
Abstract
Molecular cytogenetics and the study of genome size have been used to understand evolutionary and systematic relationships in many species. However, this approach has seldom been applied to alpine plants. A group of dysploid–polyploid high mountain Artemisia species, distributed from the European Sierra Nevada to Central Asian mountains, through the Pyrenees, the Alps and the Caucasus, is a good model to consider changes at the genome and chromosome levels. These small perennial Artemisia, found frequently in isolated populations, present highly disjunct distributions. Some are considered rare or even endangered. Here, we show results for nine species and 31 populations, including genome size (2C-values), fluorochrome banding and fluorescent in situ hybridisation of ribosomal RNA genes (rDNA). Significant intraspecific genome size variation is found in certain populations of A. eriantha and A. umbelliformis, but without taxonomic significance due to the absence of morphological or ecological differentiation. The number and position of GC-rich DNA bands is mostly coincidental with rDNA although there is an expansion of GC-rich heterochromatin in centromeres in some taxa. Ancestral character states have been reconstructed and x = 9 is inferred as the likely ancestral base number, while the dysploid x = 8 has appeared repeatedly during the evolution of Artemisia. On the basis of cytological observations, Robertsonian translocations are proposed for the appearance of dysploidy in the genus. A remarkable presence of x = 8-based species has been detected in the clade including high mountain species, which highlights the important role of dysploidy in the diversification of high mountain Artemisia. Conversely, polyploidy, though present in the alpine species, is more common in the rest of the genus, particularly in arctic species. Hypotheses on the mechanisms underpinning the relative abundance of dysploids and scarcity of polyploids in high mountain Artemisia are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
High mountain communities are systems that endure harsh conditions, including extreme cold and fluctuating temperatures, exposure to snow, ice and strong winds, poor and rocky soils, high levels of UV radiation and little oxygen availability. Moreover, elevation gradients allow large environmental changes in short spatial distances (Nieto Feliner 2011). All these factors promote local adaptations which, coupled with processes such as long-term isolation and secondary contacts, may lead to an increased biodiversity at high altitudes (Nagy and Grabherr 2009). Distribution areas of cold-adapted species were fragmented and restricted to higher elevations during interglacial periods, which created the current pattern of vicariant taxa between mountain tops in the Mediterranean Basin (Nieto Feliner 2014). In this regard, multiple glacial refugia have been identified in mountain ranges across Europe and North Africa, both in plants (Španiel et al. 2011) and animals (Canestrelli and Nascetti 2008).
Polyploidy and genome size studies in high mountain plants: state of the art
Several studies have hypothesised that high mountain plants undergo polyploidy more frequently (Löve and Löve 1949; Petit and Thompson 1999). It is thought that the ranges for polyploid species would include more extreme environments, such as high altitudes and latitudes, than their diploid relatives (Levin 2002; Weiss-Schneeweiss et al. 2013). However, the pattern stating the prevalence of polyploid taxa in alpine environments is still controversial. Besides, dysploidy (i.e. change in basic chromosome number, usually by fusion or fission, without a significant gain or loss of genetic material) has also contributed to the diversification of several alpine groups such as Phyteuma (Schneeweiss et al. 2013), Saxifraga (Mas de Xaxars et al. 2015) or Leontopodium (Russell et al. 2013) and, together with polyploidy, it is another important mechanism of karyotype evolution.
The C-value (i.e. genome size or the total amount of DNA in the unreplicated haploid or gametic nucleus) has been used in many taxonomic, systematic and evolutionary studies in a range of plant groups. Although genome size has been frequently correlated with environmental, phenotypic or ecological variables (see a review in Greilhuber and Leitch 2013), it has rarely been investigated in the context of high mountain communities. Albach and Greilhuber (2004) found significantly higher DNA amounts in alpine species from Veronica, while Mráz et al. (2009) found negative correlations with altitude and latitude in alpine populations of Hieracium alpinum L. Loureiro et al. (2013) explored genome size variation, beyond polyploidy incidence, in plants from two Spanish mountain ranges, finding no significant genome size differences or correlates. However, they stated that large genomes were difficult to find in high mountain environments.
Artemisia L. and its high mountain species
Artemisia comprises about 500 species with a circumpolar and northern hemisphere distribution, with only a few species in South America and Africa (Tkach et al. 2008). The fossil pollen record places the onset of differentiation of Artemisia back to the Early Miocene, about 19.8 Ma (Sanz et al. 2011). The main speciation and diversification centre of the genus is Central Asia, with secondary centres located in the Irano-Turanian and Mediterranean regions and in western North America (Vallès et al. 2011). Artemisia species are mostly perennial and particularly diverse in mountains, grasslands and semidesertic areas (Ling et al. 2006). The traditional classification of the genus, which relies on capitulum characters, is partially incongruent to lineages identified in molecular studies (Sanz et al. 2008). Nevertheless, the classical subgenera [Artemisia, Absinthium (Mill.) Less., Dracunculus (Besser) Rydb., Seriphidium Besser ex Less., and Tridentatae (Rydb.) McArthur] are still widely used for the circumscription of the genus.
Within Artemisia two main basic chromosome numbers are recognised, x = 9 and x = 8, the former being the most common. Abundant karyological and cytogenetic information is available for the genus (for a review see Vallès et al. 2011). Polyploidy is common in the genus, ploidy levels have been found up to 16x and many species exist in polyploid series. Genome size variation has also been widely explored in Artemisia, which has become one of the best known genera in the family from this point of view.
A suitable model for studying the implications of genome size variation, dysploidy and polyploidy in the diversity and evolution of high mountain plants is found in a group of alpine Artemisia. This group is distributed from Sierra Nevada (Iberian Peninsula) to the Central Asian Mountains, through the Pyrenees, the Alps and the Caucasus. Some of the species associated with high elevations are currently considered as rare or endangered, such as Artemisia granatensis, endemic to Sierra Nevada. These small herbaceous perennials are usually found in isolated populations growing on siliceous rocky ledges or limestone substrates, often in crevices, at elevations between 1500 and 3400 m above sea level. Some taxa, such as A. eriantha and A. umbelliformis, show a highly disjunct distribution, reflecting the geographical complexity of the Pyrenees and the Alps (Sanz et al. 2014).
In the present study, we aim to explore genome size variation, ploidy and base chromosome number changes, and heterochromatin and rDNA loci evolution in alpine Artemisia and considering a phylogenetic context. Given the wealth of information currently available on the genus, we have the opportunity to address genome dynamics and evolutionary factors in this group of alpine plants. Our findings will contribute further evidence to shape the genomic/cytogenetic characteristics of alpine species. To fulfil these goals, two main questions have been specifically addressed: (1) is there any significant difference in basic chromosome number, genome size, heterochromatic or rDNA distribution patterns in the high mountain Artemisia with respect to their lower altitude counterparts? and (2) are polyploidy and/or dysploidy more frequent in this group than in other non-high mountain Artemisia, and what is their biological significance? Additionally, and taking advantage of all available phylogenetic, cytogenetic and genome size information, we will infer the ancestral state of the studied features in this group and in the genus, and we will discuss the prevalence of ancestral states in the present-day high mountain Artemisia.
Materials and methods
Plant material
As part of an ongoing project focusing on the biogeography of two of the most abundant alpine Artemisia species, A. eriantha and A. umbelliformis (Sanz et al. 2014, Sanz et al. unpublished), samples of these as well as closely related species were collected to perform cytogenetic and genome size assays. All specimens were gathered from natural sites except one population of A. eriantha obtained via Index Seminum from the Alpine Botanical Garden of Lautaret (Hautes-Alpes, France), which had been formerly collected near the Col du Lautaret at 2300 m above sea level. Achenes were germinated in pots and cultivated in the greenhouse at the Faculty of Pharmacy, Universitat de Barcelona (UB). Vouchers have been deposited in the herbarium BCN, of the Centre de Documentació de Biodiversitat Vegetal, UB. Table 1 shows the populations studied. Figure 1 indicates the geographical locations of the populations assessed across the high mountain ranges. Seeds of Pisum sativum ‘Express Long’ and Petunia hybrida ‘PxPC6′ used as internal standard for flow cytometry measurements, were obtained from the Institut des Sciences du Végétal (CNRS, Gif-sur-Yvette, France). Root tip meristems were obtained from seedlings produced by germinating achenes on wet filter paper in Petri dishes in the dark at room temperature.

Map indicating the provenance of the populations studied. Zoomed area in the upper left corner corresponds to the Alps, where many of the populations studied are present
DNA content assessments
DNA 2C-values of the selected species were estimated using flow cytometry. As outlined above, P. sativum and P. hybrida (2C = 8.37 and 2.85 pg, respectively; Marie and Brown 1993) were used as internal standards. We have analysed 29 populations of 8 species, with special emphasis on A. eriantha (12 populations) and A. umbelliformis (10 populations), because these are two of the most widely distributed species of this group. Young healthy leaf tissues from the species to be studied and the calibration standard were placed together in a plastic Petri dish and chopped in LB01 buffer (Doležel et al. 1989) with a razor blade. The suspension of nuclei was filtered through a nylon mesh with a pore size of 70 μm and stained for 20 min with propidium iodide (60 μg/mL; Sigma-Aldrich, Madrid, Spain) and supplemented with 100 μg/mL ribonuclease A (Boehringer, Meylan, France). More details on the methodology can be found in Garcia et al. (2006). Mean values and standard deviations were calculated based on the results for five individuals per population. Results on genome size have been submitted to the GSAD database (http://www.asteraceaegenomesize.com).
Chromosome preparations
Root tips were pre-treated with 0.05 % aqueous colchicine, at room temperature, for 2.5–4 h. The material was fixed in absolute ethanol and glacial acetic acid (3:1) and then stored at 4 °C. Protoplast preparations for fluorochrome banding and fluorescent in situ hybridisation (FISH) were carried out using the air-drying technique of Geber and Schweizer (1987), with some modifications as explained in Garcia et al. (2007).
Fluorochrome banding and fluorescent in situ hybridisation
In order to reveal GC-rich DNA bands, chromomycin A3 was used, following the protocol in Garcia et al. (2007). DNA hybridisation was carried out also following Garcia et al. (2007) with minor changes: the 18S-5.8S-26S rDNA probe was obtained from the clone pTa71 (Gerlach and Bedbrook 1979) and was labelled with direct Cy3 -red- (GE Healthcare, Buckinghamshire, UK); the 5S rDNA probe was obtained from the clone pTa794 (Gerlach and Dyer 1980) and labelled with digoxigenin-11-dUTP-green-(Boehringer, Meylan, France). The preparations were counterstained with Vectashield (Vector Laboratories, Burlingame, CA, USA), a mounting medium containing DAPI. FISH preparations were observed with an epifluorescent Zeiss Axiophot microscope with different combinations of Zeiss excitation and emission filter sets (01, 07 and 15). Hybridisation signals were analysed and photographed using the highly sensitive CCD camera (Princeton Instruments, Trenton, NJ, USA), and an image analyser software (Metavue, version 4.6, Molecular Devices Corporation, Sunnyvale, CA, USA). Results on number and position of rDNA loci for each of the populations studied have been submitted to the Plant rDNA database (http://www.plantrdnadatabase.com).
Phylogenetic analyses and reconstruction of character evolution
Beyond the high mountain Artemisia, the sampling strategy for the phylogenetic analysis includes several complementary species to survey representative taxa across the different recognised subgenera in Artemisia, bearing in mind availability of previous cytogenetic and genome size (2C-values) data. Species of Argyranthemum, Nipponanthemum and Tanacetum were selected as outgroups based on phylogenetic results of Sanz et al. (2008) and Zhao et al. (2010), and the availability of complete cytogenetic and genome size information. The nuclear ITS1 + ITS2 (470 bp) and the ETS sequences (374 bp), downloaded from GenBank (Supplementary Materials S1 and S2), and the newly generated ETS sequence for A. genipi (KT954132), were manually edited and concatenated using BioEdit v. 7.1.3.0 (Hall 1999). For phylogenetic analyses, only the 3′ end of the ETS region was used due to the existence of subrepeats and indels causing alignment problems. Bayesian phylogenetic analysis was performed in MrBayes v. 3.1.2 (Ronquist and Huelsenbeck 2003) using the SYM + G model previously determined from jModeltest v. 2.1.3 (Darriba et al. 2012) under the Akaike information criterion (Akaike 1979). The Markov chain Monte Carlo (MCMC) sampling approach was used to calculate posterior probabilities (PPs). Four consecutive MCMC computations were run for 2,000,000 generations, with tree sampling every 100 generations. The first 1000 samples were discarded as the burn-in period. PPs were estimated through the construction of a 50 % majority-rule consensus tree.
The ancestral character reconstructions were performed with Mesquite v. 3.02 software (Maddison and Maddison 2015). All taxa included in these analyses were diploid in order to avoid the bias of polyploidy in the estimated nuclear DNA contents and number of rDNA sites. The 50 % majority-rule consensus tree resulting from Bayesian Inference analysis was used as the input tree file (after pruning polyploid taxa). Basic chromosome numbers (x = 8 and x = 9) and the number of rDNA sites were transformed into categorical data (discrete and ordered). Ancestral states using the “trace character history” function were then inferred using maximum likelihood under the Mk1 model, in which all changes are equally probable, following the same approach as Vaio et al. (2013). For reconstructing the ancestral holoploid genome sizes (2C), we selected squared-change maximum parsimony as implemented for continuous characters in Mesquite. This approach has been widely used to reconstruct ancestral genome size states is many plant groups (e.g. Burleigh et al. 2012).
Statistical analyses
Analyses of regression and one-way ANOVA (or alternatively the Kruskal–Wallis test), Shapiro–Wilk test for normality and Barlett’s test for equality of variances were performed with RStudio, v.0.98.1078, a user interface for R (http://www.rstudio.com). Additionally, to analyse variation of the basic chromosome number, holoploid genome sizes, number of chromomycin A3 positive (CMA+) bands and of rDNA sites in a phylogenetic context, the phylogenetically based generalised least squares (PGLS) algorithm, as implemented in the nlme package for R (Version 3.1-118), was used (Pinheiro et al. 2015). Data on holoploid genome size and ribosomal DNA loci for the complementary and outgroup species were extracted from the Genome Size in the Asteraceae Database (http://www.asteraceaegenomesize.com) and the Plant rDNA database (http://www.plantrdnadatabase.com).
Results
Genus Artemisia is well known from the molecular cytogenetic perspective, yet the large subgenus Absinthium and, in particular, the group of taxa inhabiting high mountains, had not been the focus of any cytogenetic research so far. With the present study, known genome size and molecular cytogenetic profiles of Artemisia span up to 32 and 10.4 % of the accepted taxa, respectively (531 taxa according to The Plant List Version 1.1 http://www.theplantlist.org, accessed March 13th, 2015).
Phylogenetic placement of high mountain Artemisia
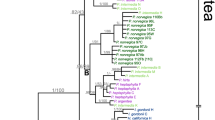
The nuclear ribosomal DNA original dataset included 73 Artemisia species selected to provide a good representation of all subgenera (Vallès et al. 2011). The concatenated aligned dataset consisted of 844 characters (bp), excluding the 5.8S gene. According to our phylogenetic reconstruction (Fig. 2 and Supplementary Material S2), all high mountain taxa, except A. atrata, are grouped in a strongly supported clade (PP = 100 %) that also includes other members of subgenera Absinthium and Artemisia. For simplicity, we have named this group ‘high mountain and allies’ (HMA clade hereafter). When polyploid species are pruned from the tree, 52 species are retained and the HMA clade still retains statistical support (PP = 99 %).

Ancestral state reconstruction of genome size (left) and basic chromosome number and number of rDNA sites (right) for diploid Artemisia taxa. The HMA clade is highlighted in pink and the strictly high mountain species in bold. Species for which no data are available indicated in striped grey. Ancestral state reconstruction was estimated by Mesquite (v.3.02) using the 50 % majority-rule consensus topology obtained by Bayesian inference phylogenetic analysis of the ITS1, ITS2 and ETS data sequence. The Bayesian clade-credibility values are indicated as grey and black squares (posterior probability >0.7 and >0.95, respectively). Blue bars depict genome sizes (2C values)
Basic chromosome numbers
Our sample contains representatives of the main basic numbers of the genus (x = 8 and x = 9) and from the diploid to the hexaploid level. For both the genus and the HMA clade x = 9 has been reconstructed as the ancestral chromosome number, while x = 8 has appeared independently in the three main clades of the phylogeny (Fig. 2). In particular, x = 8 has arisen repeatedly in the HMA clade, with 50 % of its members having this basic chromosome number. Overall, comparing the number of x = 8 species with the number of x = 9 species, the incidence of the former is significantly higher in this clade than elsewhere in the genus (F2,59 = 5.397, p = 0.007).
Genome size variation and relationship with dysploid karyotypes
Table 1 provides holoploid genome size results (2C-values) obtained in the species studied, and a plot illustrating mean genome sizes of x = 8 and x = 9 based Artemisia is presented in Supplementary Material S3. Results range from 5.6 pg for the diploid A. genipi to 18.4 pg for the hexaploid A. nitida, which means a global 3.28-fold range, with a 1.59-fold at the diploid level. The low HPCV (half peak coefficient of variation corresponding to ten samples of five different individuals) mean value (3.22 %) indicates good quality of the flow cytometric assessments. Intraspecific (interpopulation) differences have been detected in cases in which several populations were assessed, reaching up to 11.26 % in diploid A. eriantha and 8.39 % in the tetraploid A. umbelliformis. Within these species, several populations (those usually with values at the limits of the range) had significantly different genome sizes (F11,40 = 5.882, p < 0.0001 for the populations of A. eriantha and F9,40 = 10.963, p < 0.0001 for the populations of A. umbelliformis). Two populations of each for A. glacialis and A. granatensis were also measured, but differences were small, 1.48 % and 2.42 %, respectively.
Using the phylogenetically generalised least squares method (PGLS) we have observed that holoploid genome size (2C) is significantly and positively correlated with ploidy level (t 2,47 = 8.345, p = 0.0039) and chromosome number (t 2,47 = 8.358, p = 0.023), as expected, while monoploid genome size (1Cx) decreases significantly with both (t 2,47 = −1.680, p = 0.0000; t 2,47 = −1.631, p = 0.0000, respectively), indicating a certain degree of genome downsizing. There is an approximate genome size reduction of 13 and 20 % at the 4x and 6x levels, respectively.
Fluorochrome banding (GC-rich heterochromatin) and rDNA loci
Results of chromomycin A3 (CMA+) banding, indicative of GC-rich bands, and rDNA FISH are presented in Figs. 3 and 4. The number of CMA+ signals in one population of A. eriantha and A. genipi is four, while in another population of A. eriantha it is eight, all located at terminal position and overlapping with rDNA sites. Artemisia umbelliformis presents six terminal sites, although one faint centromeric signal is also visible. Artemisia melanolepis chromosomes bear five to six terminal sites, while the closely related tetraploid A. splendens displays eight CMA+ signals. The remaining species display much more CMA+ signals, some of them being particularly abundant in pericentromeric position. This is the case for A. glacialis, showing up to 20 CMA+ signals out of which 18 are pericentromeric, and A. assoana with 16–18 visible CMA+ bands, out of which at least 12 are pericentromeric. Artemisia nitida shows c. 50 CMA+ bands although few are pericentromeric in this case. DAPI after FISH has resulted in signals at both chromosome ends in most chromosomes of A. glacialis and A. assoana, and in some of A. nitida.

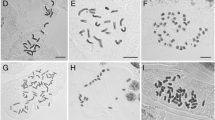
Chromomycin A3-positive (CMA+) and FISH pictures of metaphases of Artemisia species from the HMA clade. a, b A. eriantha, 2n = 18 (Urdiceto); c, d A. genipi 2n = 18; e, f A. melanolepis 2n = 16; arrows in c indicate CMA+ signals; arrow in e indicates one large chromosome; g, h A. glacialis 2n = 16; arrows in g indicate a large chromosome pair and in h a detached ribosomal DNA locus; most CMA+ signals are pericentromeric in g (i, j) A. nitida 2n = 54; arrow in i indicates probably decondensed rDNA loci; k, l A. assoana 2n = 16; arrows in k and l indicate a large chromosome pair, and asterisks in l mark loci with low rDNA copy number; m, n A. splendens 2n = 32; arrows in m mark faint CMA+ signals; o, p, q, r A. umbelliformis 2n = 34; note 8 and 6 rDNA loci in pictures p and r, respectively; arrows in q indicate faint CMA+ signals

Metaphases from Artemisia eriantha: regular metaphase (a, b) and metaphase with a likely Robertsonian translocation (c, d). Aligned chromosomes from b and d with the translocation pair underlined, and the possible mechanism of the translocation (e)
For rDNA loci, our results (Table 2) show a homogenised L-type organisation of ribosomal RNA genes, in which all rDNA sites, in all species, present overlapping 5S and 35S signals, thus both rRNA genes colocalise in the same loci. Perfect coincidence between the number of rDNA sites and the number of CMA+ bands is only seen, however, in A. eriantha (Fig. 4), A. genipi (Fig. 3c, d) and in A. splendens (Fig. 3m, n), while in another A eriantha (Fig. 3a, b, corresponding to the Lautaret population) and in A. umbelliformis (Fig. 3o, p) not all rDNA sites are located in CMA+ regions, and in A. melanolepis, A. glacialis, A. nitida and A. assoana there are more CMA+ bands than rDNA sites—in the last three cases many more (Fig. 3g, i, k). The number of sites ranges from four to eight at the diploid, six to eight at the tetraploid, and nine at the hexaploid levels. The position of signals is invariably terminal and its number is constant within a population except for A. eriantha and A. umbelliformis, in which individuals presenting six and eight rDNA sites have been found; in one A. eriantha a small and unpaired chromosome shows an rDNA site at each end (Fig. 4). As for genome size, the number of rDNA sites is positively correlated with chromosome number (F 1,62 = 82.06, p < 0.0001) and ploidy level (F 1,62 = 79.18, p < 0.0001), and both for the whole genus and for the HMA clade (the distribution of ploidy levels in Artemisia is presented in Fig. 5). Also, there is a significant (F 1,46 = 4.767, p = 0.0341) reduction of the number of rDNA sites per chromosome complement with increasing ploidy. Considering data from all the diploid and tetraploid Artemisia known until present (mean 2C = 2.63 vs. 2C = 1.93 in 2x and 4x, respectively), 26.6 % overall loci loss has been found. The number of sites per monoploid genome in Artemisia is gradually reduced at higher ploidy levels: hexaploids present a 33.5 % reduction in number of loci per monoploid genome with respect to the diploids, and the highest polyploids (decaploids) have 35.36 % less loci than the diploids. Finally, several species show considerable site size differences, such as A. assoana (Fig. 3l) and A. nitida (Fig. 4j). Besides, a detached rDNA site is observed in A. glacialis (Fig. 3h).

Distribution of ploidy levels among different Artemisia groups. Data for the whole genus are taken from Vallès et al. (2011). Arctic s. str. species are selected from Tkach et al. (2008) and chromosome numbers are extracted from the index to chromosome numbers in Asteraceae. “n” indicates sample size
Reconstruction of ancestral characters
The reconstruction of ancestral holoploid genome size onto the phylogeny (Fig. 2), based on diploid taxa, infers a holoploid genome size between 6.92 pg and 8.13 pg as the likely ancestral estimates both for the genus and for the HMA clade, in which ups and downs with respect to their putative common ancestors have occurred during the evolution of the genus. There has been an increase in the number of rDNA sites in 14 cases (45.16 %, three of them within the HMA clade, two within the high mountain) and a decrease in only two cases (6.45 %); the remaining 15 (48.39 %) retain the alleged ancestral number of signals.
Discussion
Intraspecific genome size variation
Research on genome size variation within the species level has a long tradition, from the first reports in flax (Evans et al. 1966). Nevertheless, the extent of true intraspecific variation is still a source of debate. Critical reassessments of such variations have proved them to be artefacts in some cases, having identified technical problems, methodological errors or presence of endogenous staining inhibitors as likely causes (Greilhuber 2005). However, many well-documented examples, based on accurate assessments, do suggest the existence of genuine intraspecific variation in nuclear DNA content (e.g. Suda et al. 2007; Trávnícek et al. 2013).
With regards to the high mountain Artemisia, intraspecific genome size variation has been evaluated in A. eriantha (11.26 %) and A. umbelliformis (8.39 %), the most widely distributed species through the European mountains. The percentages here reported reflect a certain degree of variation, and interpopulational differences are significant in some cases in both species. Other studies on intraspecific variation across a distribution range performed in Artemisia have detected similar percentages: 8.8 % for 17 populations in the diploid A. arborescens (Garcia et al. 2006) and 9.22 % for 45 populations in the hexaploid A. crithmifolia (Pellicer et al. 2009). Yet it is difficult to set a cut-off point from which a given percentage of variation should be considered important. Intraspecific genome size variations have been related to microevolutionary differentiations and could be taxonomically significant, especially if there is some degree of morphological or ecological differentiation (Murray 2005). Neither A. eriantha nor A. umbelliformis show a clear morphological, geographical or ecological pattern to correlate with this variation; however, the detected differences are probably genuine. Possible sources of true intraspecific genome size variation that could explain the detected differences include the presence of aneuploidy or B-chromosomes, introgression or hybridisation processes (Morgan-Richards et al. 2004) or changes in repetitive DNA content (Trávnícek et al. 2013).
Polyploidy is associated with genome downsizing and loci loss, but not necessarily with loss of GC-rich heterochromatin
Most studies have found genome downsizing on polyploids in multiple occasions, as it is the case with the alpine Artemisia. Recently, Meudt et al. (2015) found not only significant genome downsizing across Veronica but also a link with the diversification success of polyploid lineages. However, in some cases as in Nicotiana (Leitch et al. 2008, Renny-Byfield et al. 2011), despite the most common response to polyploidy is still genome downsizing, unchanged genome sizes and even genome upsizing have also been found linked to polyploidy processes in certain species.
Besides, the loci loss detected in these high mountain polyploids fits well with previous findings, particularly at the highest ploidies. Globally, most diploid Artemisia have four rDNA sites (58 %) while some have six (26 %), eight (13 %), and only in two cases nine and ten sites (http://www.plantrdnadatabase.com, accessed March 20th 2015). Regarding tetraploids, Artemisia species mostly show eight sites, consistent with our new findings for A. splendens and one population of A. umbelliformis. As with genome downsizing, loci loss is commonly observed across ploidy levels and seems to be a global response to polyploidy, however, others have found directly proportional increase (Bareka et al. 2012) or even loci gain (Hasterok et al. 2006). Factors such as qualitative and quantitative differences in transposable element (TE) composition of genomes, possible hybrid origins, and the amount of time elapsed after the polyploidisation event, may play a role on the extent of genome size loss after polyploidisation.
The amplification of certain GC-rich repetitive DNA families specific to certain species could explain the differential pattern observed in those species showing considerably more CMA+ signals, such as A. nitida. A similar finding was reported for the tetraploid A. argilosa (Garcia et al. 2007) and more recently, Olanj et al. (2015) found a striking number of CMA+ signals in Tanacetum, a genus closely related to Artemisia (reaching up to 66 at the diploid level, 2n = 18). The observed abundance of pericentromeric CMA+ signals in certain species (A. glacialis and A. assoana) while most are (sub)terminal, would also point to other kinds of repetitive GC-rich DNA amplifying differently in these taxa. Contrasting to genome downsizing and loss of rDNA loci, there is no clear pattern with respect to the pattern of change of CMA+ bands with increasing ploidy: a given species can have very different profiles, and in some cases tetraploids have less CMA+ bands than diploids. As shown previously (Garcia et al. 2007, Olanj et al. 2015), the underpinning evolutionary dynamics of GC-rich DNA is particularly elusive.
Reconstruction of ancestral characters and Artemisia cytogenetic features
Studies that address the evolution of cytogenetic traits considering a phylogenetic and temporal perspective are not common and there is no clear pattern in the direction of their changes (Leitch et al. 2008). During Artemisia’s evolution, both genome size and number of rDNA loci have undergone ups and downs, as others have previously found (Burleigh et al. 2012), this showing the dynamic nature of plant genomes. The ancestral-like condition of four rDNA sites in Artemisia is consistent with the most frequently found numbers of rDNA sites in angiosperms (http://www.plantrdnadatabase.com). The (sub) terminal position is also the most common, both in Artemisia and in angiosperms as a whole, consistently with evolutionary constraints favouring the terminal position for rDNA (Roa and Guerra 2012). Holoploid genome size has mostly remained stable or increased during Artemisia’s evolution: the reconstruction of ancestral holoploid genome size onto the phylogeny (Fig. 2) infers a value between 6.92 and 8.13 as the likely ancestral estimates both for the genus and for the HMA clade. This is considered intermediate (7 ≤ 2C < 28 pg) according to the genome size categories of plants (Leitch et al. 2005). In angiosperms, the ancestral genome size has been reconstructed as very small (2C ≤ 2.8 pg) with several independent increases and decreases taking place (Bennett and Leitch 2005).
Low incidence of polyploid taxa in the high mountain Artemisia
Polyploidy in the HMA clade is not particularly abundant when compared to the whole genus. In Artemisia, 44 % of species are only diploid, 30 % only exclusively polyploid, while 26 % are known both at diploid and polyploid levels (Vallès et al. 2011). However, in the HMA clade, while 73.08 % are only diploid, 19.23 % are exclusively polyploid and 7.69 % are known both at diploid and polyploid levels (Fig. 5). Other few high mountain species not belonging to this clade are also diploid, such as A. atrata. Nie et al. (2005) only detected 22 % of polyploid taxa across the flora of the Hengduan Mountains (Qing-Hai Tibet Plateau). More recently, Loureiro et al. (2013) found 23 % of polyploids across two Spanish high mountain ranges, showing that polyploidisation is probably not essential in determining species adaptation to such environments. Conversely, Löve and Löve (1967) and Morton (1993) found a high rate of polyploids in the alpine zone of Mt. Washington and in the Cameroon Mountains, respectively. Likewise, Vamosi and McEwen (2013) detected an increase of polyploids in high elevations of British Columbia. However, it has been suggested that a high genome size (product of polyploidisation) would not be selected in alpine environments, in principle (Baack 2004). This could have promoted a lower frequency of alpine polyploids in medium-large holoploid genome size genera such as Artemisia. Interestingly, as Tkach et al. (2008) pointed out, we confirmed a higher frequency of polyploids in arctic Artemisia species (Fig. 5). This suggests that polyploidy ranges, and their increase with latitude, could be more related with their ability to colonise new habitats, such as newly deglaciated environments (Brochmann et al. 2004), rather than with a greater cold-hardiness or higher altitudes (Stebbins 1971; te Beest et al. 2012).
Descending dysploidy by recurrent Robertsonian translocations
A pioneer study by Kawatani and Ohno (1964) suggested descending dysploidy in Artemisia on the grounds of the much larger proportion of x = 9-based taxa. Dysploidy has contributed to species differentiation in many genera, occurring once or repeatedly (Blöch et al. 2009; Schneeweiss et al. 2013). As pointed out previously, within the HMA clade there is a significant number of x = 8 (dysploid) species (12 out of 26 species) in a mostly x = 9 based genus, tribe and family (Schweizer and Ehrendorfer 1983). Within Artemisia, x = 8-based species are around 14 % of the total whose chromosome number is known (Vallès et al. 2011). Yet in the HMA clade this grows to 46 %. Our ancestral state reconstruction points to the fact that x = 9 is the ancestral condition in the genus, as in family Asteraceae, although Semple and Watanabe (2009) hypothesised x = 10 was the ancestral base number in tribe Anthemideae (to which Artemisia belongs). This would support a hypothesis of downwards dysploidy within the tribe. Descending dysploidy has been related to plants adapted to extreme habitats in some Asteraceae genera (Garnatje et al. 2004) and this might be linked to x = 8 being over-represented in the HMA clade.
Few works have shown in detail the specific process leading to a decrease in basic chromosome number. However, a likely case of Robertsonian (Rb) translocation that could account for the descending dysploidy has been found in A. eriantha (Fig. 4). In fact, similar processes had already been suggested for Artemisia on the basis of observed centromeric fragility in a large chromosome pair (Vallès and Siljak-Yakovlev 1997). Likewise, a Rb translocation could have been involved in the appearance of x = 17 in species such as A. umbelliformis. Given the distribution of x = 8 taxa across Artemisia’s phylogeny, Rb translocations may have occurred independently several times in the genus (notwithstanding any other mechanisms promoting descending dysploidy). It is possible, however, that the chromosomes involved in the translocation are not the same in all instances where x = 8 appears (i.e. different translocations affecting different chromosomes). We hypothesise that this may be the case in Artemisia since, (1) a notably larger chromosome pair is seen in several x = 8 species from the HMA clade (Fig. 4, e, f, g, h, k, l, arrows) while this is rarely observed in the other x = 8 species outside this clade and (2) holoploid genome size of x = 8 species from the HMA clade is significantly higher than that of the other x = 8 species outside this clade (Supplementary Material S3) which could be related to their specific translocation process (involving different chromosomes, which would explain the observed size differences, and perhaps different genomic processes which somehow could be related with the genome size differences detected). Therefore, the fact that only species from the HMA clade share these features (i.e. large chromosome pair and increased genome size) indicates that the event leading to x = 8 in this group would be common and may differ from other emergences of the dysploid karyotype in other groups, leading to size differences in the translocation chromosomes and genome size accumulation.
Conclusions
This work shows that Artemisia species from high mountain habitats do not present, at first sight, particular karyological features as compared with other alpine plant groups or other non-high mountain Artemisia. However, we do detect a high frequency of dysploids in the clade containing alpine Artemisia. Besides, polyploids are remarkably less abundant in the high mountains than in the rest of the genus. Thus, it seems that evolutionary processes in high mountain Artemisia have been less influenced by polyploidy and more affected by dysploidy and other mechanisms involving genome size increase, such as, for example, mobilisation of transposable elements or amplification of repetitive DNA, which still remain to be proved in these taxa. As previously reported for high mountain species from genus Phyteuma (Schneeweiss et al. 2013), descending dysploidy could be associated with lineage diversification in alpine Artemisia.
References
Akaike H (1979) A Bayesian extension of the minimum AIC procedure of autoregressive model fitting. Biometrika 66:237–242
Albach DC, Greilhuber J (2004) Genome size variation and evolution in Veronica. Ann Bot 94:897–911
Baack EJ (2004) Cytotype segregation on regional and microgeographic scales in snow buttercups (Ranunculus adoneus: Ranunculaceae). Am J Bot 91:1783–1788
Bareka P, Siljak-Yakovlev S, Kamari G (2012) Molecular cytogenetics of Bellevalia (Hyacinthaceae) species occurring in Greece. Plant Syst Evol 298:421–430
Bennett MD, Leitch IJ (2005) Nuclear DNA amounts in angiosperms—progress, problems and prospects. Ann Bot 95:45–90
Blöch C, Weiss-Schneeweiss H, Schneeweiss GM, Barfussa MHJ, Rebernig CA, Villaseñor JC, Stuessy TF (2009) Molecular phylogenetic analyses of nuclear and plastid DNA sequences support dysploid and polyploid chromosome number changes and reticulate evolution in the diversification of Melampodium (Millerieae, Asteraceae). Mol Phylogenet Evol 53:220–233
Brochmann C, Brysting A, Alsos IG, Borgen L, Grundt HH, Scheen AC, Elven R (2004) Polyploidy in arctic plants. Biol J Linn Soc 82:521–536
Burleigh JG, Barbazuk WB, Davis JM, Morse AM, Soltis PS (2012) Exploring diversification and genome size evolution in extant gymnosperms through phylogenetic synthesis. J Bot
Canestrelli D, Nascetti G (2008) Phylogeography of the pool frog Rana (Pelophylax) lessonae in the Italian peninsula and Sicily: multiple refugia, glacial expansions and nuclear–mitochondrial discordance. J Biogeogr 35:1923–1936
Darriba D, Taboada G, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
Doležel J, Binarova P, Lucretti S (1989) Analysis of nuclear DNA content in plant cells by flow cytometry. Biol Plant 31:113–120
Doležel J, Bartoš J, Voglmayr H, Greilhuber J (2003) Nuclear DNA content and genome size of trout and human. Cytom Part A 51:127–128
Evans GM, Durrant A, Rees H (1966) Associated nuclear changes in the induction of flax genotrophs. Nature 212:697–699
Garcia S, Sanz M, Garnatje T, Kreitschitz A, McArthur ED, Vallès J (2004) Variation of DNA amount in 47 populations of the subtribe Artemisiinae and related taxa (Asteraceae, Anthemideae): karyological, ecological, and systematic implications. Genome 47:1004–1014
Garcia S, Garnatje T, Twibell JD, Vallès J (2006) Genome size variation in the Artemisia arborescens complex (Asteraceae, Anthemideae) and its cultivars. Genome 49:244–253
Garcia S, Garnatje T, Hidalgo O, McArthur DE, Siljak-Yakovlev S, Vallès J (2007) Extensive ribosomal DNA (18S-5.8S-26S and 5S) colocalisation in the North American endemic sagebrushes (subgenus Tridentatae, Artemisia, Asteraceae) revealed by FISH. Plant Syst Evol 267:79–92
Garnatje T, Vallès J, Vilatersana R, Garcia-Jacas N, Susanna A, Siljak-Yakovlev S (2004) Molecular cytogenetics of Xeranthemum L. and related genera (Asteraceae, Cardueae). Plant Biol (Stuttg) 6:140–146
Geber G, Schweizer D (1987) Cytochemical heterochromatin differentiation in Sinapis alba (Cruciferae) using a simple air-drying technique for producing chromosome spreads. Plant Syst Evol 158:97–106
Gerlach W, Bedbrook J (1979) Cloning and characterisation of ribosomal RNA genes from wheat and barley. Nucleic Acids Res 7:1869–1885
Gerlach W, Dyer T (1980) Sequence organisation of the repeating units in the nucleus of wheat which contain 5S rRNA genes. Nucleic Acids Res 8:4851–4865
Greilhuber J (2005) Intraspecific variation in genome size in angiosperms: identifying its existence. Ann Bot 95:91–98
Greilhuber J, Leitch IJ (2013) Genome size and the phenotype. In: Greilhuber J, Doležel J, Wendel JF (eds) Plant Genome Diversity, vol 2. Springer, Vienna, pp 323–344
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hasterok R, Wolny E, Hosiawa M, Kowalczyk M, Kulak-Ksiazczyk S, Ksiazczyk T, Heneen WK, Maluszynska J (2006) Comparative analysis of rDNA distribution in chromosomes of various species of Brassicaceae. Ann Bot 97:205–216
Kawatani T, Ohno T (1964) Chromosome numbers in Artemisia. Bull Natl Inst Hyg Sci 82:183–193
Leitch IJ, Soltis DE, Soltis PS, Bennett MD (2005) Evolution of DNA amounts across land plants (Embryophyta). Ann Bot 95:207–217
Leitch IJ, Hanson L, Lim KY, Kovarik A, Chase MW, Clarkson JJ, Leitch AR (2008) The ups and downs of genome size evolution in polyploid species of Nicotiana (Solanaceae). Ann Bot 101:805–814
Levin DA (2002) The role of chromosomal change in plant evolution. Oxford University Press, New York
Ling YR, Humphries CJ, Shultz L, Flora of China Editorial Committee (2006) The genus Artemisia L. Flora China 20:1151–1259
Loureiro J, Castro M, Cerca de Oliveira J, Mota L, Torices R (2013) Genome size variation and polyploidy incidence in the alpine flora from Spain. Anales Jard Bot Madrid 70:39–47
Löve Á, Löve D (1949) The geobotanical significance of polyploidy. I. Polyploidy and latitude. Port Acta Biol Ser A 273–352
Löve Á, Löve D (1967) Polyploidy and altitude: Mt. Washington. Biol Zentralbl Suppl 307–312
Maddison W, Maddison D (2015) Mesquite: a modular system for evolutionary analysis. Version 2.75. http://mesquiteproject.org
Marie D, Brown SC (1993) A cytometric exercise in plant DNA histograms, with 2C values for 70 species. Biol Cell 78:41–51
Mas de Xaxars G, García-Fernández A, Barnola P, Martín J, Mercadé A, Vallès J, Vargas P, Vigo J, Garnatje T (2015) Phylogenetic and cytogenetic studies reveal hybrid speciation in Saxifraga subsect. Triplinervium (Saxifragaceae). J Syst Evol 53:53–65
Meudt HM, Rojas-Andrés BM, Prebble JM, Low E, Garnock-Jones PJ, Albach DC (2015) Is genome downsizing associated with diversification in polyploid lineages of Veronica? Bot J Linn Soc 178:243–266
Morgan-Richards M, Trewick SA, Chapman HM, Krahulcová A (2004) Interspecific hybridization among Hieracium species in New Zealand: evidence from flow cytometry. Heredity 93:34–42
Morton J (1993) Chromosome numbers and polyploidy in the flora of Cameroons Mountain. Op Bot 121:159–172
Mráz P, Chrtek J, Šingliarová B (2009) Geographical parthenogenesis, genome size variation and pollen production in the arctic-alpine species Hieracium alpinum. Bot Helv 119:41–51
Murray BG (2005) When does intraspecific C-value variation become taxonomically significant? Ann Bot 95:119–125
Nagy L, Grabherr G (2009) The biology of alpine habitats. Oxford University Press, New York
Nie Z, Wen J, Gu Z, Boufford DE, Sun H (2005) Polyploidy in the flora of the Hengduan Mountains hotspot, southwestern China. Ann Missouri Bot Gard 92:275–306
Nieto Feliner G (2011) Southern European glacial refugia: a tale of tales. Taxon 60:365–372
Nieto Feliner G (2014) Patterns and processes in plant phylogeography in the Mediterranean Basin. A review. Perspect Plant Ecol 16:265–278
Olanj N, Garnatje T, Sonboli A, Vallès J, Garcia S (2015) The striking and unexpected cytogenetic diversity of genus Tanacetum L. (Asteraceae): a cytometric and fluorescent in situ hybridisation study of Iranian taxa. BMC Plant Biol 15:174
Pellicer J, Garcia S, Garnatje T, Vallès J (2009) Changes in genome size in a fragmented distribution area: the case of Artemisia crithmifolia L. (Asteraceae, Anthemideae). Caryologia 62:152–160
Petit C, Thompson JD (1999) Species diversity and ecological range in relation to ploidy level in the flora of the Pyrenees. Evol Ecol 13:45–65
Pinheiro J, Bates D, DebRoy S, Sarkar D and R Core Team (2015) nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1-121, http://CRAN.R-project.org/package=nlme
Renny-Byfield S, Chester M, Kovařík A, Le Comber SC, Grandbastien MA, Deloger M, Nichols RA, Macas J, Novák J, Chase MW, Leitch AR (2011) Next generation sequencing reveals genome downsizing in allotetraploid Nicotiana tabacum, predominantly through the elimination of paternally derived repetitive DNAs. Mol Biol Evol msr112
Roa F, Guerra M (2012) Distribution of 45S rDNA sites in chromosomes of plants: structural and evolutionary implications. BMC Evol Biol 12:225
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Russell A, Safer S, Weiss-Schneeweiss H, Temsch E, Stuppner H, Stuessy TF, Samuel R (2013) Chromosome counts and genome size of Leontopodium species (Asteraceae: Gnaphalieae) from south-western China. Bot J Linn Soc 171:627–636
Sanz M, Vilatersana R, Hidalgo O, Garcia-Jacas N, Susanna A, Schneeweiss GM, Vallès J (2008) Molecular phylogeny and evolution of floral characters of Artemisia and allies (Anthemideae, Asteraceae): evidence from nrDNA ETS and ITS sequences. Taxon 57:66–78
Sanz M, Schneeweiss GM, Vilatersana R, Vallès J (2011) Temporal origins and diversification of Artemisia and allies (Anthemideae, Asteraceae). Collect Bot 30:7–15
Sanz M, Schönswetter P, Vallès J, Schneeweiss GM, Vilatersana R (2014) Southern isolation and northern long-distance dispersal shaped the phylogeography of the widespread, but highly disjunct, European high mountain plant Artemisia eriantha (Asteraceae). Bot J Linn Soc 174:214–226
Schneeweiss GM, Pachschwöll C, Tribsch A, Schönswetter P, Barfuss MH, Esfeld K, Weiss-Schneeweiss H, Thiv M (2013) Molecular phylogenetic analyses identify Alpine differentiation and dysploid chromosome number changes as major forces for the evolution of the European endemic Phyteuma (Campanulaceae). Mol Phylogenet Evol 69:634–652
Schweizer D, Ehrendorfer F (1983) Evolution of C-band patterns in Asteraceae-Anthemideae. Biol Zentralbl 102:637–655
Semple J, Watanabe K (2009) A review of chromosome numbers in Asteraceae with hypotheses on chromosome base number evolution. In: Funk VA, Susanna A, Suetssy TF, Bayer RJ (eds) Systematics, evolution and biogeography of Compositae. International Association for Plant Taxonomy, Viena, pp 61–72
Španiel S, Marhold K, Passalacqua NG, Zozomová-Lihová J (2011) Intricate variation patterns in the diploid-polyploid complex of Alyssum montanum-A. repens (Brassicaceae) in the Apennine Peninsula: evidence for long-term persistence and diversification. Am J Bot 98:1887–1904
Stebbins GL (1971) Chromosomal evolution in higher plants. Addison-Wesley, London
Suda J, Weiss-Schneeweiss H, Tribsch A, Schneeweiss GM, Trávníček P, Schönswetter P (2007) Complex distribution patterns of di-, tetra-, and hexaploid cytotypes in the European high mountain plant Senecio carniolicus (Asteraceae). Am J Bot 94:1391–1401
te Beest M, Le Roux JJ, Richardson DM, Brysting AK, Suda J, Kubešová M, Pyšek P (2012) The more the better? The role of polyploidy in facilitating plant invasions. Ann Bot 109:19–45
Tkach NV, Hoffmann MH, Röser M, Korobkov AA, von Hagen KB (2008) Parallel evolutionary patterns in multiple lineages of arctic Artemisia L. (Asteraceae). Evolution 62:184–198
Torrell M, Vallès J (2001) Genome size in 21 Artemisia L. species (Asteraceae, Anthemideae): systematic, evolutionary, and ecological implications. Genome 44:231–238
Trávníček P, Kirschner J, Chudáčková H, Rooks F, Štěpánek J (2013) Substantial genome size variation in Taraxacum stenocephalum (Asteraceae, Lactuceae). Folia Geobot 48:271–284
Vaio M, Gardner A, Emshwiller E, Guerra M (2013) Molecular phylogeny and chromosome evolution among the creeping herbaceous Oxalis species of sections Corniculatae and Ripariae (Oxalidaceae). Mol Phyl Evol 68:199–211
Vallès J, Siljak-Yakovlev S (1997) Cytogenetic studies in the genus Artemisia L. (Asteraceae): fluorochrome-banded karyotypes of five taxa, including the iberian endemic species Artemisia barrelieri Besser. Can J Bot 75(4):595–606
Vallès J, Garcia S, Hidalgo O, Martín J, Pellicer J, Sanz M, Garnatje T (2011) Biology, genome evolution, biotechnological issues and research including applied perspectives in Artemisia (Asteraceae). Adv Bot Res 6:349–419
Vamosi J, McEwen J (2013) Origin, elevation, and evolutionary success of hybrids and polyploids in British Columbia, Canada. Botany 91:182–188
Weiss-Schneeweiss H, Emadzade K, Jang TS, Schneeweiss GM (2013) Evolutionary consequences, constraints and potential of polyploidy in plants. Cytogenet Genome Res 140:137–150
Zhao HB, Chen FD, Chen SM, Wu GS, Guo WM (2010) Molecular phylogeny of Chrysanthemum, Ajania and its allies (Anthemideae, Asteraceae) as inferred from nuclear ribosomal ITS and chloroplast trnL-F IGS sequences. Plant Syst Evol 284:153–169
Acknowledgments
This work was supported by the Ministerio de Economía y Competividad from the Government of Spain (CGL2010-22234-C02-01,02/BOS and CGL2013-49097-C2-2-P), the Generalitat de Catalunya, government of Catalonia (“Ajuts a grups de recerca consolidats”, 2009SGR0439 and 2014SGR514) and the IRBIO (Institut de Recerca de la Biodiversitat). GMdX benefited from an ADR grant from Universitat de Barcelona and SG from a Juan de la Cierva postdoctoral contract of the Ministerio de Economía y Competitividad from the Government of Spain. We thank Cristina Roquet, María Sanz and Roser Vilatersana for their help in collecting plant material. Ricard Àlvarez, Jaume Comas, Chari González and Sonia Ruiz are thanked for their assistance in flow cytometric analyses, and Spencer Brown for supplying seeds of the internal standards used. We also thank Odile Robin with technical assistance at FISH slide preparation, Samuel Pyke for English proofreading and three anonymous reviewers for their helpful comments on earlier versions of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
35_2015_159_MOESM3_ESM.tif
Dynamite plot on mean genome sizes of x = 8 and x = 9 based Artemisia species from the HMA clade (dark blue) and from the rest of the genus (light blue). “n” indicates sample size (TIFF 27 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Mas de Xaxars, G., Garnatje, T., Pellicer, J. et al. Impact of dysploidy and polyploidy on the diversification of high mountain Artemisia (Asteraceae) and allies. Alp Botany 126, 35–48 (2016). https://doi.org/10.1007/s00035-015-0159-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00035-015-0159-x