESIMS) mode. 1H NMR spectra were recorded (Brucker Avance) DPX FT-NMR at 400 and 500 MHz and 13C NMR at 125 and 100 MHz in CDCl3 and MeOD, chemical shifts values are reported in δ (ppm) units and coupling constants values in hertz. Tetramethylsilane (TMS) was used as internal standard. Semi preparative HPLC was performed on an Agilent HPLC with a RP-18 column (250 × 4.6 mm, 5 μm; Agilent), a photodiode array detector and auto injector function (Agilent 1260 series). Fetal bovine serum was obtained from GIBCO Invitrogen Corporation USA.
Fermentation conditions
Seed inoculums of the Streptomyces sp. was prepared by growing the culture in 500 mL of modified FM 17 medium (glucose, 20 g; yeast powder, 5 g; casein, 5 g; KNO3, 10 g; CaCO3, 2 g; seawater, 1 L; pH 7.2–7.4 for 3 days at 30 ± 2 οC. These inoculums was used to seed 5 L of FM 17 media and incubated for 10 days at 30 ± 2 οC and 150 rpm.
Extraction and isolation
The microbial producer Streptomyces sp. was isolated from the marine ascidian Synoicum indicum, collected from intertidal rocky habitat of Honnavar, Karnataka, India (14.19 Long, 74.45 Lat). The specimens were frozen immediately after collection which was further washed in sterile seawater 3 times followed by moist heat treatment at 50 °C for 30 min. The sample was homogenized with sterile seawater, serially diluted till 10−5 dilutions and 0.1 ml aliquots of each dilution were plated on to Actinomycete Isolation Agar (AIA1). The plates were incubated at 30 °C for 2 to 3 weeks. Colonies were selected based on filamentous growth and the color of substrate and aerial mycelium. The individual colonies were purified and maintained in 20 % glycerol at −80 °C. Out of several strains, NIO-1034 was selected for further bioassay-guided isolation of active molecules.
The sequences of PCR products were analyzed by using Basic Local Alignment Search Tool (BLAST). Nucleotide-nucleotide BLAST was carried out with facility of National Centre for Biotechnology Information (NCBI) database, NIO-1034 showing 99 % identity with Streptomyces sp. (Ali et al. 2013). 5 litre culture broths of Streptomyces sp. extracted with ethyl acetate and concentrated under reduced pressure, afforded 250 mg of ethyl acetate extract. The extract was re-dissolved in HPLC-grade acetonitrile (4 mL) followed by filtration through a 0.45 µm filter; the resulting solution was subjected to semi-preparative HPLC. As a result, ten compounds were purified: 1 (42 mg, t R 21.4 min), 2 (32 mg, t R 22.6 min), 3 (5.2 mg, t R 25.0 min), 4 (2.5 mg, t R 30.6 min), 5 (4.3 mg, t R 32.5 min), 6 (20.3 mg, t R 35.7 min), 7 (25 mg, t R 42.3 min), 8 (9 mg, t R 44.8 min), 9 (1.2 mg, t R 50.7 min), 10 (12 mg, t R 15.6 min). The semi-preparative HPLC used was equipped with Agilent 1260 series with PDA detector. The elution performed with water (A) and acetonitrile (B) as mobile phase (0–35 min, 19 % B; 35–45 min, 19–40 % B; 45–50 min, 40 % B; 50–57 min, 40–95 % B; 57–65 min, 95 % B; 65–70 min, 95–19 %; 70–75 min, 19 % B). Eclipse XDB-C-18 (5 µm, 250 × 9.4 mm) column was used at a flow rate of 0.7 mL/min (run time 75 min; column temp. 30 °C; injection vol. 35 µL; UV detection at 215 nm). The purity of the compounds was >95 % by HPLC/UV analysis. Purified compounds were identified and characterized by IR, UV, HRESIMS, NMR (1D and 2D spectroscopy), and their comparison with those of the literature data.
Compound 1
White solid; (+)-HRESIMS ion peak at m/z 261.12353 [M+H]+ (calcd for C14H17N2O3, 261.1234); 1H NMR (500 MHz, MeOD) δ 7.04 (d, J = 8.4 Hz, 2H), 6.70 (d, J = 8.4 Hz, 2H), 4.35 (t, J = 4.0 Hz, 1H), 4.09–3.98 (m, 1H), 3.54 (dt, J = 11.8, 8.3 Hz, 1H), 3.38–3.31 (m, 1H), 3.05 (qd, J = 14.2, 4.9 Hz, 2H), 2.08 (dt, J = 15.9, 5.6 Hz, 1H), 1.85–1.74 (m, 2H), 1.28–1.16 (m, 1H); 13C NMR (126 MHz, MeOD) δ = 170.81 (C, C-1), 166.96 (C, C-6), 157.67 (C, C-4′), 132.13 (2×CH, C-2′), 127.66 (C, C-1′), 116.20 (2×CH, C-3′), 60.06 (CH, C-2), 57.90 (CH, C-7), 45.94 (CH2, C-5 ), 37.64 (CH2, C-8), 29.40 (CH2, C-3), 22.74 (CH2, C-4).
Compound 2
White solid; (+)-HRESIMS ion peak at m/z 197.12857 [M+H]+ (calcd for C10H17N2O2, 197.1285); 1H NMR (500 MHz, CDCl3) δ 6.69 (s, 1H), 4.08 (t, J = 7.6 Hz, 1H), 3.94 (s, 1H), 3.64 (dt, J = 11.8, 8.2 Hz, 1H), 3.60–3.44 (m, 1H), 2.63 (dtd, J = 13.9, 6.9, 2.4 Hz, 1H), 2.44–2.29 (m, 1H), 2.15–2.04 (m, 1H), 2.02 (dd, J = 10.6, 7.8 Hz, 1H), 1.97–1.82 (m, 1H), 1.09 (d, J = 7.2 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 170.26 (C, C-1), 165.02 (C, C-6), 60.46 (CH, C-7), 58.80 (C, C-2), 45.10 (CH2, C-5), 28.51 (CH2, C-3), 28.44 (CH, C-8), 22.34 (CH2, C-4), 19.07 (CH3, C-9), 16.07 (CH3, C-10).
Compound 3
White solid; (+)-HRESIMS ion peak at m/z 227.1391 [M+H]+ (calcd for C11H19N2O3, 227.1390); 1H NMR (500 MHz, MeOD) δ 4.52 (dd, J = 10.8, 6.4 Hz, 1H), 4.46 (d, J = 3.8 Hz, 1H), 4.16 (d, J = 5.2 Hz, 1H), 3.65 (dd, J = 12.7, 4.4 Hz, 1H), 3.43 (d, J = 12.8 Hz, 1H), 2.28 (dd, J = 13.2, 6.4 Hz, 1H), 2.12–2.08 (m, 1H), 1.98–1.84 (m, 3H), 1.56–1.47 (m, 1H), 0.96 (dd, J = 6.1, 2.5 Hz, 6H); 13C NMR (126 MHz, MeOD) δ 173.10 (C, C-1), 169.07 (C, C-6), 69.14 (CH, C-4), 58.74 (CH, C-2), 55.20 (CH2, C-5), 54.63 (CH, C-7), 39.41 (CH2, C-8), 38.19 (CH2, C-3), 25.82 (CH, C-9), 23.33 (CH3, C-10), 22.23 (CH3, C-11).
Compound 4
White solid; (+)-HRESIMS ion peak at m/z 211.1440 [M+H]+ (calcd for C11H19N2O2, 211.1441); 1H NMR (400 MHz, CDCl3) δ 6.21 (s, 1H), 4.10 (dd, J = 9.7, 6.7 Hz, 1H), 3.83–3.77 (m, 1H), 3.71 (dt, J = 11.7, 8.6 Hz, 1H), 3.59–3.48 (m, 1H), 2.48–2.37 (m, 1H), 2.09–2.00 (m, 1H), 2.00–1.96 (m, 1H), 1.96–1.91 (m, 1H), 1.92–1.82 (m, 1H), 1.58–1.52 (m, 1H), 1.26 (ddd, J = 10.6, 9.7, 4.9 Hz, 1H), 1.03 (d, J = 6.9 Hz, 3H), 0.94 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 169.20 (C, C-1), 165.15 (C, C-6), 62.97 (CH, C-7), 58.37 (CH, C-2), 45.65 (CH2, C-5), 39.66 (CH, C-8), 29.40 (CH2, C-3), 24.52 (CH2, C-9), 21.99 (CH2, C-4), 15.30 (CH3, C-11), 11.28 (CH3, C-10).
Compound 5
Yellowish oil; (+)-HRESIMS ion peak at m/z 261.1232 [M+H]+ (calcd for C14H17N2O3, 261.1234); 1H NMR (500 MHz, CDCl3) δ 7.36 (t, J = 7.3 Hz, 2H), 7.33–7.28 (m, 1H), 7.23 (d, J = 7.5 Hz, 2H), 5.72 (s, 1H), 4.58 (s, 1H), 4.47 (dd, J = 10.9, 6.1 Hz, 1H), 4.32 (d, J = 9.8 Hz, 1H), 3.78 (dd, J = 13.2, 4.3 Hz, 1H), 3.63 (d, J = 3.6 Hz, 1H), 3.58 (d, J = 13.5 Hz, 1H), 2.79 (dd, J = 14.5, 10.6 Hz, 1H), 2.36 (dd, J = 13.3, 6.1 Hz, 1H), 2.11–1.97 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 169.64 (C, C-1), 165.15 (C, C-6), 135.80 (C, C-1′), 129.33 (2×CH, C-2′), 129.13 (2×CH, C-3′), 127.63 (CH, C-4′), 68.40 (CH, C-4), 57.41 (CH, C-2), 56.16 (CH, C-7), 54.44 (CH2, C-5), 37.79 (CH2, C-3), 36.71 (CH2, C-8).
Compound 6
White solid; (+)-HRESIMS ion peak at m/z 211.1441 [M+H]+ (calcd for C11H19N2O2, 211.1441); 1H NMR (400 MHz, CDCl3) δ 6.44 (s, 1H), 4.15–4.01 (m, 1H), 3.97 (s, 1H), 3.64 (dt, J = 13.2, 8.0 Hz, 1H), 3.55 (ddd, J = 12.6, 12.0, 8.2 Hz, 1H), 2.44–2.34 (m, 1H), 2.34–2.21 (m, 1H), 2.14–2.02 (m, 1H), 2.02–1.96 (m, 1H), 1.96–1.83 (m, 1H), 1.43 (dtd, J = 14.9, 7.5, 3.5 Hz, 1H), 1.20 (tdd, J = 9.8, 8.9, 5.3 Hz, 1H), 1.07 (dd, J = 7.2, 2.1 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 170.22 (C, C-1), 165.06 (C, C-6), 60.53 (CH, C-2), 58.76 (CH, C-7), 45.12 (CH2, C-5), 35.36 (CH, C-8), 28.52 (CH2, C-3), 24.04 (CH2, C-9), 22.33 (CH2, C-4), 15.78 (CH3, C-11), 12.06 (CH3, C-10).
Compound 7
Colorless Chrystal; (+)-HRESIMS ion peak at m/z 211.14435 [M+H]+ (calcd for C11H19N2O2, 211.1441); 1H NMR (400 MHz, CDCl3) δ 6.04 (s, 1H), 4.12 (t, J = 8.2 Hz, 1H), 4.02 (dd, J = 9.5, 3.5 Hz, 1H), 3.65–3.47 (m, 2H), 3.17–3.14 (m, 1H), 2.35 (dtd, J = 9.8, 6.9, 3.0 Hz, 1H), 2.20–2.09 (m, 1H), 2.10–2.03 (m, 1H), 2.04–1.96 (m, 1H), 1.96–1.83 (m, 1H), 1.82–1.74 (m, 1H), 1.53 (ddd, J = 14.5, 9.5, 5.0 Hz, 1H), 1.00 (d, J = 6.6 Hz, 3H), 0.96 (d, J = 6.6 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 171.08 (C, C-1), 166.95 (C, C-6), 59.18 (CH, C-2), 53.60 (CH, C-7), 45.58 (CH2, C-5), 38.46 (CH2, C-8), 28.11 (CH2, C-3), 24.59 (CH, C-9), 23.17 (CH3, C-10), 22.80 (CH2, C-4), 21.46 (CH3, C-11).
Compound 8
White solid; (+)-HRESIMS ion peak at m/z 245.12868 [M+H]+ (calcd for C14H17N2O2, 245.1285); 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 1H), 7.27 (d, J = 3.5 Hz, 2H), 7.21 (d, J = 5.9 Hz, 2H), 5.87 (s, 1H), 4.21 (s, 1H), 3.64 (d, J = 9.1 Hz, 1H), 3.42 (t, J = 10.6 Hz, 1H), 3.20–3.06 (m, 2H), 3.04 (d, J = 9.8 Hz, 1H), 2.19 (s, 1H), 1.95 (s, 1H), 1.81 (dd, J = 20.3, 9.8 Hz, 1H), 1.75 (d, J = 18.6 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 169.59 (C, C-1), 164.89 (C, C-6), 135.20 (C, C-1′), 129.95 (2×CH, C-2′), 128.78 (2×CH, C-3′), 127.55 (CH, C-4′), 59.01 (CH, C-7), 57.74 (CH, C-2), 45.17 (CH2, C-5), 40.43 (CH2, C-8), 28.91 (CH2, C-3), 21.66 (CH2, C-4).
Compound 9
White solid; (+)-HRESIMS ion peak at m/z 261.1233 [M+H]+ (calcd for C14H17N2O3, 261.1234); 1H NMR (700 MHz, CDCl3) δ 7.40–7.34 (m, 2H), 7.34–7.30 (m, 1H), 7.25 (d, J = 6.8 Hz, 2H), 5.84 (s, 1H), 4.49 (dd, J = 10.6, 3.7 Hz, 1H), 3.84–3.78 (m, 1H), 3.63–3.57 (m, 1H), 3.66 (d, J = 21.4 Hz, 1H), 2.83–2.77 (m, 1H), 2.26 (dd, J = 12.1, 5.8 Hz, 1H), 2.15 (d, J = 16.1 Hz, 1H), 2.23–2.18 (m, 1H), 2.01 (s, 1H); 13C NMR (176 MHz, CDCl3) δ 168.89 (C, C-1), 166.5 (C, C-6), 132.9 (C, C-1′), 129.3 (2×CH, C-2′), 129.25 (2×CH, C-3′), 127.66 (CH, C-4′), 84.77 (CH, C-2), 55.90 (CH, C-7), 45.46 (CH2, C-5), 36.95 (CH2, C-8), 36.93 (CH2, C-3), 19.89 (CH2, C-4).
Compound 10
White solid; (+)-HRESIMS ion peak at m/z 245.12877 [M+H]+ (calcd for C14H17N2O2, 245.1285); 1H NMR (400 MHz, CDCl3) δ 7.35 (t, J = 7.2 Hz, 2H), 7.31–7.26 (m, 1H), 7.23 (d, J = 7.0 Hz, 2H), 5.81 (s, 1H), 4.28 (dd, J = 10.3, 2.9 Hz, 1H), 4.07 (t, J = 7.7 Hz, 1H), 3.71–3.62 (m, 1H), 3.62–3.57 (m, 1H), 3.59–3.49 (m, 1H), 2.80 (dd, J = 14.5, 10.4 Hz, 1H), 2.45–2.25 (m, 1H), 2.03 (ddd, J = 12.8, 9.2, 4.7 Hz, 1H), 1.99–1.93 (m, 1H), 1.93–1.84 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 169.44 (C, C-1), 165.07 (C, C-6), 136.03 (C, C-1′), 129.20 (2×CH2, C-2′), 129.16 (2×CH, C-3′), 127.46 (CH, C-4′), 59.11 (CH, C-2), 56.25 (CH, C-7), 45.40 (CH2, C-4), 36.80 (CH2, C-8), 28.33 (CH2, C-3), 22.50 (CH2, C-5).
Preparation of chemicals and drugs for in vitro and in vivo anti-inflammatory activity studies
In vitro assay
Heparinised venous blood was taken from a healthy human volunteer. Mononuclear cells were isolated in a Ficoll–Hypaque density gradient using standard procedures which separated PBMCs from whole blood. The buffy coat containing PBMCs was removed carefully following centrifugation and washed twice in phosphate buffer saline (PBS). Cells were counted and the samples were incubated with 1 × 106 cells/well in triplicate in 96 well microtiter plates. All the compounds (1–10) were tested to check their anti-inflammatory activity against TNF-α and IL-6 at three different concentrations. To do so, anti-inflammatory activity against TNF-α and IL-6 were analyzed as the percentage of inhibition of these cytokines production when human peripheral blood mononuclear cells (PBMCs) were treated with Lipopolysaccharide (LPS). Dose response study was done in LPS (mitogen) (1 μg/mL) induced PBMC with different concentrations i.e., 10, 5 and 1 µM of compounds. The results were expressed in % inhibition with respect to control. Experiments were done in triplicates and repeated three times. Anti-inflammatory effect of 1–10 dose response assays was used to determine the capacity of inhibition of these compounds upon the pro-inflammatory cytokines TNF-α and IL-6 measured by Enzyme-linked immunosorbent assay (ELISA). Data represents the mean ± SEM of the representative compounds for the inhibition of TNF-α and IL-6 tested on three different assays.
In vivo assay
Female Balb/C mice (Mus musculus) 8–10 weeks old and weighing 20–25 g, in groups of six, were used for the study. Experiments were designed to minimize the numbers of animals used. The animals were housed under standard laboratory conditions 23 ± 1 °C, 55 ± 10 % relative humidity, 12/12 h light/dark cycles, fed with standard pellet diet (Lipton India Ltd) and received water ad libitum. None of the animals was sacrificed throughout the study; however, as per the institute’s norms, the animals, after experimentation, were subjected to euthanasia by a high inhalational dose of diethyl ether and disposed of by incineration. Mice were orally treated with the test compound at different concentrations for 1 h. Then via i.p. administration 0.5 mg/kg LPS was given. After 2.5 h blood was obtained by retro-orbital plexus and allowed to clot for 30 min at R.T. Serum was collected and stored at −80 °C till ELISA. Then it was analyzed for IL-6 and TNF-α by ELISA. Cytokine analysis for IL-6 and TNF-α was performed using Mouse TNF-α ELISA and Mouse IL-6 ELISA kits purchased from Invitrogen, USA. Absorbance was read at 450 nm. The results were expressed in % inhibition with respect to control. Experiments were done in triplicates and repeated three times. Results were expressed as the mean ± SEM.
Conclusions
In summary, here we reported ten cyclic dipeptides from marine-derived bacteria Streptomyces sp. For the first time, all these compounds evaluated for their anti-inflammatory activities, and the compound 7 found to be specific inhibitor for TNF-α which inhibiting efficiently TNF-α release in a dose-dependent manner and decreased LPS induced TNF-α and IL-6 production in human PBMCs, in both in vitro and in vivo experiments.
References
Akyol S, Tanriverdi T, Hanci M (2012) Do immunologic events in degenerative disc tissue alter the peripheral immune tolerance? World Spinal Column J 3:45–49
Ali A et al (2013) Modulation of LPS induced inflammatory response by Lawsonyl monocyclic terpene from the marine derived Streptomyces sp. Immunol Lett 150:79–86
Ammon HPT (2010) Modulation of the immune system by Boswellia serrata extracts and boswellic acids. Phytomedicine 17:862–867
Cronan JM, Davidson TR, Singleton FL, Colwell RR, Cardellina JH (1998) Plant growth promoters isolated from a marine bacterium associated with Palythoa sp. Nat Prod Lett 11:271–278
De Vries DJ, Beart PM (1995) Fishing for drugs from the sea: status and strategies. Trends Pharmacol Sci 16:275–279
Debitus C et al (1998) Quinolones from a bacterium and tyrosine metabolites from its host sponge, Suberea creba from the coral sea. J Mar Biotechnol 6:136–141
Fdhila F, Vázquez V, Sánchez JL, Riguera R (2003) dd-Diketopiperazines: antibiotics active against vibrio anguillarum isolated from marine bacteria associated with cultures of pecten maximus. J Nat Prod 66:1299–1301
Gayathri B, Manjula N, Vinaykumar KS, Lakshmi BS, Balakrishnan A (2007) Pure compound from Boswellia serrata extract exhibits anti-inflammatory property in human PBMCs and mouse macrophages through inhibition of TNF-α, IL-1β, NO and MAP kinases. Int Immunopharmacol 7:473–482
Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95:2409–2415
Jahromi MM, Millward BA, Demaine AG (2000) A polymorphism in the promoter region of the gene for interleukin-6 is associated with susceptibility to type 1 diabetes mellitus. J Interferon Cytokine Res 20:885–888
Jayatilake GS, Thornton MP, Leonard AC, Grimwade JE, Baker BJ (1996) Metabolites from an Antarctic sponge-associated bacterium, Pseudomonas aeruginosa. J Nat Prod 59:293–296
Lam KS (2006) Discovery of novel metabolites from marine actinomycetes. Curr Opin Microbiol 9:245–251
Lin WC, Lin JY (2011) Five bitter compounds display different anti-inflammatory effects through modulating cytokine secretion using mouse primary splenocytes in vitro. J Agric Food Chem 59:184–192
Lovering F, Zhang Y (2005) Therapeutic potential of TACE inhibitors in stroke. Curr Drug Targets CNS Neurol Disord 4:161–168
Papa Gobbi R et al (2016) A galectin-specific signature in the gut delineates Crohn’s disease and ulcerative colitis from other human inflammatory intestinal disorders. BioFactors 42:93–105
Park YC, Gunasekera SP, Lopez JV, Mccarthy PJ, Wright AE (2006) Metabolites from the marine-derived fungus Chromocleista sp. Isolated from a deep-water sediment sample collected in the Gulf of Mexico. J Nat Prod 69:580–584
Remsing LL et al (2003) Mithramycin SK, a novel antitumor drug with improved therapeutic index, mithramycin SA, and demycarosyl-mithramycin SK: three new products generated in the mithramycin producer Streptomyces argillaceus through combinatorial biosynthesis. J Am Chem Soc 125:5745–5753
Rosler N, Wichart I, Jellinger KA (2001) Intra vitam lumbar and post mortem ventricular cerebrospinal fluid immunoreactive interleukin-6 in Alzheimer’s disease patients. Acta Neurol Scand 103:126–130
Rudi A, Kashman Y, Benayahu Y, Schleyer M (1994) Amino acid derivatives from the marine sponge Jaspis digonoxea. J Nat Prod 57:829–832
Ruiz-Alcaraz AJ et al. (2011) Peritoneal macrophage priming in cirrhosis is related to ERK phosphorylation and IL-6 secretion. Eur J Clin Invest 41:8–15
Selmaj K, Raine CS, Cannella B, Brosnan CF (1991) Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J Clin Invest 87:949–954
Smolen JS, Steiner G (2003) Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov 2:473–488
Solanki R, Khanna M, Lal R (2008) Bioactive compounds from marine actinomycetes. Indian J Microbiol 48:410–431
Such J et al (2001) Tumor necrosis factor-alpha, interleukin-6, and nitric oxide in sterile ascitic fluid and serum from patients with cirrhosis who subsequently develop ascitic fluid infection. Dig Dis Sci 46:2360–2366
Sujatha P, Bapi Raju KV, Ramana T (2005) Studies on a new marine streptomycete BT-408 producing polyketide antibiotic SBR-22 effective against methicillin resistant Staphylococcus aureus. Microbiol Res 160:119–126
Takaya Y et al (2007) Antioxidant constituents in distillation residue of awamori spirits. J Agric Food Chem 55:75–79
Williams PG et al (2007) Saliniketals A and B, bicyclic polyketides from the marine actinomycete Salinispora arenicola. J Nat Prod 70:83–88
Wu SJ, Fotso S, Li F, Qin S, Laatsch H (2007) Amorphane sesquiterpenes from a marine Streptomyces sp. J Nat Prod 70:304–306
Yuan G, Wahlqvist ML, He G, Yang M, Li D (2006) Natural products and anti-inflammatory activity. Asia Pac J Clin Nutr 15:143–152
Acknowledgments
The authors are grateful to the University grant commission (UGC-New Delhi) and CSIR for providing financial support having the Grant No. ITR-0001 for the work of YN, and SM. YK, SG and VK acknowledge AcSIR for their enrolment in Ph. D program. VK also acknowledge DST for INSPIRE Fellowship. We are also thankful to Deeαpika Singh for recording the NMR experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Nalli, Y., Gupta, S., Khajuria, V. et al. TNF-α and IL-6 inhibitory effects of cyclic dipeptides isolated from marine bacteria Streptomyces sp. Med Chem Res 26, 93–100 (2017). https://doi.org/10.1007/s00044-016-1730-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-016-1730-8