
Abstract
Heteroaromatic derivatives (3a–f) have been synthesized and evaluated for their activity against four cancer cell lines. Among the studied compounds, 1-(7-Chloroquinolin-4-yl)-2-[(1H-pyrrol-2-yl)methylene]hydrazine (3e) exhibited an excellent cytotoxic activity against the referred lines, and especially on melanoma cells (MDAMB-435). In this case, compound 3e is four times more active than the standard substance Doxorubicin. Together with other results from our group, 7-chloro-4-quinolinylhydrazones derived from chloroquine could be considered a relevant finding toward the rational design of new leads for antitumor compounds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the field of drug discovery, the quinoline nucleus, found in many synthetic and natural products, is an important class of heterocyclic compound, since it presents a wide range of pharmacological activities, such as antiviral (Font et al., 1997), antibacterial (Kaminsky and Meltzer, 1968), antifungal (Musiol et al., 2006), antiobesity (Warshakoon et al., 2006), and anti-inflammatory (Sloboda et al., 1991) and antimalarial (Foley and Tilley, 1998) properties. Such a variety is well illustrated by the availability of a large number of drugs containing this heterocyclic class.
Quinoline derivatives are particularly relevant in antimalarial drug research. Quinine, an alkaloid which was originally isolated from the bark of the cinchona tree in Peru, was the first effective treatment for malaria (Foley and Tilley, 1998). This substance played the role of starting point to design new antimalarial drugs, such as, for instance, Chloroquine (Fig. 1, CQ), that is reputed as the drug of choice for malaria, due to its effectiveness, low toxicity, and reduced costs. In view of these characteristics, the therapeutic activity of CQ in other diseases, such as lupus erythematosus, rheumatoid arthritis and amoebic hepatitis (Augustijns et al., 1992), HIV-1/AIDS and chikungunya fever (Savarino et al., 2006), has been widely studied.

7-chloro-4-quinolinylhydrazones derivatives designed by molecular hybridization between hydrazone scaffold (A) existing in bisantrene and 7-chloro-quinoline nucleus (B) found in CQ
Furthermore, as CQ was recently found to present activities against different types of human cancers (Chuandong et al., 2006; Kim et al., 2010), the drug emerged as a potential anticancer agent. Thanks to these properties, CQ could be considered a lead compound to design new antitumor drugs. Based on this hypothesis, our research group has recently studied two series of mono and polysubstituted 7-chloro-4-quinolinylhydrazone derivatives designed by molecular hybridization, which have shown promising cytotoxic activities (Fig. 1).
Therefore, in the present study, we decided to synthesize and evaluate a series of heteroaromatic 7-chloro-4-quinolinylhydrazones against cancer cell lines. Hence, four heterocyclic nuclei were selected (furan, thiophene, pyrrole, and imidazole) based on isosteric replacements: (i) substitution of oxygen atom from the furane ring (3b) by sulfur (3d) or nitrogen (3e); (ii) substitution of –CH=by –N=in pyrrole ring (3e) to provide an imidazole ring (3f). Besides, a nitro group has been introduced on furan (3a) and thiophene (3c) rings, in an attempt to analyze the influence of this group on the biological activity of said series, since nitro compounds have been recently studied as radio sensitizers in the antitumor therapy (Krause et al., 2005).
Materials and methods
Chemistry
Melting points were determined on a Buchi apparatus and are uncorrected. Infrared spectra were recorded in a Thermo Nicolet Nexus 670 spectrometer, as potassium bromide pellets and frequencies are expressed in cm−1. Mass spectra (ESI assay in solution of ammonium chloride) were recorded in Micromass ZQ Waters mass spectrometer. NMR spectra were recorded in a Bruker Avance 500 spectrometer operating at 500.00 MHz (1H), in deuterated dimethylsulfoxide. Chemical shifts are reported in ppm (δ) relative to tetramethylsilane and J-coupling in Hertz (Hz). Proton spectra were typically obtained at room temperature. For TLC plates, coated with silica gel, were run in chloroform/methanol (9:1) mixture and spots were developed in ultraviolet and solution of ninhidrine (0.2% (w/v) in ethanol).
General procedures for synthesis of 7-chloro-4-quinolinylhydrazones derivatives (3a–f)
The 7-chloro-4-quinolinylhydrazones derivatives (3a–f) were obtained by the reaction between 7-chloro-4-hydrazinoquinoline (1.03 mmols) and the appropriate heteroaromatic aldehyde (2a–f) (1.24 mmols) in ethanol (5 ml). After stirring for 12–24 h at room temperature, the resulting mixture was concentrated under reduced pressure. Then, the residue was filtered under vacuum and purified by washing with cold Et2O (3 × 10 ml), leading to the pure derivatives (3a–f) as solids in 78–92% yields.
1-(7-Chloroquinolin-4-yl)-2-[(5-nitro-furan-2-yl)methylene]hydrazine (3a)
Yield: 85%; Mp: 238–240°C; 1H NMR [500 MHz (FIDRES ± 0.15 Hz), DMSO-d 6 ] δ: 11.70 (1H, br, NH), 8.35 (2H, m, H2 and H5), 8.30 (1H; s; N=C–H), 7.81 (2H, d, J = 3,7 Hz, H7′ and H6), 7.55 (1H, br, H8); 7.22–7.26 (2H, m, H3 e H8′). IRλmax (cm−1; KBr pellets): 3,158 (N–H); 1,571 (C=N). MS/ESI: m/z [M − H]+.: 315. Anal. Calcd for C14H9ClN4O3: C 53.09, H 2.86, N 17.69. Found: C 52.93, H 2.78, N 17.74.
1-(7-Chloroquinolin-4-yl)-2-[(5-nitro-thiophen-2-yl)methylene]hydrazine (3c)
Yield: 92%; Mp: 189–190°C; 1H NMR [500 MHz (FIDRES ± 0.15 Hz), DMSO-d 6 ] δ: 11.91 (1H, br, NH), 8.55 (2H, m, H2 and H5), 8.22 (1H; s; N=C–H), 8.08 (1H, d, J = 4.4 Hz, H7′); 7.53 (1H, d, J = 1.9 Hz, H8); 7.48–7.51 (2H, m, H8′ e H6); 6.98 (1H, d, J = 7.4 Hz, H3). IRλMax (cm−1; KBr pellets): 3,182 (NH); 1,585 (C=N). MS/ESI: m/z [M + H]+.: 333. Anal. Calcd for C14H9ClN4O2S: C 50.53, H 2.73, N 16.84. Found: C 50.36, H 2.68, N 16.91.
Biological assays
Cytotoxicity against cancer cell lines
Compounds 3a–f (0.009–5 μg/ml) were tested for their cytotoxic activity against three four cancer cell lines: SF-295 (glioblastoma), HCT-8 (colon), MDA-MB-435 (melanoma), HL60 (leukaemia) (National Cancer Institute, Bethesda, MD). All cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C with 5% CO2. Each compound was dissolved with DMSO, until reaching a concentration of 1 mg/ml. The final concentration of DMSO in the culture medium was kept constant, below 0.1% (v/v). Compounds 3a–f were incubated with the cells for 72 h. The negative control received the same amount of DMSO (0.001% in the highest concentration). The cell viability was determined by reduction of the yellow dye 3-(4,5-dimethyl-2-thiazol)-2,5-diphenyl-2H-tetrazolium bromide (MTT) to a blue formazan product as described by Mosmann (Ahmed et al., 1994).
Cell membrane disruption
The test was performed in 96-well plates using a 2% mouse erythrocyte suspension in 0.85% NaCl containing 10 mM CaCl2 (Sharma and Sharma 2001). The compounds 3a–f were diluted as mentioned above, and tested at 250 μg/ml. After incubation at room temperature for 30 min, centrifugation, and removal of the supernatant, the released hemoglobin was measured by spectrophotometry at 540 nm. DMSO was used as a negative control, and Triton X-100 (1%) as positive control. After incubation at room temperature for 1 h, centrifugation, and removal of the supernatant, the released hemoglobin was measured by spectrophotometry at 540 nm. (EC50 is the calculated effective concentration that induced lysis on 50% that of the Triton X-100).
Results and discussion
All the 7-chloro-4-quinolinylhydrazones derivatives 3a–f were synthesized by our research group (Scheme 1). Compounds 3a–f were obtained through reaction between 7-Chloro-4-hydrazinoquinoline 1 and the appropriate aldehydes 2a–f, as described in “Materials and Methods” section (Table 1). In general, 1H NMR spectra showed the imine proton (N=C–H) as a singlet at 8.81–8.29 ppm. Furthermore, IR spectra presented N–H and N=C stretching vibrations at 3,197–3,247 and 1,612–1,576 cm−1, respectively.

Synthesis of heteroaromatic 7-chloro-4-quinolinylhydrazone derivatives 3a–f
All synthesized compounds 3a–f were tested in vitro against three cancer cells at 25 μg/ml, by using MTT assay (Table 2) (Ahmed et al., 1994), and then, according to their growth inhibition (GI) percentage in, at least, one cell line, they were classified as active (100% GI), moderately active (75% < GI < 100%), or inactive (GI < 50%).
Among all tested compounds, only the 1-(7-Chloroquinolin-4-yl)-2-[(1H-pyrrol-2-yl)methylene]hydrazine (3e) was found to be active against all cancer cell lines, with a GI higher than 90%. Furthermore, derivative 3f was moderately active on MDAMB-435 cells, while all other compounds were inactive against all cancer cell lines at 25 μg/ml.
In view of the above data, the compound 3e, that presented a GI above 90%, was selected for in vitro anticancer activities evaluation against four human cancer cell lines, by using MTT assay (Table 3) (Ahmed et al., 1994). The concentrations that induce 50% inhibition of cell growth (IC50) in μg/ml are presented in Table 3. This compound was not able to disrupt the cell membrane integrity of erythrocytes in mouse model (data not shown) (Sharma and Sharma, 2001).
These results showed that compound 3e exhibited an excellent cytotoxic activity against the four cancer cell lines, mainly on melanoma cells (MDAMB-435). In this case, compound 3e is four times more active than the standard substance Doxorubicin, which could be considered a relevant finding toward the rational design of new leads for antitumor compounds.
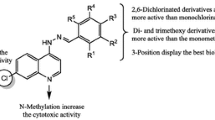
In general, when compared to the monosubstituted and polysubstituted 7-chloro-4-quinolinylhydrazones previously evaluated by our research group (Montenegro et al., 2011), the heteroaromatic derivatives 3a-f showed worst antitumor activities than these other two series, except for 3e. Furthermore, in a previous study (Montenegro et al., 2011), the presence of nitro group was important to trigger the biological activity of monosubstituted quinolinylhydrazones (IC50 ranging from 0.7967 to 1.200 μg/ml). This behavior was already expected, since nitro compounds have been recently studied as radio sensitizers in antitumor therapy, due to the bioreduction capacity of the nitro group, which releases intermediates in the redox process (Krause et al., 2005). However, in contrast with these findings, the presence of nitro group in heteroaromatic series did not lead to improved anticancer activities (please refer to 3a vs. 3b, and 3c vs. 3d, as shown in Table 3).
Conclusion
Six 7-chloro-4-quinolinylhydrazones were synthesized and evaluated against four different types of cancer cell lines. Among them, derivative 1-(7-Chloroquinolin-4-yl)-2-[(1H-pyrrol-2-yl)methylene]hydrazine (3e) could be considered a relevant finding toward the rational design of new leads for antitumor compounds, since it is four times more active than the standard substance Doxorubicin on melanoma cells (MDAMB-435). Further studies for the purpose of obtaining more information about structure–activity relationship, as well as researches to elucidate the molecular mechanisms of cytotoxicity presented by these compounds are still in progress.
References
Ahmed SA, Gogal RM Jr, Walsh JE (1994) A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: An alternative to [3H]thymidine incorporation assay. J Immunol Methods 170:211–224
Augustijns P, Geusens P, Verbeke N (1992) Chloroquine levels in blood during chronic treatment of patients with rheumatoid arthritis. Eur J Clin Pharmacol 42:429–433
Chuandong F, Wang W, Zhao B, Zhang S, Miao J (2006) Chloroquine inhibits cell growth and induces cell death in A549 lung cancer cells. Bioorg Med Chem 14:3218–3222. doi:10.1016/j.bmc.2005.12.035
Fattorusso C, Campiani G, Kukreja G, Persico M, Butini S, Romano MP, Altarelli M, Ros S, Brindisi M, Savini L, Novellino E, Nacci V, Fattorusso E, Parapini S, Basilico N, Taramelli D, Yardley V, Croft S, Borriello M, Gemma S (2008) Design, synthesis, and structure-activity relationship studies of 4-quinolinyl- and 9-acrydinylhydrazones as potent antimalarial agents. J Med Chem 51:1333–1343. doi:10.1021/jm7012375
Ferreira ML, Gonçalves RSB, Cardoso LNF, Kaiser CR, Candéa ALP, Henriques MGO, Lourenço MCS, Bezerra FAFM, Souza MVN (2010) Synthesis and antitubercular activity of heteroaromatic isonicotinoyl and 7-chloro-4-quinolinyl hydrazone derivatives. TheScientificWorldJOURNAL 10:1347–1355. doi:10.1100/tsw.2010.124
Foley M, Tilley L (1998) Quinoline antimalarials: mechanisms of action and resistance and prospects for new agents. Pharmacol Ther 79:55–87. doi:10.1016/S0163-7258(98)00012-6
Font M, Monge A, Ruiz I, Heras B (1997) Structure-activity relationships in quinoline Reissert derivatives with HIV-1 reverse transcriptase inhibitory activity. Drug Des Discov 14:259–272
Kaminsky D, Meltzer RI (1968) Quinolone antibacterial agents: Oxolinic acid and related compounds. J Med Chem 11:160–163. doi:10.1021/jm00307a041
Kim EL, Wüstenberg R, Rübsam A, Schmitz-Salue C, Warnecke G, Bücker EM, Pettkus N, Speidel D, Rohde V, Schulz-Schaeffer W, Deppert W, Giese A (2010) Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro Oncol 12:389–400
Krause W, Jordan A, Scholz R, Jimenez JLM (2005) Iodinated nitroimidazoles as radiosensitizers. Anticancer Res 25:2145–2151
Montenegro RC, Lotufo LV, Moraes MO, Pessoa CO, Rodrigues FAR, Bispo MLF, Cardoso LNF, Kaiser CR, Souza MVN (2011) Synthesis and antitumoral evaluation of 7-chloro-4-quinolinylhydrazones derivatives. Med Chem 7:599–604
Musiol R, Jampilek J, Buchta V, Silva L, Niedbala H, Podeszwa B, Palka A, Majerz-Maniecka K, Oleksyn B, Polanski J (2006) Antifungal properties of new series of quinoline derivatives. Bioorg Med Chem 14:3592–3598. doi:10.1016/j.bmc.2006.01.016
Savarino A, Di Trani L, Donatelli I, Cauda R, Cassone A (2006) New insights into the antiviral effects of chloroquine. Lancet Infect Dis 6:67–69
Sharma P, Sharma JD (2001) In vitro hemolysis of human erythrocytes—by plant extracts with antiplasmodial activity. J Ethnopharmacol 74:239–243
Sloboda AE, Powell D, Poletto JF, Pickett WC, Gibbons JJJr, Bell DH, Oronsky AL, Kerwar SS (1991) Antiinflammatory and antiarthritic properties of a substituted quinoline carboxylic acid: CL 306, 293. J Rheumatol 18:855–860
Warshakoon NC, Sheville J, Bhatt RT, Ji W, Mendez-Andino JL, Meyers KM, Kim N, Wos JA, Mitchell C, Paris JL, Pinney BB, Reizes O, Hu XE (2006) Design and synthesis of substituted quinolines as novel and selective melanin concentrating hormone antagonists as anti-obesity agents. Bioorg Med Chem Lett 16:5207–5211
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Montenegro, R.C., Lotufo, L.V., de Moraes, M.O. et al. 1-(7-Chloroquinolin-4-yl)-2-[(1H-pyrrol-2-yl)methylene]hydrazine: a potent compound against cancer. Med Chem Res 21, 3615–3619 (2012). https://doi.org/10.1007/s00044-011-9894-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-011-9894-8