
Abstract
Benzimidazole is an interesting heterocyclic compound because it is found in various drugs such as bifeprunox, pimozide, droperidol, etc. The efficacy of substituted benzimidazoles in the treatment of psychoses is well known. Substituted benzimidazole moieties are established pharmacophores in schizophrenia chemotherapy. Benzimidazole derivatives have proven to be of great potential in determining a wide range of activity by binding to various receptors. In addition, piperazinyl moieties linked to benzimidazole enhances the activity at the DA receptors to a greater extent. Various routes have been adopted for the synthesis of derivatives having comparable anti-psychotic activity at various receptors. Dopamine receptors have the potential to mediate activity to varying extent through the binding of ligands, upon it. In this review, binding affinity values of various substituted parent compounds to various dopamine receptors as well as few 5-HT receptors with antipsychotic activity have been tabulated and the synthetic scheme of the same has been shown. This review is summarized to know about various benzimidazolo-piperazinyl ligands that have potential to bind at the dopamine receptors.
Similar content being viewed by others
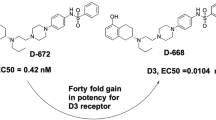
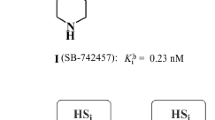
Avoid common mistakes on your manuscript.
Introduction
Benzimidazole is a heterocyclic aromatic organic compound. It is an important pharmacophore and a privileged structure in medicinal chemistry. This compound is bicyclic in nature which consists of the fusion of benzene and imidazole rings. At present, it is a moiety of choice which possesses many pharmacological properties. The most prominent benzimidazole compound in nature is N-ribosyl-dimethylbenzimidazole, which serves as an axial ligand for cobalt in vitamin B12 (Barker et al., 1960; Walia et al.,
2011).
Piperazines are another class of organic heterocyclic compounds that consists of a six-membered ring containing two nitrogen atoms. The piperazines are a broad class of chemical compounds, many with important pharmacological properties, which contain a core piperazine functional group (Merck Index, 1989; Ashford’s Dictionary of Industrial Chemicals, 2011; Chisholm, 1911).
Dopamine is a predominant catecholamine neurotransmitter in the brain, where it controls a variety of functions including locomotor activity, cognition, emotion, positive reinforcement, food intake and endocrine regulation. This catecholamine also plays multiple roles in the periphery as a modulator of cardiovascular function, catecholamine release, hormone secretion vascular tone, renal function, and gastrointestinal motility. Dopaminergic systems have been the focus of research since past 30 years mainly because several pathological conditions, such as Parkinson’s disease, schizophrenia, Tourette syndrome, hyperprolactinemia, substance addiction, affective disorders, and attention-deficit hyperactivity disorders, have been linked to dysregulation of dopaminergic transmission.
Dopamine receptor antagonists have been developed to block hallucinations and delusions that occur in schizophrenia patients, whereas dopamine receptor agonists are effective in alleviating the hypokinesia of Parkinson’s disease. However, desensitization of DA receptors can induce extrapyramidal effects resulting from DA depletion, and high doses of agonists can cause psychoses. The therapies of disorder resulting from dopamine imbalances are thus associated with several side effects. The complementary DNAs of the five distinct DA receptor subtypes (D1–D5) have been infact isolated and characterized by application of gene-cloning procedure. A classical requirement to elucidate the functional role of individual receptor subtypes is identification of selective agonists and antagonists (Missale et al., 1998).
Wide range of antipsychotic drugs have been synthesized until today which have shown their varying affinity toward D1, D2, D3, D4, 5-HT1A, and 5-HT2A receptors. Drugs may or may not have affinity toward all the above receptors, but they have higher selectivity toward a specific receptor where they act as agonist or antagonist. This in turn depends on the nature of the parent molecule and the type of substituent attached to the parent moiety. Dopamine receptor antagonism plays a key role in mediation of antipsychotic effect. Novel antipsychotic derivatives have been synthesized to act at a specific receptor and exert their action against various indications of psychoses, i.e., schizophrenia, persistent delusional disorders, chronic hallucinatory psychoses, bipolar disorder, personality disorder, schizoaffective disorders, and Schizotypy disorders (Kebabian and Calne, 1979; Missale et al., 1998). The review will start with a general study on structure of dopamine receptors. The detailed account of the structures of various benzimidazole piperazinyl compounds and their interaction with enzymes has been summarized. Finally, the synthetic scheme of each compound along with chemistry has been discussed.
Structure of dopamine receptors
Analysis of the primary structure of the cloned DA receptors revealed that they are members of the seven transmembrane (TM) domain G protein-coupled receptor family and share most of their structural characteristics (Fig. 1). Members of this family display considerable amino acid sequence conservation within TM domains (Probst et al., 1992).

Representation of a general dopamine receptor structure (D1-like). The active phosphorylation sites are represented on the –COOH terminal and the third intracellular loop (I3) whereas the active glycosylation sites are represented on –NH2 terminal (Strange 1996). E1–E3:extracellular loops, I2–I3:intracellular loops, 1–7:transmembrane domains
Analysis of DA receptor structure pointed to similarities and dissimilarities between D1-like and D2-like receptors (Civelli et al., 1993; Gingrich and Caron, 1993; Jackson and Westlind-Danielsson, 1994; O’Dowd, 1993). Members of the same family share considerable homology. The D1 and D5 receptors share an 80 % identity in their TM domains. The D2 and D3 receptors have a 75 % identity in their TM domains, and the D2 and D4 receptors share a 53 % identity in the TM domains. Fig. 1 shows structural features of the D1-like receptors, whereas the D2-like receptors are characterized by a shorter COOH-terminal tail and by a bigger third intracellular loop (Missale et al., 1998).
The NH2-terminal stretch has a similar number of amino acids in all the receptor subtypes and carries a variable number of consensus N-glycosylation sites. The D1 and D5 receptors possess two such sites, one in the NH2 terminal and the other one in the second extracellular loop. The D2 receptor has four potential glycosylation sites, the D3 has three, and the D4 possesses only one (Civelli et al., 1993; Gingrich and Caron, 1993; Jackson and Westlind-Danielsson, 1994; O’Dowd, 1993).
The COOH terminal is about seven times longer for D1-like receptors than for the D2-like receptors (Fig. 1). Likewise, as in all G protein-coupled receptors, DA receptors possess two cysteine residues in extracellular loops 2 and 3 (Civelli et al., 1993; Gingrich and Caron, 1993; Jackson and Westlind-Danielsson, 1994; O’Dowd, 1993), which have been suggested to form an intramolecular disulfide bridge to stabilize the receptor structure (Dohlman et al., 1990; Fraser, 1989). The D2-like receptors have a long third intracellular loop, a feature which is common to receptors interacting with inhibitory, Gi proteins to inhibit AC, whereas the D1-like receptors are characterized by a short third loop as in many receptors coupled to stimulatory, Gs protein (Civelli et al., 1993; Gingrich and Caron, 1993; O’Dowd, 1993).
The D1 and D5 receptor’s third intracellular loop and their –COOH terminus are similar in size but divergent in their sequence. In contrast, the small cytoplasmic loops 1 and 2 are highly conserved so that any difference in the biology of these receptors can be probably related to the third cytoplasmic loop and the –COOH terminal tail (Civelli et al., 1993; Gingrich and Caron, 1993; O’Dowd, 1993). The external loop between TM4 and TM5 is considerably different in the two receptor subtypes, being shorter (27 amino acids) in the D1 receptor than in the D5 receptor (41 amino acids). The amino acid sequence of this loop, in addition, is divergent in the D5 and in its rat counterpart D1b (Sunahara et al., 1991; Tiberi et al., 2004).
Site-directed mutagenesis for catecholamine receptors (Kjelsberg et al., 1992; Strader et al., 1989; Strader et al., 1988) and protein modeling with the β2-, α2-, and D2 receptors (Hibert et al., 1992; Hibert et al., 1993; Trumpp-Kallmeyer et al., 1992) suggested that the agonist binding likely occurs within the hydrophobic TM domains (Fig. 1). Highly conserved residues are present in the core of the protein and define a narrow binding pocket that most probably corresponds to the agonist binding site (Hibert et al., 1993). In particular, an aspartate residue in TM3 is most probably involved in binding the amino group of the catecholamine side chain (Strader et al., 1988; Hibert et al., 1993). Two serine residues in TM5 have been shown to be hydrogen bond donors to bind the hydroxyl groups of the catechol moiety for the β2- (Strader et al., 1989), α2- (Wang et al., 1991), D2 (Cox et al., 1992; Mansour et al., 1992), and D1 (Tomic et al., 2004) receptors. A phenylalanine in TM6 is highly conserved in all receptors interacting with catecholamine neurotransmitters and can make a stabilizing orthogonal interaction with the aromatic moiety of the ligad. A highly conserved aspartate residue in TM2 has been shown to play a crucial role in β2-adrenergic (Strader et al., 1988; Hibert et al., 1993 Chung et al., 1988), α2-adrenergic (Wang et al., 1991), and D1 (Tomic et al., 1993) and D2 dopaminergic (Neve et al., 1991) receptor activation and to affect agonist binding (Hibert et al., 1993; Tomic et al., 2004; Sidhu et al., 1992). It has been suggested that the interaction between this aspartate and the agonist is allosteric and can be modulated by Na/or H/ (Hibert et al., 1992; Horstman et al., 1990; O’Dowd et al., 1989). A number of cytoplasmic residues, such as the DRY sequence in the second intracellular loop or the alanine residue in the third intracellular loop of the α-adrenoceptor, also play a role in receptor activation (Kjelsberg et al., 1992; Strader et al., 1988).
Benzimidazole piperazinyl (arylpiperazines) affinity
Example 1
Within the scope of the program aimed at the discovery of new dopaminergic ligands, a series of benzimidazoles were synthesized (Soskic and Joksimovic, 1998). The most active compounds were obtained by connecting benzimidazole ring through a flexible spacer with N-arylpiperazines, structure of one such example that produces ligand in such a way is presented in Chart 1. It was observed that the affinity of the obtained ligands for the binding to the D2 dopamine receptor depends on both the structure of benzimidazole and arylpiperazine part of the molecule and the spacer itself (Roglic et al., a, b). In order to obtain a better structure-dopaminergic activity relationship and to evaluate the influence of the spacer in this type of ligands on their binding affinity, various benzimidazolopiperazinyl derivatives have been synthesized, evaluated, and docking analyses of a series of new compounds has been performed (Klocker et al., 2003a, b); therefore, ligand–receptor interaction can be effected on several levels.
Chemistry (Scheme 1)
The derivatives 1–13 and 20–43 (Table 1) were synthesized as outlined in Scheme 1. Compounds with propylene spacer were prepared according to previously described strategy (Roglic et al., 2001a) starting from 4-(3-chloropropyl)-2-nitroaniline (1). Ethylenoxy compounds were obtained starting from 4-(2-chloroethoxy)-2-nitroaniline (17) as a parent compound (Sukalovic et al., 2005). Briefly, 1-(2-chloroethoxy)-4-nitrobenzene (14) was prepared by alkylating 4-nitrophenol with 1, 2-dichloroethane. A simultaneous reduction and acetylation with zinc dust in acetanhydride/acetic acid mixture afforded acetanilide (15). Nitration of 15 in boiling 20 % nitric acid gave o-nitroacetanilide (16) that was further hydrolyzed to o-aniline 17 with boiling HCl. Compound 17 readily alkylates N-phenyl-piperazine in DMF in the presence of K2CO3 and K i at elevated temperature affording 2-nitro-4-[2-(4-arylpiperazin-1-yl)ethoxy]-anilines (18). Further reduction with Ra–Ni/hydrazine afforded o-phenylenediamines 19. Target benzimidazoles (4–8 and 20–24), benzimidazole- 2-thiones (9–13 and 25–32), benzimidazole-2-ones (33–40) and benzotriazoles (41–43) were prepared as described earlier (Soskic and Joksimovic, 1998).

Synthesis of 5-[3-(4-arylpiperazine-1-yl)propyl]-1H-benzimidazole, 5-[2-(4-arylpiperazine-1-yl)ethoxy]-1H-benzimidazole and their analogs. a Arylpiperazine, K2CO3, DMF. b Ra–Ni, N2H4, EtOH, DCE. c HCOOH, reflux. d CS2, KOH, EtOH. e 1,1′-carbonyldiimidazole and f NaNO2, AcOH. Obtained yields are claimed in parentheses

Example 2: selective dopamine D3 ligands-II
Compound B had high affinity for DA D3 receptors (K i = 1.5 nM) with much weaker affinity for D2 receptors (K i = 406 nM) (Svensson et al., 1994). Thus, arylpiperazine analogs of compound 3 were prepared to explore the potency and selectivity of this novel lead for DA D3 receptors (Svensson et al., 2003).
Chemistry (Scheme 2)
The synthesis of compound 3 and analogs is shown in Scheme 2 (Wright et al., 1994) Step (i) could be performed with 2 equivalents of the piperazine instead of using triethylamine to neutralize the HBr formed. However, the piperazines were often in short supply and the use of triethylamine allowed all of the piperazine to be converted to product.

i Et3N, CH2CI2, reflux, 2 h. ii NaH, DMF, 60 °C, 12 h. iii 1,2-diaminobenzene, NaHSO3, MeOH, reflux, 4 h
The importance of the phenylpiperazine group for DA D3 binding potency was examined via preparation and testing of the analogs shown in Table 2. Replacement of phenylpiperazine with methylpiperazine (compound 1) greatly reduces DA binding. This result clearly separates the SAR of this series from that of the previously described dimeric series. In that series, dialkylamines were the most active; phenylpiperazine was inactive. As the phenyl was important for DA D3 binding, a Topliss analysis of substituents on the phenyl ring was undertaken (the aryl piperazine starting materials are all known).

The 4-chlorophenyl analog 2 had weaker DA binding affinity than the phenyl parent B. The Topliss analysis thus suggested that 4-methoxyphenyl analog 3 although had weaker DA D3 binding, but improved D2 binding. In this case, the Topliss analysis proposed the 3-chlorophenyl analog 4. This compound also had weaker affinity for DA D3 receptors. The Topliss analysis, along with several other analogs not reported here concluded that aromatic substitution on the phenylpiperazine group of B will not enhance DA D3 binding.
An unexpected result was seen with 2-substituted phenyl compounds 5–7. These had high affinity for DA D3 receptors, but were disappointingly non-selective due to strong binding to D2 receptors. The 2-pyridylpiperazine and 2-pyrimidylpiperazine analogs 8 and 9 were both reasonably potent at DA D3 receptors but were again less interesting due to moderate affinity for D2 receptors.
From this study, compound 3 showed optimal affinity and selectivity for DA D3 receptors and was evaluated further.
Example 3
During the last decade, the strategy focused on drug design and synthesis of mixed dopaminergic–serotonergic compounds was followed with a possible atypical neuroleptic potential. One of the synthetic approaches was the design and synthesis of heterocyclic arylpiperazines with a specific structure of heteroaryl group that mimics catechol moiety of the dopamine (benzimidazole, substituted benzimidazoles, benztriazoles, and 1,4-dihydroquinoxaline-2,3-diones). Variations of heterocyclic and aryl groups had produced over 100 new compounds that were screened for their affinities at bovine brain D1, D2, and 5-HT1A receptors by in vitro radioligand binding assays (Dukic et al., 1997a, b; Kostic-Rajacic et al., 1998) The six most active compounds (Table 3) were selected for further pharmacological examinations.

All compounds expressed a higher affinity for the 5-HT1A receptors comparing to clozapine for up to one order of magnitude (Table 3). Only two compounds (2 and 3) showed a higher affinity for 5-HT2A than D2 receptors, and this was more prominent for compound 2 (K i of 5.10and 51.3nM at 5-HT2A and D2 receptors, respectively). Compound 2 expressed a comparable affinity to clozapine for the D2, 5-HT2A, and α1-adrenergic receptors, and somewhat lower affinity for the 5-HT2C receptors. It exhibited a poor affinity at the D1 receptor and no binding potency at the 5-HT3 receptor. On the other hand, compound 2 showed a higher affinity for the 5-HT1A receptor than clozapine (K i values of 90.2 and 415 nM, respectively) and a certain potential to inhibit in vitro synaptosomal 5-HT uptake (IC50 = 260 nM). However, the inhibitory strength of compound 2 for 5-HT reuptake, although higher than that of compounds 1 and 3, seems to be fairly low to take into account its possible antidepressive ability by this mechanism, proposed for some new atypical APDs (Meltzer et al., 2003).
Example 4: chemistry
Drug design strategies toward novel dopamine D2 agonists have been based predominately around the endogenous neurotransmitter dopamine (DA, compound 1). (Kaiser and Jain, 1985; Seyfried and Boettcher, 1990; Wikstrom, 1992). Traditional dopamine agonists have a close resemblance to DA (1), most having the ‘3-OH-phenethylamine’ DA pharmacophore (2) or a bioisosteric surrogate, embedded within their molecular structure, e.g., compound 3 (Wikstrom et al., 1989). Studies from various laboratories have resulted in the emergence of a new generation of dopaminergic agents that no longer rely upon the ‘3-OH-phenethylamine’ framework (Mewshaw et al., 1998a, b, c).
In the Scheme 3, the synthesis and structure–activity relationships of various indole, indolone, benzimidazolone, and benzimidazole derivatives (i.e., 5–7 and 9–13) based on the 3-OH-Nl-phenylpiperazine DA D2 template (4) is disclosed. The initial efforts resulted in the identification of several phenolic D2 agonist prototypes (e.g., 4) (Mewshaw et al., 1998a, b, c) that can be used as templates for the design of bioisosteric analogs. As part of a program to discover compounds that could be potentially useful as antipsychotic drugs, embarkment on exploiting these phenolic prototypes by preparing several heterocyclic bioisosteric analogs is done.

Synthesis and structure-activity relationships of various analogs based on the 3-OH-Nl-phenylpiperazine moiety
Shown in Tables 4 and 5 are the affinities of the target compounds (i.e., 5–7 and 9–13) for the D2-1ike receptors. The affinities of compounds for the D2 receptors in rat striatal membranes were determined for both the agonist state (high affinity state, D2High) and the antagonist state (low affinity state, D2Low). The D2High state was labeled with [3H] quinpirole (in the absence of GTP and sodium) and the D2Low state was labeled with [3H] spiperone (using ketanserin to exclude 5-HT2 receptor binding) in the presence of GTP The ratio K i (D2Low)/K i (D2High) was used as a preliminary and reliable estimate of the compounds’ intrinsic activity as determined by other assays (Lahti et al., 1992; Wasik et al., 1996). The D2 partial agonist (S)-3-PPP [(K i (D2Low)]/K i (D2High) = 33], was used as a benchmark from which a compound’s estimated intrinsic activity was compared. Affinity for the human cloned receptors was determined using membranes from CHO cells labeled with [3H] spiperone.


As shown in Table 4, replacement with the phenol moiety of 4 with the indolone bioisostere (i.e., compound 5) resulted in a tenfold increase in affinity for the D2High receptor and a similar predicted intrinsic activity ratio [(K i (D2Low)/K i (D2High) = 14]. Indolone 5 was observed to have high affinity for all the D2-like receptors and was one of the most potent compounds identified in this study. Though the 5-chloro derivative (6) had unimpressive affinity for the D2High receptor, it was observed to exhibit selectivity for the hD4.4 receptor. Benzimidazolone 7 had similar affinity to its phenol analog (4) with a slight preference for the hD4.4 receptor. Indole, 9 (Table 4) was found to have similar D High2 affinity as its phenol prototype (i.e., compound 4); however, a lower predicted intrinsic activity than 4 was observed [9; (K i (D Low2 )/K i (D High2 ) = 5]. Introduction of either a bromine or chlorine into compound 9 (i.e., 10 and 11) resulted in compounds having selectivity for the hD4.4 receptor. Though benzimidazole 12 had good affinity for the D2-High receptor, introduction of the trifluoromethyl group led to a 19-fold increase in D2-High affinity. In fact, the benzimidazole (13) had similar affinity as its indolone analog (5) for the D2-High receptor, revealing that the indolone and 2-CF3-benzimidazole groups can both serve as surrogate phenol bioisosteres.
Consistent with the predicted low intrinsic activity, in vivo studies showed both 5 and 13 to reduce spontaneous locomotor activity (ED50 = 0.12 mg/kg sc for 13; 5 decreased activity at 0.01, 0.03, 1, and 3 mg/kg sc) and to inhibit apomorphine-induced stereotypy (S) and climbing (C) in mice (ED50s for 13: S = 13.8 mg/kg sc, C = 0.4 mg/kg sc; for 5: S = 1.5 mg/kg sc, C = 2.3 mg/kg sc). Unlike 5, benzimidazole 13 induced catalepsy at 10 mg/kg sc in mice, which correlates with their predicted intrinsic activity ratios (6 vs 14).
Conclusion
It is concluded that among the aryl piperazines, benzimidazolo-piperazines together are effective in binding to the dopamine receptors as well as 5-HT receptors. By varying the substitutions on benzimidazolo-piperazinyl moiety, the compound affinity and preference to various dopamine receptor subtypes are altered and thereby their binding constants are altered. For suitable binding, there is a requirement of a suitable distance between hydroxyl (if present) and a distant nitrogen atom which can be studied through docking analysis. Also, benzimidazolones (indoles and indolones) piperazinyl class of derivatives has been found to possess varying D2 and D4 affinity. Also, the long alkoxy chain separating the benzimidazole and piperazine has given derivatives with increased D3 receptor affinity. There is a wide scope for the synthetic scientists to synthesize more and more compounds with different substitutions using benzimidazole and piperazine as a basic moiety. Thus, investigation and probing of the boundaries of the D2 agonist pharmacophoric criteria is being continued by identifying potential antipsychotic agents that belong to this new generation of dopaminergic agents which in turn could possess optimum antipsychotic activity.
References
Ashford’s Dictionary of Industrial Chemicals (2011), 3rd edn, 7332
Barker HA, Smyth RD, Weissbach H, Toohey JI, Ladd JN, Volcani BE (1960) Isolation and properties of crystalline Cobamide coenzymes containing the Benzimidazole or 5,6-dimethylbenzimidazole. J. Biol. Chem. 235(2):480–488
Chisholm H (ed) (1911) Piperazin. Encyclopedia Britannica (11th edn). Cambridge University Press, Cambridge
Chung FZ, Wang CD, Potter PC, Venter JC, Fraser CM (1988) Site-directed mutagenesis and continuous expression expression of human b adrenergic receptors: identification of a conserved aspartate residue involved in the agonist binding and receptor activation. J. Biol. Chem. 263:4052–4055
Civelli O, Bunzow JR, Grandy DK (1993) Molecular diversity of the dopamine receptors. Annu. Rev. Pharmacol. Toxicol. 32:281–307
Cox BA, Henningsen RA, Spanoyannis A, Neve RL, Neve KA (1992) Contributions of the conserved serine residues to the interactions of ligands with dopamine D2 receptors. J. Neurochem. 59:627–635
Dohlman HG, Caron MG, Deblasi A, Frielle T, Lefkowitz RJ (1990) A role of the extracellular disulfide bonded cysteines in the ligand binding function of the b2 adrenergic receptor. Biochemistry 29:2335–2342
Dukic S, Kostic- Rajacic S, Dragovic D, Soskic V, Joksimovic J (1997a) J Pharm Pharmacol 49:1036
Dukic S, Vujovic M, Soskic V, Joksimovic J (1997b) Arzneimforsch 47:239
Fraser C (1989) Site-directed mutagenesis of beta-adrenergic receptors. J Biol Chem 264:9266–9270
Gingrich JA, Caron MG (1993) Recent advances in the molecular biology of the dopamine receptors. Ann Rev Neurosci 16:32–299
Hibert MF, Trumpp-Kallmeyer S, Bruinvels A, Hoflack J (1992) Three-dimensional models of neurotransmitter G-binding protein-coupled receptors. Mol Pharmacol 40:8–15
Hibert MF, Trumpp-Kallmeyer S, Hoflack J, Bruinvels A (1993) This is not G protein-coupled receptor. Pharmacol Sci 14:7–12
Jackson DM, Westlind-Danielsson A (1994) Dopamine receptors: molecular biology, biochemistry and behavioural aspects. Pharmacol Ther 64:291–369
Kaiser C, Jain T (1985) Pied Res Rev 5:145
Kebabian JW, Calne DB (1979) Multiple receptors for dopamine. Nature 277:93–96
Kjelsberg MA, Cotecchia S, Ostrowski J, Caron MG, Lefkowitz RJ (1992) Constitutive activation of the a1-beta adrenergic receptor by all the amino acid substitutions at a single site. Evidence for a region that constrains receptor activation. J Biol Chem 267:1430–1433
Klocker J, Karpfen A, Wolschann P (2003a) Chem Phys Lett 367:566–575
Klocker J, Karpfen A, Wolschann P (2003b) J Mol Struct (Theochem) 635:141–150
Kostic-Rajacic S, Soskic V, Joksimovic (1998) J Arch Pharm (Weinheim) 331:22
Lahti RA, Figur LM, Peircey MF, Ruppel PL, Evans (1992) D L Mol Pharm 42:432
Mansour A, Meng F, Meador-Woodruff JH, Taylor LP, Civelli O, Akil H (1992) Site-directed mutagenesis of the human dopamine D2 receptor. Eur J Pharmacol 227:205–214
Meltzer HY, Li Z, Kaneda Y, Ichikawa (2003) J Prog Neuropsychopharmacol 27:1159
Merck Index (1989) (11th edn) 7431
Mewshaw RE, Husbands M, Gildersleeve ES, Webb MB, Shi X, Mazandarani H, Cockett M, Ochalski R, Brennan JA, Abou-Gharbia M, Marquis K, McGaughey GB, Coupet J, Andree TH (1998a) New generation dopaminergic agents: discovery of 3-OH-phenoxyethylamine and 3-OH-N1-phenylpiperazine dopaminergic templates. Bioorg Med Chem Lett 8:295
Mewshaw RE, Verwijs A, Shi X, McGaughey GB, Nelson JA, Mazandarani H, Brennan JA, Marquis KL, Coupet J, Andree TH (1998b) New generation dopaminergic agents: heterocyclic bioisosteres that exploit the 3-OH–N1–phenylpiperazine dopaminergic template. Bioorg Med Chem Lett 8:2675–2680
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG (1998) Dopamine receptors: from structure to function. Physiol Rev 78(1):189–225 (Printed in USA)
Neve KA, Cox BA, Henningsen AR, Spanoyannis A, Neve RL (1991) Pivotal role for aspartate-80 in the regulation of dopamine D2 receptor affinity for the drugs and inhibition of the adenylyl cyclase. Mol Pharmacol 39:733–749
O’Dowd BF (1993) Structure of dopamine receptors. J Neurochem 60:804–816
O’Dowd BF, Hnatowich M, Caron MG, Lefkowitz RJ, Bouvier RJ (1989) Palmitoylation of the human b2 adrenergic receptor: mutation of CYS 341 in the carboxy tail leads to an uncoupled, non-palmitoylated form of the receptor. J Biol Chem 264:7564–7569
Probst WC, Snyder LA, Schuster DI, Brosius J, Sealfon (1992) Sequence and alignment of the G-protein coupled receptor superfamily. DNA Cell Biol 11:1–20
Roglic G, Andric D, Kostic-Rajacic S, Dukic S, Soskic V (2001a) Arch Pharm Pharm Med Chem 334:375–380
Roglic G, DuKic-Stefanovic S, Andric D, Kostic-Rajacic S, Soskic V (2001b) Pharmazie 56:803–807
Seyfried CA, Boettcher H (1990) Drugs Future 15:819
Sidhu A, Vachvanichsanong P, Jose PA, Felder RA (1992) Persistent defective coupling of dopamine receptors to Proteins after solubilization from the Kidney proximal tubules of the hypertensive rats. J Clin Invest 89:789–793
Soskic V, Joksimovic J (1998) Curr Med Chem 5:493–512
Strader CD, Sigal IS, Candelore MR, Rouds E, Hill WS, Dixon RAF (1988) Conserved aspartic acid residues 79 and 113 of the b-adrenergic receptor have different roles in receptor function. J Biol Chem 263:10267–10271
Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RAF (1989) Identification of two serine residues involved in agonist activation of the beta-adrenergic receptor. J Biol Chem 264:13478–13572
Strange PG (1996) Dopaminereceptors: Studies and function. Advances in Drug Research 28:313–351
Sukalovic V, Andric Deana, Roglic G, Sladjana Kostic-Rajacic S, Schrattenholz A, Soskic V (2005) Synthesis, dopamine D2 receptor binding studies and the docking analysis of 5-[3-(4-arylpiperazin-1-yl)propyl]-1H-benzimidazole, 5-[2-(4-arylpiperazin-1-yl)ethoxy]-1H-benzimidazole and their analogs. Eur J Med Chem 40:481–493
Sunahara RK, Guan HC, O’Dowd BF, Seeman P, Laurier LG, NG G, George SR, Torchia J, Van tol HHM, Niznik HB (1991) Cloning of the gene for a human dopamine D5 receptor with higher affinity for dopamine than D1. Nature 350:614–619
Svensson TH (2003) Prog Neuropsychopharmacol Biol Psychiatry 27:1145
Svensson K, Carlsson A, Huff RM, Kling-Petersen T, Waters N (1994) Behavioral and neurochemical data suggest functional differences between dopamine D2 and D3 receptors. Eur J Pharmacol 263:235–243
Tiberi M, Jarvie KR, Silvia C, Falardeau P, Gingrich JA, Godinot N, Bertrand L,Yang-feng TL, Fremeau RT, Tomic M, Kundakovic M, Butorovic B, Janac B, Andric D, Roglic G, Ignjatovic D, Kostic-Rajacic S (2004)
Tomic M, Seeman P, George SR, O’Dowd B (1993) Dopamine D1 receptor mutagenesis: role of amino acids in the agonist and antagonist binding. Biochem Biophys Res Commun 191:1020–1027
Tomic M, Kundakovic M, Butorovic B, Janac B, Andric D, Roglic G, Ignjatovic D, Kostic-Rajacic S (2004) Pharmacological evaluation of selected arylpiperazines with atypical antipsychotic potential. Bioorg Med Chem Lett 14:4263–4266
Trumpp-Kallmeyer S, Hoflack J, Bruinvels A, Hibert M (1992) Modeling of the G protein-coupled receptors: application to the dopamine, adrenaline, serotonin, acetylcholine and mammalian opsin receptors. J Med Chem 35:3448–3462
Wang CD, Buck MA, Fraser CM (1991) Site-directed mutagenesis of alpha-2A adrenergic receptors: identification of the amino acids involved in ligand binding and receptor activation by agonists. Mol Pharmacol 40:168–179
Wasik T, Cockett M, Andree TH (1996) Soc Neurosci 22:831 Abstract
Wikstrom H (1992) Prog Med Chem 29:185
Wikstrom H, Sanchez D, Lindberg P, Arvidsson LE, Hacksell U, Johansson A, Nilsson JLG, Hjorth S, Carlsson AJ (1989) Pied Chem 25:925
Walia R, Md Hedaitullah, Naaz SF, Khalid I, Lamba HS (2011) Benzimidazole derivatives—an overview. IJRPC 1(3):565–574
Wright JL, Caprathe BW, Downing DM, Glase SA, Heffner TG, Jaen JC, Johnson SJ, Kesten SR, MacKenzie RG, Meltzer LT, Pugsley TA, Smith SJ, Wise LD, Wustrow DJJ (1994) Binding assays carried out in triplicate at cloned human D2L and D3 receptors transfected into CHO-KI cells versus [3H] spiperone. Med Chem 37:3523
Wright J, Heffner T, Pugsley T, MacKenzie R, Wise L (1995) Discovery of the selective dopamine D3 ligands: II. 2-[4-[3-(4-Aryl-1-piperazinyl)propoxy]phenyl]benzimidazole partial agonists. Bioorg Med Chem Lett 5(21):2547–2550
Acknowledgments
The authors thank Dr. Anwar R. Shaikh and Dr. Prashant Naik, Department of Pharmaceutical Chemistry, M.E.T’s Institute of Pharmacy, Nashik and Dr. Paraag Gide, Principal, M.E.T’s Institute of Pharmacy, Nashik for their excellent technical assistance and for critical proof-reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jain, Z.J., Kankate, R.S., Chaudhari, B.N. et al. Action of benzimidazolo-piperazinyl derivatives on dopamine receptors. Med Chem Res 22, 520–530 (2013). https://doi.org/10.1007/s00044-012-0055-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-012-0055-5