
Abstract
Dorema glabrum (Apiaceae) is a monocarpic plant distributed in southern Caucasus. The gum-resin of this species is traditionally used as diuretic and anti-diarrheal and for the treatment of bronchitis and catarrh. In the present study, free radical-scavenging activity and total phenolic content of the essential oil together with n-hexane, chloroform, ethyl acetate, and methanol extracts of D. glabrum roots were evaluated in DPPH and Folin-Ciocalteu assays, respectively. Methanol extract with the highest free radical-scavenging activity (IC50 = 74.2 ± 6.6 μg ml−1) and total phenolic content (186.7 ± 8.6 mg GAE/g) was subjected to phytochemical investigation using different chromatographic methods on the Si gel (normal and reversed-phase) and Sephadex LH-20 columns. Chemical constituents of the roots oil were also analyzed using GC and GC–MS. Two new phloroacetophenone glycosides, azerosides A (1) and azerosides B (7), along with nine known phenolic compounds, echisoside (2), pleoside (3), hyrcanoside (4), 7,8-dihydroferulic acid-4-O-β-d-glucopyranoside (5), Lavandoside (6), 6,7,8-trihydroxycoumarin (8), chlorogenic acid (9), 4,5-Di-O-caffeoylquinic acid (10), and cynarin (11), were isolated and identified from D. glabrum roots. Among the isolated compounds, 8–11 exhibited potent free radical-scavenging activity (IC50 values of 1.8–2.7 μg ml−1) in comparison with BHT (IC50 = 19.5 ± 0.8 μg ml−1). Twenty-six compounds were also identified in the roots oil, among them myristicin (14.1 %) and elemicin (11.7 %), two bioactive phenylpropanoid derivatives, were main compounds. This study introduces D. glabrum as a source of phloroacetophenone glycosides and caffeoylquinic acid derivatives and suggests it as an appropriate candidate for further pharmacological and toxicological studies.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Iran with about 120 genera is one of the main distribution centers of the Apiaceae (alt. Umbelliferae) family plants in the world (Ghahreman, 2002; Mozaffarian, 2007). The genus Dorema from this family comprises about 12–14 species, mainly distributed in southwestern and central Asia (Rechinger, 1987; Pimenov, 1988). Some of Dorema species exude gum-resins with medicinal properties, among them the gum-resin “ammoniacum” from D. ammoniacum is a well-known therapeutic agent which is used as an antispasmodic and expectorant and for the treatment of skin inflammatory diseases (Amanzadeh, 2002).
Dorema glabrum Fisch. & C.A. Mey. is one of the seven Dorema species from the flora of Iran which is also found in Azerbaijan Republic and Armenia (Mozaffarian, 2007). In Nakhichevan region (Azerbaijan Republic), the gum-resin of this species is used by Azeri people as a diuretic and anti-diarrheal agent, as well as for the treatment of bronchitis and catarrh (Mirbabayev et al., 1993).
Previous biological surveys have shown anti-lipidemic and antioxidant effects of the aerial parts (Dehghan et al., 2009) and cytotoxic activity of the fruits of D. glabrum (Amirkhiz et al., 2013). The essential oil of the roots of this plant has also been reported to contain δ-cadinene and β-bisabolene as the main compounds with a weak free radical-scavenging activity (IC50 = 2.2 mg ml−1) (Asnaashari et al., 2011).
Unfortunately, decrease in natural population of D. glabrum during recent decades, which is most likely caused by extensive medicinal uses and seed germination problems in Dorema genus plants, has led it to be considered as an endangered species (Ibadullayeva et al., 2011; Irvani et al., 2012).
Previous reports on phytochemical constituents of Dorema species roots are limited to the isolation of some sesquiterpene derivatives from D. kopetdaghense (Iranshahi et al., 2007) and some phloroacetophenone glycosides from D. hyrcanum (Nurmukhamedova and Nikonov, 1976) and D. aitchisonii (Bukreeva and Pimenov, 1991). In this study, we report essential oil composition and isolation of the eleven free radical-scavenging compounds from D. glabrum roots including two new phloroacetophenone glycosides, namely azerosides A and B.
Experimental section
Plant material
The roots of D. glabrum were collected from the rocky slopes of “Ghaflankuh” mountains located in East-Azerbaijan (northwest of Iran) in June 2012. A voucher specimen of the plant (No. 2120 MPIH) was deposited in the herbarium of Institute of Medicinal Plants, ACECR, Karaj, Iran.
Extraction
The air-dried and ground roots (2.4 kg) were macerated successively with n-hexane, chloroform, ethyl acetate, and methanol (3 × 5 L each) at the room temperature. The obtained extracts were then concentrated using a rotary evaporator under 45 °C.
Essential oil extraction
The air-dried and comminuted roots (100 g) were subjected to hydrodistillation for 4 h using a Clevenger-type apparatus to produce pale yellow oil in 0.3 % (v/w) yield. The obtained oil was dried over anhydrous sodium sulfate and stored in 4 °C until analysis.
DPPH free radical-scavenging activity assay
Free radical-scavenging potentials of the samples were evaluated using 2, 2-diphenyl-1-picryl-hydrazyl (DPPH) method described by Delazar et al., (2012). Briefly, the stock sample solution (5 mg ml−1 in methanol) was diluted twofold with methanol to get concentrations ranging from 5 to 9.5 × 10−3 mg ml−1. DPPH (Sigma) was prepared in the concentration of 80 μg ml−1 in methanol. Diluted solutions (1 ml each) were mixed with 1 ml of DPPH solution and were kept 30 min at 25 °C in dark for any reaction to take place.
UV absorptions of the solutions were recorded on a Cecil CE7250 spectrophotometer at 517 nm. Butylated hydroxytoluene (BHT), a synthetic antioxidant, was used as a positive control. All tests were performed in triplicate, and IC50 values were reported as Mean ± SD.
Determination of total phenolic content
Total phenolic content (TPC) of the extracts was measured by a colorimetric method using Folin-Ciocalteu reagent as described by Moradi-afrapoli et al., (2012). Briefly, 1.5 ml of tenfold distilled-water diluted Folin-Ciocalteu reagent (Merck) was added to 200 μl of extract solution (500 μg ml−1 in methanol) and allowed to stand at the room temperature for 5 min. Sodium bicarbonate solution (60 g l−1, 1.5 ml) was then added to the mixture and stored 90 min at 22 °C. The absorptions of the final solution were recorded on a Cecil CE7250 spectrophotometer at 725 nm.
The TPCs were quantified using a calibration curve obtained from absorbance measuring of the known gallic acid concentrations (50–200 μg ml−1 in methanol) as standard. The experiment was performed in triplicate, and the results were expressed as milligrams of gallic acid equivalents (GAE) per gram of dry extracts as Mean ± SD.
Isolation and purification of compounds
Methanol extract having the highest free radical-scavenging activity and TPC (Table 1) was subjected to phytochemical analysis to identify the compounds involved in its free radical-scavenging activity.
Insoluble white precipitate was appeared after adding the methanol (100 ml) to the methanol extract (100 g). One gram of the filtered insoluble material (7 g in total) was moved on a reversed-phase (RP-18) column (230–400 mesh, fully endcapped, Fluka) and eluted with CH3CN-H2O (1:9) to get one (12 mg) and two (680 mg). Fifty grams of the concentrated supernatant (the soluble part) was chromatographed on a Sephadex LH-20 (Fluka) column, eluted with MeOH to afford four fractions (A–D). Si gel column chromatography (230–400 mesh, Merck) of the fraction A (10 g) eluted with a gradient mixture of MeOH-CHCl3 (2:8 → 4:6) yielded twelve fractions (A1–12). Compound 3 (58 mg) was obtained from the fraction A1 (173 mg) by chromatography on a RP-18 column, eluted with CH3CN-H2O (2:8). RP-18 column chromatography of the fraction A4 (360 mg) (CH3CN-H2O, 2:8) afforded three fractions (A4a–4c). Fraction A4b (60 mg) was purified on a RP-18 column (CH3CN-H2O, 1.5:8.5) to get 4 (53 mg). Si gel column chromatography of the fraction A5 (435 mg) eluted with MeOH-CHCl3 (2.5:7.5) yielded four fractions (A5a–5d). Fraction A5a (77 mg) was moved on a RP-18 column (CH3CN-H2O, 2:8) to get 5 (35 mg) and 6 (7 mg). RP-18 column chromatography of the fraction A11 (270 mg) with CH3CN:H2O (0.5:9.5 → 1.5:8.5) resulted in the isolation of 7 (36 mg). A portion of fraction B (2 g) was moved on a RP-18 column (CH3CN-H2O, 0.5:9.5 → 2:8) to obtain nine fractions (B1–9). Fraction B1 (415 mg) was rechromatographed over a RP-18 column (CH3CN-H2O, 0.5:9.5) to get 8 (8 mg) and 9 (162 mg). Fraction B6 (320 mg) moved on a Sephadex LH-20 column and eluted with MeOH-H2O (8:2) gave 10 (110 mg). Compound 11 (94 mg) was obtained from the fraction B7 (360 mg) by chromatography on Si gel column, eluted with H2O-HCOOH-CH3COOH-EtOAc (2.4:1:1:63).
In all steps, column chromatography was monitored by thin layer chromatography (Pre-coated Si gel GF254 and Si gel 60 RP-18 F254s plates, Merck), and fractions with similar spots detected under UV (254 and 366 nm) or after spraying anisaldehyde/sulfuric acid reagent were combined.
The structures of the isolated compounds were elucidated by 1H-NMR, 13C-NMR, HMBC, and HSQC spectral analysis (Bruker Avance 400 DRX, 400 MHz for 1H and 100 MHz for 13C), as well as by comparing with data published in the literature. In the case of new compounds, melting points were measured on an Electro-thermal melting point apparatus. The optical rotations were determined on a Perkin-Elmer 241 polarimeter. UV spectra were recorded on a Cecil CE7250 spectrophotometer. IR spectra were obtained on a Nicolet Magna 550-FT-IR spectrometer. EIMS spectra were acquired on a Hewlett-Packard model 5973 HP system. Elemental analyses were carried out on Costech 4010 CHNS/O Elemental Combustion System.
Sugar analysis
The type of sugar moieties was confirmed, if needed, by a chromatographic method described by Yassa et al., (2007). Briefly, about 10 mg of compound was hydrolyzed in acidic condition (HCl (2 N), 10 ml) with heating for 45 min on a steam bath. The obtained solution was cooled and extracted by diethyl ether to remove aglycon portion. The aqueous phase was then chromatographed alongside with glucose, galactose, rhamnose, mannose, and xylose on paper (Whatman, No. 1) in ethyl acetate-pyridine-water (12:5:4) as solvent system. The chromatogram was then sprayed with a solution of p-anisidine hydrochloride/sodium hydrosulfite reagent and heated for 10 min until the appearance of brown spots of sugars.
GC and GC–MS analysis of the essential oil
The essential oil was analyzed on a Hewlett-Packard 6890 gas chromatograph with HP-5MS column (30 m × 0.25 mm id, 0.25 μm film thickness) equipped with a mass detector (Hewlett-Packard model 5973 HP). The flow rate of helium (carrier gas) was 1 ml min−1. The initial oven temperature was 40 °C and was then raised at a rate of 3 °C per minute to 250 °C. The injection temperature was 250 °C, and the oil sample (1 μl) was injected with a split ratio of 1:90. The mass spectra were obtained by electron ionization at 70 eV. The retention indices (RI) of the compounds were calculated using a homologous series of n-alkanes injected in conditions equal to the samples.
Identification of the compounds was carried out using computer matching with the Wiley7n.L library, and also by comparison of the RI and fragmentation pattern of the mass spectra with those for standard compounds published in the literature (Adams, 2007).
The essential oil was also analyzed on an Agilent HP-6890 gas chromatograph coupled with a FID detector for calculation of the relative amounts of the separated compounds. The FID detector temperature was 290 °C, and the operation was performed under the same conditions as described for GC–MS analysis.
Results and discussion
Among the tested extracts, methanol extract was found to contain the highest free radical-scavenging capacity (IC50 = 74.2 ± 6.6 μg ml−1) and TPC (186.7 ± 8.6 mg GAE/g) in DPPH and Folin-Ciocalteu assays, respectively (Table 1).
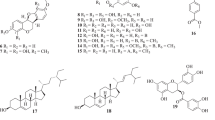
Phytochemical analysis of this bioactive extract using different chromatographic methods yielded to the isolation of eleven phenolic compounds. The isolated compounds were identified as 2-O-[β-d-glucopyranosyl-(1″ → 6′)-β-d-glucopyranosyl]-phloroacetophenone (azeroside A) (1), 2-O-[β-d-glucopyranosyl-(1″ → 6′)-β-d-glucopyranosyl]-4-O-methyl-phloroacetophenone (echisoside) (2) (Bukreeva and Pimenov, 1991), 2-O-β-d-glucopyranosyl-4-O-methyl-phloroacetophenone (pleoside) (3) (Nurmukhamedova and Nikonov, 1976; Chevalley et al., 2001), 2-O-[α-d-glucopyranosyl-(1″ → 6′)-β-d-glucopyranosyl]-4-O-methyl-phloroacetophenone (hyrcanoside) (4) (Nurmukhamedova and Nikonov, 1976; Singh and Bharate, 2006), 7,8-dihydroferulic acid-4-O-β-d-glucopyranoside (5) (Kraus and Spiteller, 1997; Tezuka et al., 2001), Ferulic acid-4-O-β-d-glucopyranoside (6) (Kurkin et al., 2008; Morikawa et al., 2012), 2-O-[β-d-glucopyranosyl-(1″ → 6′)-β-d-glucopyranosyl-(1″′ → 6″)-β-d-glucopyranosyl]-4-O-methyl-phloroacetophenone (azeroside B) (7), 6,7,8-trihydroxycoumarin (8) (Nykolov et al., 1993; Kayser and Kolodziej, 1995), 5-O-caffeoylquinic acid (5-CQA, chlorogenic acid) (9) (Chan et al., 2009), 4,5-Di-O-caffeoylquinic acid (4,5-diCQA) (10) (Basnet et al., 1996), and 1,5-Di-O-caffeoylquinic acid (1,5-diCQA, cynarin) (11) (Carnat et al., 2000).
The structures of these compounds were elucidated using their NMR spectroscopic data (1H-NMR, 13C-NMR, HMBC, and HSQC) and by comparing with those reported in the literature (Fig. 1).

Structures of the isolated compounds from the methanol extract of D. glabrum roots
Among the isolated compounds, azeroside A (1) and azeroside B (7) report in this study for the first time. This is also the first report on the isolation of 5–11 from Dorema genus plants. Compounds 5 and 8 are two rare compounds which have been reported from few natural sources (Nykolov et al., 1993; Kayser and Kolodziej, 1995; Kraus and Spiteller, 1997; Tezuka et al., 2001).
The 1H-NMR spectrum of 1 was found similar to echisoside (2), a known phloroacetophenone glycoside previously reported from the roots of D. aitchisonii (Bukreeva and Pimenov, 1991), with an additional broad singlet resonance at δ 8.38 and without any resonance characteristic for methoxy group at δ 3.7–4. These differences suggested that the methoxy group of 2 has been substituted by a hydroxy group in 1. Therefore, structure of 1-{2-[β-d-glucopyranosyl-(1 → 6)-β-d-glucopyranosyloxy]-4,6-dihydroxyphenyl}-1-ethanone was suggested for compound 1 (Fig. 1), supporting with 13C-NMR spectral data, and was named azeroside A. The EIMS spectrum of 1 revealed [M + H]+ ion peak at m/z 493, corresponding to the molecular formula C20H28O14 (M = 492). An ion peak at m/z 168 was also found in EIMS spectrum of 1, assignable to the 1-[2,4,6-trihydroxyphenyl]-1-ethanone (phloroacetophenone) fragment as aglycon of 1. The monoglycoside analog of azeroside A, Myrciaphenone A, has been reported from Curcuma comosa and Corymbia maculata with choleretic and antileishmanial activities (Suksamrarn et al., 1997; Sidana et al., 2013).
Pleoside (domesticoside) (3) has previously been isolated from the roots of D. hyrcanum (Nurmukhamedova and Nikonov, 1976). This compound has also been identified as an antifungal principle of the roots of Ribes rubrum (Chevalley et al., 2001).
Hyrcanoside (4) is a stereoisomer of 2, previously reported from the roots of D. hyrcanum (Nurmukhamedova and Nikonov, 1976). The appearance of the anomeric proton resonances at δ 5.00 (1H, d, J = 7.1 Hz) and 4.81 (1H, d, J = 2.8 Hz) in 1H-NMR spectra of this compound and identification of only glucose as a result of paper chromatography following acidic hydrolyses of this compound were indicative of α (1 → 6) linkage of its two glucopyranosyl moieties.
1H and 13C-NMR spectra of 7 showed a methoxy phloroacetophenone derivative structure similar to 2–4, containing three glucopyranosyl moieties. The 4-O-methyl-phloroacetophenone skeleton was confirmed as aglycon of 7 from long-range correlations (2 J HC or 3 J HC) of δ 3.81 (3H, s, OCH3) and δ 6.37 (1H, d, J = 2, H3) with δ 165.73 (C4), δ 2.66 (3H, s, H8) with δ 203.46 (C7), as well as δ 6.37 (1H, d, J = 2, H3) and δ 6.12 (1H, d, J = 2, H5) with δ 105.71 (C1) in HMBC spectrum of 7 (Fig. 2). The EIMS spectrum of 7 also displayed an ion peak at m/z 182, representing the 4-O-methyl-phloroacetophenone (C9H2O4) fragment as aglycon of 7. The anomeric carbon resonances of glucosyl moieties were assigned at δ 100.8 (C1′), 103.31 (C1″), and 103.61(C1″′) from their direct correlations (1 J HC) with δ 5.04 (1H, d, J = 7.3, H1′), 4.20 (1H, d, J = 7.7, H1″), 4.18 (1H, d, J = 7.7, H1″′), respectively, observed in HSQC spectrum of 7. Finally, detection of 3 J H,C correlations of H1″ (δ 4.20) with C6′ (δ 68.70), and H1″′ (δ 4.18) with C6″ (δ 69.18) in HMBC spectrum of 7 resulted in elucidation of 1-{2-[β-d-glucopyranosyl-(1 → 6)-β-d-glucopyranosyl-(1 → 6)-β-d-glucopyranosyloxy]-6-hydroxy-4-methoxyphenyl}-1-ethanone structure for compound 7, a new compound which was named azeroside B (Fig. 1).

Selected HMBC correlations (C → H) for Azeroside B (7)
All of the five isolated phloroacetophenone glycosides (1–4, 7) exhibited a moderate free radical-scavenging activity in DPPH assay, which reports in this study for the first time (Table 1).
7,8-dihydroferulic acid-4-O-β-d-glucopyranoside (5) is a rare phenolic compound with anti-inflammatory activity reported from Zanthoxylum bungeanum (Kraus and Spiteller, 1997) and Picea glauca (Tezuka et al., 2001). This compound was differentiated from 6 by the resonance patterns of H7 and H8 in their 1H-NMR spectra. The resonances of these two protons were assigned at δ 7.72 (1H, d, J = 16.2 Hz, H7) and 6.32 (1H, d, J = 16.2 Hz, H8) for 6, whereas they were revealed as two multiplets at lower chemical shifts at δ 2.75 (2H, m, H7) and 2.45 (2H, m, H8) in the 1H-NMR spectrum of 5.
6,7,8-trihydroxycoumarin (8) is another isolated rare compound previously reported from Pelargonium sidoides (Nykolov et al., 1993) and Fraxinus ornus (Kayser and Kolodziej, 1995). This compound has also been isolated in glycoside form (6,7,8-trihydroxycoumarin-O-rhamnopyranoside) from Sarcandra glabra (Li et al., 2011).
Compounds 9–11 are classified as caffeoylquinic acid derivatives. These natural polyphenolic compounds are widely distributed in plant families and have been considered for their various biological effects such as antioxidant, hepatoprotective, hypocholesterolemic, anti-carcinogenic, anti-inflammatory, and anti-PAF activity (Schutz et al., 2004; Zhao et al., 2006).
Isolated caffeoylquinic acid derivatives (9–11), along with 6,7,8-trihydroxycoumarin (8) exhibited potent free radical-scavenging activity in DPPH assay (IC50 values of 1.8–2.7 μg ml−1), about nine to ten times stronger than BHT (IC50 = 19.5 ± 0.8 μg ml−1), a synthetic commercial antioxidant (Table 1). Considering to the well-recognized role of free radicals and reactive oxygen species (ROS) in pathogenesis of many diseases such as cancers, atherosclerosis, rheumatoid arthritis, and neurodegenerative diseases, natural antioxidants (either in diet or in supplement form) have recently received special attention for their potential role in the prevention of such diseases (Valko et al., 2007). In food industries, natural antioxidants could also be appropriate substitutes for synthetic antioxidants such as BHT and butylated hydroxyanisole (BHA), which have been questioned for their safety (Barlow, 1990).
Chromatographic and spectroscopic data
1-{2-[β-d-glucopyranosyl-(1 → 6)-β-d-glucopyranosyloxy]-4,6-dihydroxyphenyl}-1-ethanone; 2-O-[β-d-glucopyranosyl-(1″ → 6′)-β-d-glucopyranosyl]-phloroacetophenone; azeroside A (1)
White solid; mp 209–210 °C; Rf: 0.40 (CHCl3-MeOH, 8:2); [α] 25D −62.5 (c 0.002, MeOH); UV (MeOH) λ max (log ε): 205 (0.75), 282.5 (0.43) nm; IR (KBr) ν max: 3,473, 3,406, 2,926, 1,626, 1,596, 1,428, 1,398, 1,290–1,029, 929, 594 cm−1; 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): 13.78 (1H, s, OH6), 8.38 (1H, s, OH4), 6.22 (1H, d, J = 2, H3), 5.87 (1H, d, J = 2, H5), 4.92 (1H, d, J = 7.3, H1′), 4.25 (1H, d, J = 7.1, H1″), 3–4 (12H, m, H2′–6′,2″–6″), 2.64 (3H, s, H8); 13C-NMR (DMSO-d 6, δ/ppm): 202.44 (C7), 165.82 (C4), 165.82 (C6), 160.96 (C2), 104.35 (C1), 103.26 (C1″), 100.64 (C1′), 96.93 (C5), 94.71 (C3), 76.83 (C3″), 76.61 (C3′), 76.36 (C5″), 75.94 (C5′), 73.59 (C2″), 73.07 (C2′), 69.98 (C4″), 69.30 (C4′), 68.03 (C6′), 60.87 (C6″), 32.84 (C8); EIMS, 40 eV, m/z (%): 493 [M + H]+ (2), 168 [Aglycon] (40), 153 [Aglycon -CH3] (100); Anal. Calcd. for C20H28O14: C, 48.78; H, 5.73; O, 45.49. Found: C, 47.13; H, 5.48; O, 44.81.
2-O-[β-d-glucopyranosyl-(1″ → 6′)-β-d-glucopyranosyl]-4-O-methyl-phloroacetophenone; echisoside (2)
White amorphous solid; Rf: 0.45 (CHCl3-MeOH, 8:2); 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): 13.67 (1H, s, OH6), 6.34 (1H, d, J = 2, H3), 6.12 (1H, d, J = 2, H5), 5.04 (1H, d, J = 7.3, H1′), 3.99 (1H, d, J = 7.0, H1″), 3.81 (3H, s, OCH3), 2.90–3.75 (12H, m, H2′–6′,2″-6″), 2.66 (3H, s, H8); 13C-NMR (DMSO-d 6, δ/ppm): 203.50 (C7), 165.72 (C4), 165.53 (C6), 160.54 (C2), 105.82 (C1), 103.65 (C1″), 100.45 (C1′), 95.05 (C5), 93.50 (C3), 76.90 (C3″), 76.72 (C3′), 76.63 (C5″), 75.55 (C5′), 73.53 (C2″), 73.11 (C2′), 70.06 (C4″), 69.66 (C4′), 68.89 (C6′), 61.03 (C6″), 55.72 (OCH3), 33.13 (C8) (Bukreeva and Pimenov, 1991).
2-O-β-d-glucopyranosyl-4-O-methyl-phloroacetophenone; pleoside (3)
White amorphous solid; Rf: 0.85 (CHCl3-MeOH, 8:2); 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): 13.72 (1H, s, OH6), 6.28 (1H, d, J = 2, H3), 6.13 (1H, d, J = 2, H5), 5.00 (1H, d, J = 7.4, H1′), 3.80 (3H, s, OCH3), 3.10–3.75 (6H, m, H2′–6′), 2.66 (3H, s, H8); 13C-NMR (DMSO-d 6, δ/ppm): 203.47 (C7), 165.60 (C4), 165.53 (C6), 160.59 (C2), 106.12 (C1), 100.64 (C1′), 94.96 (C5), 93.43 (C3), 77.26 (C3′), 76.70 (C5′), 73.09 (C2′), 69.64 (C4′), 60.62 (C6′), 55.59 (OCH3), 33.09 (C8) (Chevalley et al., 2001).
2-O-[α-d-glucopyranosyl-(1″ → 6′)-β-d-glucopyranosyl]-4-O-methyl-phloroacetophenone; hyrcanoside (4)
White solid; Rf: 0.65 (CHCl3-MeOH, 8:2); 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): 6.27 (1H, br s, H3), 6.13 (1H, br s, H5), 5.00 (1H, d, J = 7.1, H1′), 4.81 (1H, d, J = 2.8, H1″), 3.80 (3H, s, OCH3), 3–4 (12H, m, H2′–6′,2″-6″), 2.66 (3H, s, H8); 13C-NMR (DMSO-d 6, δ/ppm): 203.46 (C7), 165.61 (C4), 165.47 (C6), 160.56 (C2), 109.23 (C1″), 105.83 (C1), 100.60 (C1′), 95.08 (C5), 93.36 (C3), 78.69 (C3″), 76.59 (C3′), 75.88 (C5′), 75.53 (C5″), 73.28 (C2″), 73.01 (C2′), 69.83 (C4″), 69.83 (C4′), 67.65 (C6′), 63.13 (C6″), 55.63 (OCH3), 33.06 (C8) (Nurmukhamedova and Nikonov, 1976).
7,8-dihydroferulic acid-4-O-β-d-glucopyranoside (5)
White crystals; Rf: 0.70 (CHCl3-MeOH, 8:2); 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): 7.04 (1H, d, J = 8.5, H5), 6.07 (1H, d, J = 2.5, H2), 6.49 (1H, dd, J = 8.5, 2.5, H6), 4.80 (1H, d, J = 7.3, H1′), 3.70 (3H, s, OCH3), 3.0–3.6 (6H, m, H2′-6′), 2.75 (2H, m, H7), 2.45 (2H, m, H8); 13C-NMR (DMSO-d 6, δ/ppm): 174.64 (C9), 158.70 (C4), 156.14 (C3), 129.82 (C5), 121.70 (C1), 106.53 (C2), 101.54 (C6), 100.90 (C1′), 77.12 (C3′), 76.73 (C5′), 73.34 (C2′), 69.88 (C4′), 60.77 (C6′), 54.96 (OCH3), 34.33 (C7), 24.76 (C8) (Kraus and Spiteller, 1997; Tezuka et al., 2001).
Ferulic acid-4-O-β-d-glucopyranoside; lavandoside (6)
White crystals; Rf: 0.70 (CHCl3-MeOH, 8:2); 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): 7.72 (1H, d, J = 16.2, H7), 7.53 (1H, d, J = 8.7, H5), 6.68 (1H, d, J = 2.4, H2), 6.53 (1H, dd, J = 8.7, 2.4, H6), 6.32 (1H, d, J = 16.2, H8), 4.92 (1H, d, J = 7.3, H1′), 3.70 (3H, s, OCH3), 3.0–3.6 (6H, m, H2′-6′); 13C-NMR (DMSO-d 6, δ/ppm): 168.65 (C9), 161.97 (C3), 161.97 (C4), 156.99 (C7), 138.25 (C1), 129.21 (C6), 116.16 (C8), 107.99 (C5), 77.21 (C3′), 76.83 (C5′), 73.27 (C2′), 69.77 (C4′), 60.67 (C6′), 55.31 (OCH3) (Kurkin et al., 2008).
1-{2-[β-d-glucopyranosyl-(1 → 6)-β-d-glucopyranosyl-(1 → 6)-β-d-glucopyranosyloxy]-6-hydroxy-4-methoxyphenyl}-1-ethanone; 2-O-[β-d-glucopyranosyl-(1″ → 6′)-β-d-glucopyranosyl-(1″′ → 6″)-β-d-glucopyranosyl]-4-O-methyl-phloroacetophenone; azeroside B (7)
White solid; mp 156–157 °C; Rf: 0.20 (CHCl3-MeOH, 8:2); [α] 25D −42.5 (c 0.002, MeOH); UV (MeOH) λmax (log ε): 223.1 (0.5), 282.5 (0.6) nm; IR (KBr) ν max: 3,483, 3,437, 2,926, 1,635, 1,590, 1,401, 1,295-1,082, 987, 853 cm−1; 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): 13.69 (1H, s, OH6), 6.37 (1H, d, J = 2, H3), 6.12 (1H, d, J = 2, H5), 5.04 (1H, d, J = 7.3, H1′), 4.20 (1H, d, J = 7.7, H1″), 4.18 (1H, d, J = 7.7, H1″′), 3.81 (3H, s, OCH3), 2.9–4 (18H, m, H2′–6′,2″–6″,2″′–6″′), 2.66 (3H, s, H8); 13C-NMR (DMSO-d 6, δ/ppm): 203.46 (C7), 165.73 (C4), 165.52 (C6), 160.47 (C2), 105.71 (C1), 103.61 (C1″′), 103.31 (C1″), 100.38 (C1′), 94.89 (C5), 93.64 (C3), 67–77(C2′–5′, 2″–5″, 2″′–5″′), 69.18 (C6″), 68.70 (C6′), 60.92 (C6″′), 55.67 (OCH3), 33.08 (C8); EIMS, 40 eV, m/z (%): 182 [Aglycon] (35), 167 [Aglycon -CH3] (100); Anal. Calcd. for C27H40O19: C, 48.50; H, 6.03; O, 45.46. Found: C, 47.24; H, 6.87; O, 44.76.
6,7,8-trihydroxycoumarin (8)
Brown solid; Rf: 0.90 (CHCl3-MeOH, 8:2); 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): 7.47 (1H, d, J = 8.3, H4), 6.05 (1H, d, J = 8.3, H3), 6.02 (1H, s, H5); 13C-NMR (DMSO-d 6, δ/ppm): 172.76 (C2), 166.81 (C7), 164.79 (C9), 161.25 (C6), 161.25 (C8), 131.47 (C4), 112.54 (C10), 104.87 (C3), 102.11 (C5) (Nykolov et al., 1993; Kayser and Kolodziej, 1995).
5-O-caffeoylquinic acid; chlorogenic acid (9)
Yellow amorphous solid; Rf: 0.55 (CHCl3-MeOH, 8:2); 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): 7.45 (1H, d, J = 15.8, H7′), 7.05 (1H, d, J = 2.0, H2′), 6.98 (1H, dd, J = 8.3, 2.0, H6′), 6.76 (1H, d, J = 8.3, H5′), 6.26 (1H, d, J = 15.8, H7′), 5.16 (1H, m, H5), 3.93 (1H, m, H3), 3.48 (1H, dd, J = 9.8, 2.7, H4), 1.6–2.0 (4H, m, H2,6); 13C-NMR (DMSO-d 6, δ/ppm): 176.29 (C7), 166.36 (C9′), 148.60 (C4′), 145.76 (C3′), 144.65 (C7′), 125.39 (C1′), 121.21 (C6′), 115.79 (C5′), 114.65 (C2′), 114.48 (C8′), 75.18 (C1), 73.34 (C4), 71.63 (C3), 71.48 (C5), 38.10 (C2,6) (Chan et al., 2009).
4,5-Di-O-caffeoylquinic acid (10)
Yellow amorphous solid; Rf: 0.75 (CHCl3-MeOH, 8:2); 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): δ 7.48 (1H, d, J = 16.0, H7″), 7.43 (1H, d, J = 16.0, H7′), 7.02 (1H, d, J = 2.0, H2′), 7.03 (1H, d, J = 2.0, H2″), 6.97 (1H, dd, J = 8.0, 2.0, H6′), 6.95 (1H, dd, J = 8.0, 2.0, H6″), 6.75 (1H, d, J = 8.1, H5′), 6.74 (1H, d, J = 8.1, H5″), 6.25 (1H, d, J = 16.0, H8′), 6.16 (1H, d, J = 16.0, H8″), 5.36 (1H, m, H5), 4.97 (1H, dd, J = 7.7, 2.8, H4), 4.19 (1H, m, H3), 1.8–2.2 (4H, m, H2,6); 13C-NMR (DMSO-d 6, δ/ppm): 174.87 (C7), 166.03 (C9″), 165.61 (C9′), 148.44 (C4′,4″), 145.53 (C7′,7″), 145.50 (C3′,3″), 125.37 (C1′,1″), 121.54 (C″), 121.45 (C6′), 115.75 (C5″), 115.69 (C5′), 114.65 (C2″), 114.62 (C2′), 113.77 (C8″), 113.55 (C8′), 73.66 (C1), 73.46 (C4), 67.64 (C3), 66.50 (C5), 37.44 (C2), 37.40 (C6) (Basnet et al., 1996).
1,5-Di-O-caffeoylquinic acid; cynarin (11)
Yellow amorphous solid; Rf: 0.70 (CHCl3-MeOH, 8:2); 1H-NMR (DMSO-d 6, δ/ppm, J/Hz): δ 7.48 (1H, d, J = 16.0, H7′), 7.45 (1H, d, J = 16.0, H7″), 7.07 (1H, d, J = 2.0, H2′), 7.04 (1H, d, J = 2.0, H8″), 6.99 (2H, dd, J = 8.1, 2.0, H6′,6″), 6.78 (1H, d, J = 8.0, H5′), 6.77 (1H, d, J = 8.0, H5″), 6.22 (2H, d, J = 16.0, H8′), 6.20 (2H, d, J = 16.0, H8″), 5.23 (1H, m, H5), 4.06 (1H, m, H3), 3.60 (1H, dd, J = 8.1, 2.8, H4), 2.0–2.4 (4H, m, H2,6); 13C-NMR (DMSO-d 6, δ/ppm): 173.76 (C7), 166.38 (C9′), 165.20 (C9″), 148.76 (C4′), 148.12 (C4″), 145.87 (C3′), 145.83(C3″), 144.87 (C7′), 143.74 (C7″), 125.74 (C1″), 125.37 (C1′), 121.33 (C6′), 120.31 (C6″), 115.96 (C8″), 115.96 (C5′), 115.84 (C5″), 114.84 (C2′), 114.81 (C2″), 114.33 (C8′), 82.06 (C1), 72.35 (C4), 70.41 (C5), 68.83 (C3), 37.82 (C6), 34.86 (C2) (Carnat et al., 2000).
GC and GC–MS analyses of the essential oil obtained from the roots of D. glabrum resulted in identification of twenty-six compounds, representing 82.8 % of the total oil. The results showed that the oil was dominated by the presence of oxygenated non-terpenes (37.9 %), of which myristicin (14.1 %) and elemicin (11.7 %) were main compounds (Table 2).
Myristicin and elemicin have been identified as responsible for hallucinogenic effects of Myristica fragrans (nutmeg) (Braun and Kalbhen, 1973). Furthermore, hepatoprotective (Srivastava et al., 2001), anti-inflammatory (Morita et al., 2003), and insecticidal (Lee and Park, 2011) properties of myristicin, and antifungal (Rossi et al., 2007) and antibacterial (Tavares et al., 2008) activities of elemicin have previously been shown during biological investigations.
Our study reports these two bioactive phenylpropanoid derivatives as the main compounds of the essential oil of D. glabrum roots, whereas a previous study on the essential oil of this species roots collected from the Jolfa region (northwest of Iran) has reported δ-cadinene (12.8 %) and β-bisabolene (7.5 %) as its main compounds, without the detection of any myristicin and elemicin in its chemical composition (Asnaashari et al., 2011). Myristicin and elemicin, however, have been identified in high levels in essential oils of the roots of Ferula species, the genus classified in the same tribe (Scandiaceae) as Dorema genus (Kurzyna-Młynik et al., 2008; Sahebkar and Iranshahi, 2011).
In DPPH assay, the oil showed very weak free radical-scavenging activity (IC50 = 1,830 ± 23.1 μg ml−1) which seems to be related to the absence or low concentrations of antioxidant principles such as oxygenated mono/sesquiterpenes and phenolic compounds in its composition.
Conclusion
The results of our study introduce D. glabrum as a source of natural phenolic antioxidants, specially phloroacetophenone glycosides and caffeoylquinic acid derivatives.
This report also emphasizes the necessity of conservation of D. glabrum, as a valuable genetic source of potentially active natural products and highlights it as an appropriate candidate for further biological and pharmacological studies.
References
Adams RP (2007) Identification of essential oil components by gas chromatography/mass spectrometry. Allured Publishing Corporation, Carol Stream
Amanzadeh Y (2002) Ammoniacum gum. In: Committee Editorial (ed) Iranian Herbal Pharmacopoeia, vol 2. Ministry of Health and Medical Education Publications, Tehran, pp 766–771
Amirkhiz MB, Rashtchizadeh N, Nazemieh H, Abdolalizadeh J, Mohammadnejad L, Baradaran B (2013) Cytotoxic effects of alcoholic extract of Dorema glabrum seed on cancerous cells viability. Adv Pharm Bull 3:403–408
Asnaashari S, Dadizadeh E, Talebpour AH, Eskandani M, Nazemiyeh H (2011) Free Radical Scavenging Potential and Essential Oil Composition of the Dorema glabrum Fisch. CA Mey Roots from Iran. BioImpacts 1:241–244
Barlow SM (1990) Toxicological aspects of antioxidants used as food additives. In: Hudson BJF (ed) Food antioxidants. Elsevier, Amasterdam, pp 253–307
Basnet P, Matsushige K, Hase K, Kadota S, Namba T (1996) Four di-O-caffeoyl quinic acid derivatives from propolis, Potent hepatoprotective activity in experimental liver injury models. Biol Pharm Bull 19:1479–1484
Braun U, Kalbhen D (1973) Evidence for the biogenic formation of amphetamine derivatives from components of nutmeg. Pharmacology 9:312–316
Bukreeva T, Pimenov M (1991) 2, 6-Dihydroxy-4-methoxyacetophenone 2-O-β-D-gentiobioside from the roots of Dorema aitchisonii. Chem Nat Com 27:638–639
Carnat A, Heitz A, Fraisse D, Carnat AP, Lamaison JL (2000) Major dicaffeoylquinic acids from Artemisia vulgaris. Fitoterapia 71:587–589
Chan E, Lim Y, Ling S, Tan S, Lim K, Khoo M (2009) Caffeoylquinic acids from leaves of Etlingera species (Zingiberaceae). LWT-Food Sci Technol 42:1026–1030
Chevalley I, Marston A, Hostettmann K (2001) Liquid chromatography-Electrospray mass spectrometry for detection and isolation of an antifungal acetophenone from Ribes rubrum (Saxifragaceae). Chromatographia 54:274–277
Dehghan G, Fatholahi G, Sheikhzadeh N, Ahmadiasl N (2009) Hypocholesteremic and antioxidant effects of Dorema glabrum extract in rats fed high cholesterol diet. J Iranian Chem Soc 6:115–143
Delazar A, Delnavazi MR, Yassa N, Parkhideh S, Delazar N, Nahar L, Sarker SD (2012) Essential oil composition and isolation of free radical-scavenging phenolic glycosides from the aerial parts of Ajuga chamaepitys growing in Iran. Rev Bras Farmacogn 22:399--305
Ghahreman A (2002) Cormophytes of Iran (Plant systematics), vol 2. University Publications, Tehran, p 670
Ibadullayeva S, Movsumova N, Gasymov H, Mamedli T (2011) Protection of some rare and endangered vegetable plants in the flora of the Nakhichevan AR. Inter J Biodivers Conserv 3:224–229
Iranshahi M, Shaki F, Mashlab A, Porzel A, Wessjohann LA (2007) Kopetdaghins AE, sesquiterpene derivatives from the aerial parts and the roots of Dorema kopetdaghense. J Nat Prod 70:1240–1243
Irvani N, Solouki M, Omidi M, Saidi A, Zare A (2012) Seed germination and dormancy breaking in Dorema ammoniacum D., an endangered medicinal plant. Trakia J Sci 10:9–15
Kayser O, Kolodziej H (1995) Highly oxygenated coumarins from Pelargonium sidoides. Phytochemistry 39:1181–1185
Kraus C, Spiteller G (1997) Comparison of phenolic compounds from galls and shoots of Picea glauca. Phytochemistry 44:59–67
Kurkin V, Lamrini M, Klochkov S (2008) Lavandoside from Lavandula spica flowers. Chem Nat Com 44:169–170
Kurzyna-Młynik R, Oskolski AA, Downie SR, Kopacz R, Wojewodzka A, Spalik K (2008) Phylogenetic position of the genus Ferula (Apiaceae) and its placement in tribe Scandiceae as inferred from nrDNA ITS sequence variation. Plant Syst Evol 274:47–66
Lee JY, Park W (2011) Anti-inflammatory effect of myristicin on RAW 264.7 macrophages stimulated with polyinosinic-polycytidylic acid. Molecules 16:7132–7142
Li X, Zhang Y, Zeng X, Yang L, Deng Y (2011) Chemical profiling of bioactive constituents in Sarcandra glabra and its preparations using ultra-high-pressure liquid chromatography coupled with LTQ Orbitrap mass spectrometry. Rapid Commun Mass Spectrom 25:2439–2447
Mirbabayev NF, Gasanov GG, Knight DW (1993) Plants of the Republic of Azerbaijan with potential medicinal applications. Pharm Biol 31:47–54
Moradi-Afrapoli F, Asghari B, Saeidnia S, Ajani Y, Mirjani M, Malmir M, Bazaz RD, Hadjiakhoondi A, Salehi P, Hamburger M (2012) In vitro α-glucosidase inhibitory activity of phenolic constituents from aerial parts of Polygonum hyrcanicum. DARU J Pharm Sci 20:37
Morikawa T, Imura K, Miyake S, Ninomiya K, Matsuda H, Yamashita C, Muraoka O, Hayakawa T, Yoshikawa M (2012) Promoting the effect of chemical constituents from the flowers of Poacynum hendersonii on adipogenesis in 3T3-L1 cells. J Nat Med 66:39–48
Morita T, Jinno K, Kawagishi H, Arimoto Y, Suganuma H, Inakuma T, Sugiyama K (2003) Hepatoprotective effect of myristicin from nutmeg (Myristica fragrans) on lipopolysaccharide/d-galactosamine-induced liver injury. J Agr Food Chem 51:1560–1565
Mozaffarian V (2007) Flora of Iran, No.54: Umbelliferae. Publication of Research Institute of Forests and Rangelands, Tehran, pp 368–374
Nurmukhamedova M, Nikonov G (1976) Glycosides of Dorema hyrcanum. Chem Nat Com 12:92–93
Nykolov N, Iossifova T, Vassileva E, Kostova I, Stoev G (1993) Reverse-phase high pressure liquid chromatographic analysis of hydroxycoumarins in plant extracts. Quantitative determination of hydroxycoumarins in Fraxinus ornus. Phytochem Analysis 4:86–88
Pimenov M (1988) Monografitcheskaya reviziya roda Dorema D. Don (Umbelliferae). Biull Mosk Ova Ispyt Prir (Biol) 93:76–90
Rechinger K (1987) Dorema. In: Hedge IC, Lamond JM, Rechinger KH (eds) Umbelliferae, Flora Iranica, vol 162. Akademische Druck-und Verlagsanstalt, Graz, pp 379–385
Rossi PG, Bao L, Luciani A, Panighi J, Desjobert JM, Costa J, Casanova J, Bolla JM, Berti L (2007) (E)-methylisoeugenol and elemicin: antibacterial components of Daucus carota L. essential oil against Campylobacter jejuni. J Agr Food Chem 55:7332–7336
Sahebkar A, Iranshahi M (2011) Volatile constituents of the genus Ferula (Apiaceae): A review. J Ess Oil Bearing Plants 14:504–531
Schutz K, Kammerer D, Carle R, Schieber A (2004) Identification and quantification of caffeoylquinic acids and flavonoids from artichoke (Cynara scolymus L.) heads, juice, and pomace by HPLC-DAD-ESI/MS. J Agr Food Chem 52:4090–4096
Sidana J, Neeradi D, Choudhary A, Singh S, Foley WJ, Singh IP (2013) Antileishmanial polyphenols from Corymbia maculata. J Chem Sci 125:765–775
Singh IP, Bharate SB (2006) Phloroglucinol compounds of natural origin. Nat Pro Rep 23:558–591
Srivastava S, Gupta M, Prajapati V, Tripathi A, Kumar S (2001) Insecticidal activity of myristicin from Piper mullesua. Pharm Biol 39:226–229
Suksamrarn A, Eiamong S, Piyachaturawat P, Byrne LT (1997) A phloracetophenone glucoside with choleretic activity from Curcuma comosa. Phytochemistry 45:103–105
Tavares AC, Goncalves MJ, Cavaleiro C, Cruz MT, Lopes MC, Canhoto J, Salgueiro LR (2008) Essential oil of Daucus carota subsp. halophilus: Composition, antifungal activity and cytotoxicity. J Ethnopharmacol 119:129–134
Tezuka Y, Irikawa S, Kaneko T, Banskota AH, Nagaoka T, Xiong Q, Hase K, Kadota S (2001) Screening of Chinese herbal drug extracts for inhibitory activity on nitric oxide production and identification of an active compound of Zanthoxylum bungeanum. J Ethnopharmacol 77:209–217
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39:44–84
Yassa N, Saeidnia S, Pirouzi R, Akbaripour M, Shafiee A (2007) Three phenolic glycosides and immunological properties of Achillea millefolium from Iran, population of Golestan. DARU J Pharm Sci 15:49–52
Zhao Y, Zhao J, Li X, Zhou C, Sun H, Hao X, Xiao P (2006) Advances in caffeoylquinic acid research. China J Chin Mater Med 31:869–874
Acknowledgments
The authors are grateful to Dr. Hassan Sereshti (Department of Chemistry, Tehran University, Tehran, Iran) for EIMS analyses and to the Central Research Laboratories of Shahid-Beheshti University of Medical Sciences (Tehran, Iran) for Elemental analyses. This research was supported by Tehran University of Medical Sciences and Health Services grant (No. 14101).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Delnavazi, MR., Hadjiakhoondi, A., Delazar, A. et al. Azerosides A and B: Two new phloroacetophenone glycosides from the roots of Dorema glabrum Fisch. & C.A. Mey. Med Chem Res 24, 787–796 (2015). https://doi.org/10.1007/s00044-014-1138-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-014-1138-2