
Abstract
We do some research about multi-target-directed AChE inhibitors of Tacrine–Nimodipine dihydropyridine. 3D-QSAR models have been built using CoMFA and CoMSIA methods, based on compounds which are very selective and potent AChEIs and show an excellent neuroprotective profile and a moderate Ca2+ channel blockade effect. These studies indicated that the QSAR models were statistically significant and high predictable (receptor-based research CoMFA model, q 2 = 0.686, r 2 = 0.948; CoMSIA model, q 2 = 0.756, r 2 = 0.907 in all of the models no. of components = 6). Consequently, based on these results, our models would offer help to better comprehend the structure–activity relationships existent for this class of compounds and also facilitate the design of novel inhibitors with good chemical property.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a neuro degenerative, irreversible disorder that is clinically characterized by a progressive loss of several cognitive abilities. It is the most common type of dementia in western societies, which has been causing profound economic and social impact as the aging population increases (Guttman et al., 1999). Although the etiology of AD is not well known, there are diverse factors such as amyloid-β (Aβ) deposits, oxidative stress, and low levels of acetylcholine (ACh) that are thought to play significant roles in the disease (Scarpini et al., 2003). The primary approach for treating AD has therefore focused on increasing the levels of acetylcholine in the brain using acetylcholinesterase inhibitors (AChEI) such as tacrine, donepezil, galantamine, and rivastigmine (Lahiri et al., 2004). On the other hand, it is well known that Ca2+ overload is the main factor initiating the processes leading to cell death. Several lines of evidence show that calcium dysfunction involved in the pathogen of AD Selkoe (1989). Since 1,4-dihydropyridines (DHPs) selectively block L-type voltage-dependent Ca2+ channels (VDCCs), we consider the pharmacological study of new multipotent hybrid molecules is meaningful, based on an AChEI and a DHP such as tacrine and nimodipine. A large diversity of multi-target-directed AChE inhibitors of Tacrine–Nimodipine DHP have been synthesized. Some of them show an excellent neuroprotective profile, and a moderate blocking effect of L-type voltage-dependent calcium channels. They represent a new family of molecules with potential therapeutic application for the treatment of Alzheimer’s disease (Marco-Contelles et al., 2006b). In addition to there ability of inhibiting AChE, they can be also modulators or blockers of neuronal voltage-dependent Ca2+ channels (de los Rios et al., 2002). In order to search for ligands with higher affinity and specificity, it will be greatly helpful to establish a reasonable pharmacophore model to guide the structural modification in future synthesis of potent ligand. The use of Comperative Molecular Field Analysis (CoMFA) and Comperative Molecular Similarity Indices Analysis (CoMSIA), two commonly used 3D-QSAR methodologies, were applied to a large data set of AChE inhibitors to gain insights into how steric, electrostatic, hydrophobic, and hydrogen-bonding interactions influence their activity. In CoMFA and CoMSIA studies, the conformation determination is so important that it affects the quality of a model. In most cases, a bound complex is not available, and therefore a computation method has to be deployed to determine conformations of a set of molecules (Thaimattam et al., 2005).
Materials and methods
Data set
In this report, we collected a large data set of these new tacrine–dihydropyridine hybrids that inhibit acetylcholinesterase, calcium entry, and exhibit neuroprotection properties from seven independently published data. These sources are from the same lab (Marco-Contelles et al., 2006a; Marco et al., 2001; Leon et al., 2008; Marco et al., 2004; Leon et al., 2005). 3D-QSAR models have been built using CoMFA and CoMSIA methods, which contain both ligand-based QSAR and receptor-based QSAR. The total set of 72 compounds was randomly divided into training set and test set comprising of 60 and 12 molecules, respectively. The training set was used to construct 3D-QSAR models, and the test set was used for the models validation. To derive the CoMFA and CoMSIA models, all the activity values of the AChE inhibitor reported as IC50 in the literatures were converted to pIC50 (−logIC50). Molecular structures and their pIC50 values were presented in Table 1.

Molecular structures
A reasonable conformation for the receptor is of great significance in providing a realistic pharmacophore for 3D-QSAR model building. Thus, the X-ray crystal structure 1acj was selected from the Protein Data Bank (PDB, http://www.rcsb.org/pdb) because it contains a co-crystallized ligand that is contained in our data set and this ligand is structurally most similar to the other compounds in our inhibitor set. The structures of the investigated ligands were built using SYBYL 7.0. Then, the optimization of the structure using Tripos force field with Gasteiger–Huckel charges and minimization using Conjugated Gradient Method till the root-mean-square deviation (RMSD) of 0.01 kcal/mol achieved were carried out. All the compounds were subjected to minimization until converged to a maximum derivative of 0.01 kcal/mol, and Partial atomic charges were calculated using the Gasteiger–Huckel method.
The obtained structure was used as the template to construct the 3D structures of all the other compounds. The side chains of all the compounds were subjected to a systematic search routine of rotatable bonds in 10° increments from 0° to 359° and rotatable rings in default parameters. Energies of conformations were computed, and the 25 of lowest energy structures were selected. Then, the 25 structures are optimized using the semi-empirical method. The lowest energy structures are used in alignment. The partial atomic charges required to estimate the electrostatic interaction were computed by semi-empirical molecular orbital methods using MOPAC with an AM1 Hamiltonian (keyword: NOMM).
There are chiral carbons in B and C systems (Fig. 1). Take C13 as an example, C13 is docked in binding pocket using AutoDock1.4.5. We let the bond rotate 360°C between the carbon and benzene ring C–C. The generated dlg files divided the docked structures into a number of clusters; the structures with the similar RMSD are in the same cluster. Through the cluster analysis, we found that R conformations account for the majority. In addition, S configuration has a greater steric hindrance with amino acids, such as GLY117, GLY118, GLY119, and TRP84. So the R configuration is better to combine protein, which is in agreement with the published articles (Durdagi et al., 2007).

Chiral carbons in B and C systems (For case of C 13 )
All CoMFA and CoMSIA were performed using the QSAR option of SYBYL7.0. The charges were determined using the Gasteiger–Huckel method. The grid spacing was set at 2 Å, and the region was calculated automatically by the program. In CoMFA studies, a default sp3-carbon atom having a charge of +1 and a van der Waals radius of 1.52 Å was used as a probe to calculate steric (Lennard-Jones 6–12 potential) field and electrostatic (Coulombic potential) field energies with a distance-dependent dielectric at each lattice point. The use of CoMFA and CoMSIA approaches together to provide better ability of visualization and interpretation of the obtained correlations in terms of field contributions (Politi et al., 2009). CoMSIA technique was also performed in our studies to obtain more information to design novel selective drugs. CoMSIA calculates also steric and electrostatic fields, and in addition hydrophobic, hydrogen-bond donor and acceptor fields. The analysis was carried out using a common probe atom with 1 Å VD radius, +1.0 charge, attenuation factor α of 0.3, grid spacing 2 Å. Because of a Gaussian type distance dependence of the physico-chemical properties, singularities were avoided at atomic positions and no arbitrary cutoffs were necessary.
The initial PLS analysis was performed using the ‘‘leave-one-out’’ cross-validation method for all 3D-QSAR analysis (Fig. 2). PLS methodology is used to correlate AChE inhibitor activities with the CoMFA and CoMSIA values. A minimum column filtering value of 2.00 kcal mol/1 was set to improve the signal-to-noise ratio by omitting those grid points whose energy variation was below this threshold (Politi et al., 2009). The predictive value of the models was evaluated first by leave-one-out (LOO) cross-validation. The cross-validated coefficient q 2 was calculated according to the following equation:
where Y pred, Y actu, and Y mean are predicted, actual, and mean values of the target property (pIC50), respectively.
PRESS is the sum of squared deviation between predicted and actual activity values for every molecule in the test set, derived from the LOO method. The optimum number of components (ONC) corresponding to the lowest PRESS value was used for deriving the final PLS regression models. In addition to the q 2 and ONC, the conventional correlation coefficient (r 2), standard error of estimate (SE), and F-ratio (F) were also computed.

The alignment of public skeleton (For case of Group c)
The autodocking studies of the database composed of all 72 compounds were performed with the AUTODOCK4.0 software (Fig. 3). AutoDock 4.0 is based on a Lamarckian genetic algorithm (LGA) method. Basically, this program determines total interaction energies between random pairs of ligands and various selected portions of protein to determine docking poses (Huey et al., 2007; Morris et al., 1998). The Autodock 4.0 Software with the following parameters: AUTODOCK1.4.5 (ADT) is prepared for ligands and receptor, Grid Box of the active site is automatic calculated using PDBBOX script. The autodock search parameters select Genetic Algorithm. The number of GA_Run is 50, the Population Size 50, and the maximum number of Evals is Medium. Other parameters are default values. The generate files are calculated by AUTOGRID 4.0 and AUTODOCK 4.0. The analysis of results is clustered by the Clusterings of the Analyze model.

Alignment of 72 inhibitors using Align Database
Results and discussion
CoMFA and CoMSIA Models
The models generated by CoMFA and CoMSIA techniques are summarized in Tables 2 and 3. The best models were selected based on better values of press, cross-validated q 2, non-cross-validated r 2, and the standard deviations (SD) values, according to the actual situation.
Ligand-based align
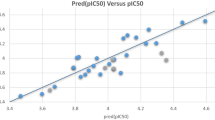
A CoMFA model contained steric and electrostatic fields was obtained, which provided satisfied values of q 2 (0.844) and r 2 (0.961) with number of optimum component (NOC) = 6. The standard error of prediction was 0.599. For CoMSIA analysis, descriptors of five physicochemical field properties were used to correlate with the changes of ligands affinities. The CoMSIA method defines explicit hydrophobic, hydrogen-bond donor and acceptor descriptors in addition to steric and electrostatic fields used in CoMFA. We derived a model with high q 2 value of 0.728 for six components and a conventional r 2 value of 0.882. CoMFA and CoMSIA analysis results are summarized in Table 4. These data indicate that a reliable CoMSIA model was successfully constructed from Fig. 4. As it was shown, most of the molecules were drawn on or near the diagonal line, which indicated that the predicted pIC50 values models are in good agreement with the actual data. So both CoMFA and CoMSIA models are persuasive. The predicted activities of all the 72 compounds for both of CoMFA and CoMSIA models were also listed in Table 4.

Correlation between the actual and predicted activities of 3D-QSAR models for the training set and test set. (a CoMFA model; b CoMSIA model. Filled square: compounds of the training set; empty triangle: compounds of the test set)
Receptor-based analysis
Before docking the derivatives of the total set into the TcAChE allosteric binding site with AUTODOCK 4.0 package, the docking protocol was validated. The tacrine extracted from the crystal structure of complex was docked back into the active binding groove of TcAChE. The RMSD between the predicted conformation and the observed X-ray crystallographic conformation of compound tacrine is 0.2540 Å. Thus, molecular autodocking produced good solution when compared to the existing X-ray structure and suggested that an overall inhibitor alignment could be used in 3D-QSAR analyses. The docked conformation for each molecule was chosen on the basis of the Grid Score which was based on an estimation of van der Waal attractive, van der Waal dispersive, and Columbic electrostatic energies (Cao et al., 2008). The results of CoMFA and CoMSIA analysis of the training set are summarized in Table 5, and the predicted versus experimental activities are displayed in Fig. 5a and b. As it was shown, most of the molecules were drawn on or near the diagonal line, which indicated that the predicted pIC50 values models are in good agreement with the actual data. The CoMFA model was obtained with LOO cross-validation q 2 and non-cross-validated r 2 values of 0.686 and 0.948, respectively. For CoMSIA analysis, we derived a model with high q 2 value of 0.756 for six components and a conventional r 2 value of 0.907.

Correlation between the actual and predicted activities of 3D-QSAR models for the training set and test set. (a CoMFA model; b CoMSIA model. Filled square: compounds of the training set; empty triangle: compounds of the test set)
All of the constructed models possessed good internal and external consistency and showed statistical significance and predictive abilities. So both CoMFA and CoMSIA models are persuasive.
The comparison of the predictive index of QSAR models based on both align database and auto docking alignment methods for the test set is shown in Table 6. The values of r 2 (in Table 6) of QSAR models both align database and flexible autodocking alignment based on same test set are high. And therefore predictabilities of QSAR models are good. The QSAR model based on autodocking alignment is more similar with real situation.
Comparison of the methods for the predictabilities of QSAR models based on both align database and autodocking alignment
In conventional ligand-based QSAR, it is through minimizing the molecules and selecting those with lowest energy that the active conformations are obtained. However, there is no guarantee that the minimum energy conformation is certainly the active conformation in vivo. As a result, the newly designed molecules based on such a model probably fail to bind the receptor even if the QSAR model is in good correlation with the experimental activity. While structure-based conformation determination by autodocking takes into account features of the binding pocket, and thus derived models are more reliable. So the receptor-based QSAR study has the advantages over ligand-based QSAR allowing for much more differences in various ligands’ structure which aimed at the same target.
Graphics interpretation of receptor-based QSAR models
In order to visualize the information of the derived 3D-QSAR, Compounds C2 and C8 are selected as reference structures in Fig. 6a–g.

a CoMFA electrostatic field contour maps (red: disfavored areas of positive potential, blue: favored areas of positive potential); b CoMFA steric field contour maps (green: favored; yellow: disfavored); c CoMSIA electrostatic field contour maps (red: disfavored areas of positive potential, blue: favored areas of positive potential); d CoMSIA steric field contour maps (green: favored; yellow: disfavored); e CoMSIA hydrophobic field contour maps (yellow: favored, white regions: disfavored); f CoMSIA H-bond donor field contour maps (cyan: favored, purple: disfavored); g CoMSIA H-bond acceptor field contour maps (magenta: favored, red: disfavored). Compounds C 2 and C 8 were chosen as the reference molecules in the graphics. The cutoff levels of contour are set at 80:20 (Color figure online)
As a result, the CoMFA electrostatic contour map is shown in Fig. 6a. The red contour near 3-position of the DHP (see the molecule in Table 1, same as below) is observed indicating the increase of electronegative can enhance the activity. According to the molecules C 2 and C 11 (see Table 1), the two molecules have the same structure except in 3-position of the DHP, while the 3-position of the DHP is ester and cyclone, respectively, and the activity (pIC50) increases (C 2 7.28 > C 11 6.27) by the increasing electro-negativity (ester > cyclone). In a similar way, molecule C6 and C12, C9 and C13 which also posses the same situation except in 3-position of the DHP, the value of pIC50 increases (C 6 7.04 > C 12 6.64, C 9 7.35 > C 13 6.66). This phenomenon gives us a further proof that we can improve the IC50 by means of increasing the electro-negativity of 3-position of the DHP.
The CoMFA steric contour plots are shown in Fig. 6b. The yellow contours near the 3-position of the DHP and the right of pyridine ring suggest that any bulky substituent at this site is likely to decrease the activity (i.e., C 6 , C 12 ). The green and yellow polyhedrons observed near the cyclanes of pyridine suggest that the substitution with moderate volume might be preferred at this site (i.e., B 11 B 13 B 16 ). The CoMSIA electrostatic and steric contour plots shown in Fig. 6c and d are consistent to that of CoMFA.
The CoMSIA hydrophobic contour plot is shown in Fig. 6e. The yellow polyhedron near the 4-position benzene ring indicates a hydrophobic group substitution is favored at this site. In addition, a white polyhedron found at benzene ring indicates any hydrophilic group substitution would be preferred at this site. So in the data set most of the substitutions are polar group such as Pyridyl, Pyrrolyl, and Thienyl.
The CoMSIA hydrogen-bond donor contour plot is presented in Fig. 6f. The cyan contour near the group DHP indicates hydrogen-bond donor group might be favored. Originally, the amino-pyridine ring is a good hydrogen-bond donor. However, the effects of steric of 4-substitution make it loss the role of hydrogen-bonding donor, such as benzene ring.
The CoMSIA hydrogen-bond acceptor contour plot is presented in Fig. 6g. The red contour near the group substitution at 3-position indicates hydrogen-bond acceptor group might be favored. For example, C 9 has two hydrogen-bonds, whose activity is the highest among the compounds.
To sum up, near the group substitution at 3-position of DHP, small volume is favored. It would be best to retain carbonyl for it being good hydrogen-bond acceptor. Or it will be good that carbonyl is replaced by the other larger electronegative group. DHP is better than dihydropyrane and pyridine ring, because it is a good hydrogen-bond donor. The strong electron-withdrawing group is not very favorable for the 4-position of the DHP. Hydrophobic and poor electron-withdrawing group are favorable, such as methoxy group, which is a good hydrogen-bond receptor also.
Conclusions
Due to the constraints, a reliable autodocking study was performed on a series of potent multi-target-directed AChE inhibitors susceptible to be potential drugs against Alzheimer’s disease. Rational CoMFA and CoMSIA models were obtained with a high predictive performance for the inhibitor training set and test set. The results of the docking studies allowed us the identification of key features of the AChE inhibitors that are responsible for their high potency. Analysis of model parameters and contour maps revealed the factor that influenced the activity of those compounds for acetylcholinesterase. The binding conformations of 72 derivatives into the acetylcholinesterase binding pocket were predicted by the program AUTODOCK 4.0. Available 3D-QSAR models were also obtained based on the flexible docking alignment. The 3-position of the DHP suggests that we can improve the IC50 by means of increasing the electro-negativity and introducing small volume substituent at this site. Similarly, as to the 4-position of the benzene ring of DHP, hydrophobic is favorable, such as methoxy group, which is a good hydrogen-bond receptor also. DHP ring is the best structure among the dihydropyran and pyridine ring, which is a good hydrogen-bond donor. This conclusion is consistent with the activity (pIC50) of compounds C9, C8, C6, C2, and so on, which have good activity.
The correlation of the results obtained from autodocking and QSAR studies lead to better understanding of the structural requirements for enhanced activity. The robust and predictive results can be used as a guideline to design and predict new and more potent acetylcholinesterase inhibitors.
References
Cao HY, Cao R, Zhang HB, Zhen XF, Gao DB (2008) Non-nucleoside inhibitors of NS5B polymerase binding to allosteric sites: 3D QSAR and molecular docking studies. Curr Med Chem 15:1462–1477. doi:10.2174/092986708786848460
de los Rios C, Marco J, Carreiras MC, Chinchon PM, Garcia A, Villarroya M (2002) Novel tacrine derivatives that block neuronal calcium channels. Bioorg Med Chem 10:2077–2088. doi:10.1016/S0968-0896(01)00378-9
Durdagi S, Kapou A, Kourouli T, Andreou T, Nikas SP et al (2007) J Med Chem 50:2875–2885. doi:10.1021/jm0610705
Guttman R, Altman RD, Nielsen NH (1999) Alzheimer disease. Report of the Council on Scientific Affairs. Arch Fam Med 8:347–353. doi:10.1001/archfami.8.4.347
Huey R, Morris GM, Olson AJ, Goodsell DS (2007) A semiempirical free energy force field with charge-based desolvation. J Comput Chem 28:1145–1152. doi:10.1002/jcc.20634
Lahiri DK, RogersJ T, Greig NH, Sambamurti K (2004) Rationale for the development of cholinesterase inhibitors as anti-Alzheimer agents. Curr Pharm Des 10:3111–3119. doi:10.2174/1381612043383331
Leon R, Marco-Contelles J, Garcia A, Villarroya M (2005) Synthesis, acetylcholinesterase inhibition and neuroprotective activity of new tacrine analogues. Bioorg Med Chem 13:1167–1175. doi:10.1016/j.bmc.2004.11.020
Leon R, de los Rios C, Marco-Contelles J, Huertas O, Barril X, Luque FJ, Lopez M, Garcia A, Villarroya M (2008) New tacrine–dihydropyridine hybrids that inhibit acetylcholinesterase, calcium entry, and exhibit neuroprotection properties. Bioorg Med Chem 16:7759–7769. doi:10.1016/j.bmc.2008.07.005
Marco JL, de los Rios C, Carreiras MC, Banos JE, Badia A, Vivas NM (2001) Synthesis and acetylcholinesterase/butyrylcholinesterase inhibition activity of new tacrine-like analogues. Bioorg Med Chem 9:727–732. doi:10.1016/S0968-0896(00)00284-4
Marco J, de los Rios C, Garcia A, Villarroya M, Carreiras MC, Martins C, Eleuterio A, Morreale A, Orozco M, Luqu FJ (2004) Synthesis, biological evaluation and molecular modelling of diversely functionalized heterocyclic derivatives as inhibitors of acetylcholinesterase/butyrylcholinesterase and modulators of Ca2+ channels and nicotinic receptors. Bioorg Med Chem 12:2199–2218. doi:10.1016/j.bmc.2004.02.017
Marco-Contelles J, Leon R, de Rios C, Guglietta A, Terencio J, Lopez M, Garcia A, Villarroya M (2006a) Novel multipotent tacrine–dihydropyridine hybrids with improved acetylcholinesterase inhibitory and neuroprotective activities as potential drugs for the treatment of Alzheimer’s disease. J Med Chem 49:7607–7610. doi:10.1021/jm061047j
Marco-Contelles J, Leon R, de los Rios C, Garcia A, Lopez M, Villarroya M (2006b) New multipotent tetracyclic tacrines with neuroprotective activity. Bioorg Med Chem 14:8176–8185. doi:10.1016/j.bmc.2006.09.025
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ (1998) Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19:1639–1662. doi:10.1002/(SICI)1096-987X(19981115)19:14<1639:AID-JCC10>3.0.CO;2-B
Politi A, Durdagi S, Moutevelis-Minakakis P, Kokotos G, Papadopoulos MG, Mavromoustakos T (2009) Application of 3D QSAR CoMFA/CoMSIA and in silico docking studies on novel renin inhibitors against cardiovascular diseases. Eur J Med Chem 44:3703–3711. doi:10.1016/j.ejmech.2009.03.040
Protein Data Bank http://www.rcsb.org/pdb
Scarpini E, Scheltens P, Feldman H (2003) Tretament of Alzheimer’s disease: current status and new perspectives. Lancet Neurol 2:539–547. doi:10.1016/S1474-4422(03)00502-7
Selkoe DJ (1989) Biochemistry of altered brain proteins in Alzheimer’s disease. Annu Rev Neurosci 12:463–490. doi:10.1146/annurev.ne.12.030189.002335
Thaimattam R, Daga P, Banerjee R, Iqbal J (2005) 3D-QSAR studies on c-Src kinase inhibitors and docking analyses of a potent dual kinase inhibitor of c-Src and c-Abl kinases. Bioorg Med Chem 13:4704–4712. doi:10.1016/j.bmc.2005.04.065
Acknowledgment
We are grateful to National Natural Science Foundation of China (No. 21071021).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, N., Liu, C., Zhao, L. et al. 3D-QSAR study of multi-target-directed AchE inhibitors based on autodocking. Med Chem Res 21, 245–256 (2012). https://doi.org/10.1007/s00044-010-9516-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-010-9516-x