
Abstract
Stream bacterial communities are shaped by a combination of local and regional processes, such as environmental filtering, biotic interactions and dispersal, but biotic interactions have received comparatively less attention. Here, we investigated stream bacterial alpha and beta diversity within taxonomic and phylogenetic contexts around Qiandao Lake in China. We further examined abiotic and biotic factors on bacterial communities by explicitly considering biotic variables including macroinvertebrate species richness, total cover of periphyton and submerged macrophytes. For alpha and beta diversity, there were consistently high correlations between taxonomic and observed phylogenetic metrics. Taxonomic and phylogenetic diversity could be explained by abiotic and biotic variables, though the former showed a stronger influence. Using a null model to break down the association between species phylogeny and co-occurrence, we found non-significant correlations for alpha and beta diversity between taxonomic and phylogenetic diversity, that is, the standardized effect size of phylogenetic diversity (ses.PD) or Unifrac dissimilarity (ses.Unifrac). Variations in taxonomic and phylogenetic diversity at both alpha and beta levels were mainly explained by pure effect of abiotic variables. Biotic variables, such as macroinvertebrate species richness and total cover of periphyton, significantly explained the variations in ses.PD and ses.Unifrac by 19.4% and 18.9%, respectively. Our findings provide an evidence that biotic variables play a non-negligible role in structuring bacterial communities and help to better understand the potential role of biological interactions across trophic levels in streams.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A central question in biogeographical and ecological research is the relationship between local environmental factors and community distributions (Alfonso and Miguel 2008; Hill et al. 2001). Compared to studies on macroorganisms such as plants and birds, microorganisms such as bacteria have been increasingly studied only in the past decade (Hill et al. 2001; Lindström and Langenheder 2012). For stream systems, most studies on microorganisms have historically investigated microscopic algae and how local factors, such as pH and coarse particulate organic matter (CPOM), affect community structure across various spatial scales (Heino et al. 2010; Verleyen et al. 2009). However, studies on the patterns of and processes structuring stream bacterial communities have been increasing in recent years (Fegel et al. 2016; Hotaling et al. 2017, 2019).
Bacterial community composition in streams is contained by both local environmental variables and spatial factors. For example, pH, dissolved organic carbon and nitrogen can predict much of the variation among bacterial communities in streams (Fierer et al. 2007) while leaving large part of the variation unexplained (Lindström and Langenheder 2012; Martiny et al. 2006). Such unexplained variation may indicate that there are unmeasured yet important environmental variables. Less well documented in the study of stream bacteria communities is the importance of biotic variables that connect species to each other at an array of spatial scales such as predation, mutualism, competition, and host-parasite interactions (Amin et al. 2012). Studies revealing have demonstrated biotic variables to be more important than abiotic variables for explaining biodiversity in streams and in soil ecosystems (Carlson et al. 2010; Johnson and Daniel 2010; Vandegehuchte et al. 2010). Therefore, biotic variables should not be ignored, since they can indicate additional local control and improve the performance of predictive bacterial community assembly models.
Since the development of community phylogeny over a decade ago, the phylogenetic community structure of macroorganisms and microbes has been widely examined (Jones and Hallin 2010; Roeder et al. 2015; Webb et al. 2002). Phylogenetic measures can reveal differences in alpha or beta diversity between two communities and have proven valuable in studies of macroorganisms, lending insight into the relative importance of evolutionary and ecological forces in shaping communities (Cavender-Bares and Wilczek 2003; Webb et al. 2002). Furthermore, null model analysis is an important research tool in community ecology, which moves beyond conventional statistical analyses, and provide a benchmark for patterns that might be expected in the absence of species interactions (Gothe et al. 2013). Previous studies have applied null models to examine the phylogenetic structure of the bacterial community (Langenheder et al. 2017; Mykra et al. 2016; Pigot and Etienne 2015; Wang et al. 2012), as phylogenetic relatedness could affect community assembly via functional traits (Gotelli 2000; Webb 2000b). The application of null models is based on the fact that co-occurring species can be more closely (clustered) or distantly (overdispersed) related than expected by chance, and may further indicate the importance of trait-based assembly mechanisms in promoting (e.g., environmental filtering) or inhibiting (e.g., competition) coexistence amongst closely related and ecologically similar species (Pigot and Etienne 2015; Webb et al. 2002).
Here, we investigated the spatial patterns of bacterial communities in streams around Qiandao Lake in China. Specifically, we examined bacterial taxonomic and phylogenetic diversity and explored their relationships with abiotic and biotic variables by including macroinvertebrates, total cover of periphyton and submerged macrophytes. The main aims were as follows: first, we explored the correlations between taxonomic and phylogenetic diversity such as alpha and beta diversity; second, we investigated the effects of abiotic and biotic variables on alpha and beta diversity at the taxonomic and phylogenetic levels, and further evaluated the relative importance of abiotic, biotic, and spatial variables in explaining the variation of these diversity metrics. We hypothesized that the macroinvertebrate community will explain variation in the bacterial community, because macroinvertebrate could increase the substrate utilization of bacteria in the processing of detritus.
Materials and methods
Field sampling
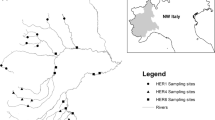
Between 28 May and 04 June 2013, stream biofilm and benthic macroinvertebrates were sampled in 24 streams around Qiandao Lake, Zhejiang Province, China (Fig. 1). Each study site was divided into five or ten cross-sections, depending on the stream width. Ten stones were selected randomly from riffle/run habitats along these sections, and biofilm was scraped off the stones for subsamples from a predefined area (9 cm2) using a sterilized sponge (for bacteria). The subsamples were subsequently pooled into a composite sample from each site. The samples were frozen at − 18 °C immediately after sampling in the field. Macroinvertebrates were sampled by taking five 0.1-m2 Surber samples (500-µm mesh) from random locations from riffle/run habitats. These samples were pooled at each site and stored in 70% ethanol in the field. We also obtained the stream water samples and 0.45-μm filtered water samples, and preserved them at − 18 °C until the chemical analyses were conducted (Wang et al. 2011).

Map of Qiandao Lake (Zhejiang Province, China) with the sampling sites indicated
Abiotic and biotic variables: in the field
Several environmental characteristics that are important for stream organisms were measured at each site. The latitude, longitude and altitude of the sampling sites were logged using a GPS unit. Overhead canopy cover (OHC) was measured at 20 locations at equidistant points along the study reach from the center of the stream. Depth, current velocity and wetted width were measured at five equidistant points along the thalweg of each study reach. Substrate size composition was assessed using the ‘Wolman Walk’ method, by selecting and measuring (beta axis) 100 stones at 1-m intervals 45° to the stream bank in a zigzag manner (Wolman 1954) and grouping into Wentworth scale size classes (Wentworth 1922). The percentage of these classes was then converted into a single substrate size index (SI) by summing their midpoint values weighted by their proportion (bedrock was assigned a nominal value of 400 mm for use in the calculation) (Quinn and Hickey 1990). Water conductivity, pH, temperature and total dissolved solids (TDS) were spot-measured at each site using a YSI Professional Plus Multiparameter Meter (YSI Inc., Ohio, USA). Pfankuch Stability Index was used as an indicator of channel stability (Region and Pfankuch 1975). The bottom component of the Pfankuch Stability Index was used in particular to indicate stream bed stability, which is considered most relevant to benthic communities (Schwendel et al. 2012).
Periphyton biomass was assessed from measures of chlorophyll a taken from five randomly chosen stones in riffles at each site. These were then transported in the dark and on ice prior to being frozen. Chlorophyll a was extracted by using 90% acetone at 5 °C for 24 h in the dark. Absorbances were then read at 750, 665 and 664 nm on a spectrophotometer. These absorbance values were then converted to pigment concentration by following Steinman et al. (2017), corrected for stone surface area using the method of Graham et al. (1988), and halved because only the upper stone surface is available for periphyton growth.
The percentage of periphyton cover was visually assessed and broken into four categories following Tonkin et al. (2014). Assessment was along ten transects of equidistant spacing along the sampling reach. Categories of periphyton cover assessed were: absent, thin film (< 0.5 mm), mat (0.5–5 mm), sludge, cyanobacteria, and green filamentous and other filamentous algae, as well as bryophyte and macrophyte cover. Along these transects we also calculated visually the percentage of CPOM (> 1 mm) cover and fine sediment (< 2 mm) cover in the following two categories: total fine sediment cover, and fine sediment cover on substrate < 2 mm thick. The percentage of debris jams was visually assessed along the full sampling reach.
Abiotic variables and biotic attributes: laboratory processing
We obtained abiotic variables for several important local habitat factors for stream macroinvertebrate communities in the laboratory. We measured ammonium (NH4+–N), nitrate (NO3−–N), nitrite (NO2−–N), and phosphate (PO4−–P) using a continuous flow analyser (Skalar SA 1000, Breda, The Netherlands). Total nitrogen (TN) and total phosphorus (TP) were analysed by peroxydisulphate oxidation and the spectrophotometric method (Association et al. 1915).
For biotic attributes, we used the following biotic attributes as predictor variables for bacterial communities: chlorophyll a of periphyton biomass, species richness of macroinvertebrates, total cover of periphyton, and submerged macrophytes. Post-sampling, macroinvertebrates were sorted and identified to the lowest possible taxonomic level in the laboratory using the standard keys (usually genus or species, but certain difficult-to-identify species, such as chironomid midges, were left at higher taxonomic levels), following Morse et al. (1994). We recorded the number of individuals for each taxa at each site. Further, we calculated the species richness and obtained the first two axes of principal coordinates analysis of the community structure (that is, inv.pcoa1 and inv.pcoa2) using the pcoa function in R (R Core Team 2018) as biotic variables.
Bacterial community analysis
DNA was extracted from freeze-dried biofilm material using a PowerSoil DNA Isolation Kit (MOBIO Laboratories). The quality of DNA extractions were assessed based on the absorbance ratios of 260/280 nm above 1.8, whereas DNA concentrations were require to be larger than 10 ng/uL. Bacterial 16S rRNA genes were amplified in triplicate using bacterial universal primers [515F, 5′-GTGCCAGCMGCCGCGGTAA-3′ and 806R, 5′-GGACTACHVGGGTWTCTAAT-3′] targeting the V4 region. Spacers of different lengths (0–7 bases) were added between the sequencing primer and the target gene primer in each of the 8 forward and reverse primer sets. To ensure that the total length of the amplified sequences did not vary with the primer set used, the forward and reverse primers were used in a complementary fashion so that all of the extended primer sets had exactly 7 extra bases as the spacer for sequencing phase shift. Barcodes were added to the reverse primer between the sequencing primer and the adaptor (Wang et al. 2017).
Positive PCR products were confirmed by agarose gel electrophoresis. PCR products from triplicate reactions were combined and quantified with PicoGreen (Eugene, OR, USA). PCR products from samples to be sequenced in the same MiSeq run were pooled at equal molality to maximize the even-sequencing efforts for all samples. The pooled mixtures were purified with a QIAquick Gel Extraction Kit (QIAGEN Sciences, Germantown, MD, USA) and requantified with PicoGreen. Sample libraries for sequencing were prepared according to the MiSeq Reagent Kit Preparation Guide (Illumina, San Diego, CA, USA). The sequencing databases were generated by Illumina MiSeq PE250 platform.
Sequences were analysed using Quantitative Insights Into Microbial Ecology (QIIME) pipeline version 1.8.0 (Caporaso et al. 2010). Chimaeras were removed with Uchime against the ChimeraSlayer reference database in the Broad Microbiome Utilities, representative sequences from each OTU were aligned to the Greengenes imputed core reference alignment V.201308 (DeSantis et al. 2006) using PyNAST (Caporaso et al. 2009).
Sequences that were longer than 200 bp were denoised with the Denoiser algorithm (Reeder and Knight 2010) and clustered into operational taxonomic units (OTUs) at 97% similarity level with the seed-based UCLUST algorithm (Edgar 2010). The taxonomic identity of each representative sequence was determined using the RDP Classifier (Wang et al. 2007), and the chloroplast and archaeal sequences were removed. Each sample was rarefied to 13,000 sequence reads using the ‘rrarefy’ function in the VEGAN package (Oksanen et al. 2013) in R. The sequences were deposited in MG-RAST database under the accession number 90910.
Statistical analyses
We considered both observed taxonomic and phylogenetic dimensions for bacterial diversity and composition.
-
1)
For alpha diversity, we used species richness and Faith’s phylogenetic diversity [PD, (Faith 1992)]. Species richness is the number of different species in a sample and Faith’s PD is the sum of total phylogenetic length of OTUs in each sample. To break down the association between species phylogeny and co-occurrence (Hardy 2008), we computed the standardized effect size of PD (ses.PD) by comparing the observed PD value from the mean of the null distribution (999 null iterations) based on random shuffling of OTU labels across the phylogenetic tips. Negative ses.PD values and low quantiles (P < 0.05) indicate that co-occurring species are more closely related than expected by chance (clustering), whereas positive values and high quantiles (P > 0.95) indicate that the co-occurring species are less closely related than expected by chance (overdispersion) (Webb 2000a).
We used linear models to explore the relationship between species richness and PD or ses.PD. and Pearson correlations to explore the relationships between species richness, PD or ses.PD and environmental variables.
-
2)
For community composition, observed taxonomic beta diversity was determined using Sorensen dissimilarity indices based on OTU occurrence between a given pair of samples. The observed phylogenetic beta diversity was determined by unweighted Unifrac (Lozupone and Knight 2005). The Sorensen index is a taxonomy-based approach that assesses community differences at a single level of taxonomic resolution by defining OTUs at 97% similarity. The phylogeny-based Unifrac index measures the overall degree of phylogenetic divergence between sets of communities, which allows the comparison of community phylogenies in a more integrated manner than the taxonomy-based approaches (Lozupone and Knight 2005). Further, to break down the association between species phylogeny and co-occurrence, the standardized effect size of phylogenetic beta diversity (ses.Unifrac) was computed as the number of standard deviations by which observed Unifrac departed from the mean of the null distribution (999 null iterations) based on random shuffling of OTU labels across the tips of phylogeny (Fine and Kembel 2011; Hardy 2008).
We used a distance-based approach (Martiny et al. 2006) akin to distance-decay analysis in which community dissimilarity is related to spatial, abiotic or biotic distance among sampled communities. The distance-decay relationship was analysed for the Sorensen, Unifrac or ses.Unifrac beta diversity metrics using a Gaussian generalized linear model, and the significance was determined using Mantel tests (Pearson’s correlation) on 999 permutations. We further used distance-based redundancy analysis (db-RDA) (Legendre and Anderson 1999) to explore the environmental variables that were important in explaining Sorensen, Unifrac and ses.Unifrac. Environmental distance was measured as Euclidean distance using all environmental variables standardized with a mean of zero and a standard deviation of one. For spatial variables, principal coordinates of neighborhood matrices [pcnm, (Borcard and Legendre 2002)] were used to represent original spatial distance matrices as sets of orthogonal eigenvectors (Langenheder et al. 2017).
-
3)
We grouped the explanatory variables into three components: abiotic, biotic and spatial. Variation in alpha diversity and beta diversity was partitioned among the three components using linear models (Anderson and Cribble 1998; Borcard et al. 1992). By generating models with the three sets of explanatory variables, we estimated the proportions of variation in bacterial diversity explained by the pure effects of spatial, abiotic and biotic variables and by the intersections of these three components (Langenheder et al. 2017).
The above analyses were conducted in the R environment with the packages vegan V2.3–3 (Oksanen et al. 2013), and picante V1.2–0 (Kembel 2009).
Results
-
i.
Physical and chemical properties of study sites
For the chemical factors, there was a relatively large range of nutrient concentrations in streams water, such as TN ranging from 0.058 to 10.279 mg L−1 and TP from 0.007 to 0.599 mg L−1. For physical factors, water temperatures ranged from 16.1 to 23.5 °C and electrical conductivity fluctuated between 12.3 and 142.5 µS cm−1. The Pfankuch Stability Index, which is considered most relevant to benthic communities, varied widely across sampled streams, ranging from 51 to 101. For a summary of site characteristics see Supplementary Table S1.
-
ii.
Bacterial community composition
A total of 913,071 reads were obtained from the 24 samples. Each sample contained 13,632–77,537 reads. Most stream samples around Qiandao Lake were dominated by three major bacterial phyla: Proteobacteria, Cyanobacteria, Bacteroidetes (Fig. S1). Proteobacteria and Cyanobacteria represented the majority of the community compositions of all streams except for one site QD08, which was dominated by Proteobacteria, Cyanobacteria and Verrucomicrobia. Nevertheless, the relative abundance of Proteobacteria was particularly high in QD16, and low in QD07 and QD08.
Species richness showed a significant correlation with PD (r = 0.91, P < 0.01; Fig. 2a) but not with phylogenetic structure (ses.PD; r = 0.045, P > 0.05; Fig. 2b). For community composition, there were also significant correlations between the taxonomic (Sorensen) and observed phylogenetic beta diversity (Unifrac) (Mantel r = 0.64, P < 0.01; Fig. 2c), but there was no correlation with the standardized effect size of Unifrac (ses.Unifrac) (Mantel r = 0.019, P > 0.05; Fig. 2d).
-
iii.
Explanatory power of abiotic factors
Species richness and PD showed significant (P < 0.05) positive relationships with physical variables (Fig. 3a, b). Among the abiotic explanatory variables, CPOM showed the highest positive correlations with species richness and PD. In contrast to PD, species richness was highly correlated with other chemical factors, such as TN and NH4+.
The db-RDA analyses showed that both Sorenson, Unifrac and ses.Unifrac were correlated with pH and Pfankuch index (Fig. 4). The Sorenson index was correlated with other abiotic variables such as fine sediment layer (on stones < 2 mm thick) and fine sediment cover. The variation explained was the highest for ses.Unifrac, at 57.8%, followed by 43.5% for Sorensen dissimilarity and 27.1% for Unifrac.
We further applied variation partitioning to examine the relative importance of abiotic variables to bacterial diversity and community composition. For species richness and PD, the pure effects of abiotic components were dominant, with explained variation percentages of 34.5% and 41.5%, respectively (Fig. 5a, b). For community composition, abiotic variables showed pure effects on Sorenson and Unifrac, with explained variation percentages of 8.2% and 12.2%, respectively (Fig. 5d, e).
-
iv.
Explanatory power of biotic factors

Correlation of alpha and beta diversity among taxonomic and phylogenetic metrics. a Relationship between species richness and observed phylogenetic alpha diversity (PD). b Relationship between species richness and standardized effect size of phylogenetic alpha diversity (ses.PD). c Relationship between taxonomic beta diversity (Sorensen) and observed phylogenetic beta diversity (Unifrac). d Relationship between taxonomic beta diversity (Sorensen) and standardized effect size of phylogenetic beta diversity (ses.Unifrac)
Species richness and PD were showed negatively correlated with biotic variables (Fig. 3a, b) such as invertebrate community composition (inv.pcoa2) and total cover of periphyton. Among these biotic explanatory variables, inv.pcoa2 showed the highest negative correlations with species richness and PD. Phylogenetic structure ses.PD was negative, indicating strong phylogenetic clustering, and only showed a significant (P < 0.05) negative correlation with one biotic variable: the species richness of macroinvertebrates (Fig. 3c).

Pearson correlation analyses of the relationships among alpha diversity and abiotic and biotic factors. Filled circles represent significant variables (P < 0.05). These alpha diversity metrics are a species richness, b observed phylogenetic alpha diversity (PD), and c standardized effect size of phylogenetic alpha diversity (ses.Unifrac). CPOM coarse particulate organic matter, TDS total dissolved solids, Sand.fine the ratio of sand to fine particles, SI substratum particle size, OHC Overhead canopy cover, Fine.sedi.stones fine sediment layer (on stones < 2 mm thick), Fine.sedi.cover fine sediment cover, Total cover total cover of periphyton, Chl.a chlorophyll a of periphyton biomass, Inv.observed species richness of macroinvertebrates, Inv.pcoa1, inv.pcoa2 the first two axes of principal coordinate analysis for macroinvertebrates
The db-RDA analyses showed that biotic variables such as total cover of periphyton had substantial effects on both Sorenson, Unifrac and ses.Unifrac (Fig. 4). The Unifrac and ses.Unifrac was correlated with other biotic variables such as macroinvertebrate community structure and chl.a, respectively. When spatial, abiotic, or biotic distances were considered, they showed significant relationships (P < 0.01) for Sorensen and Unifrac, but not for ses.Unifrac (Fig. S2).

Ordination plots of the relationships between beta diversity and abiotic and biotic factors based on distance-based redundancy analysis (db-RDA). These beta diversity metrics are a taxonomic beta diversity (Sorensen), b observed phylogenetic beta diversity (Unifrac) and c standardized effect size of phylogenetic beta diversity (ses.Unifrac). Pfankuch Pfankuch index, Total cover total cover of periphyton, Fine.sedi.stones fine sediment layer (on stones < 2 mm thick), Fine.sedi.cover fine sediment cover, Inv.pcoa2 the second axes of principal coordinate analysis for macroinvertebrates, Chl.a chlorophyll a of periphyton biomass, PCNM1, PCNM8 the first and eighth pcnm vector of principal coordinates of neighbourhood matrices analysis for spatial variables
We further applied variation partitioning to examine the relative importance of biotic variables to bacterial diversity and community composition. The percent of variation in species richness explained by biotic variables was 2.5%, which was lower than that explained by PD (Fig. 5a, b). For community composition, biotic variables showed pure effects on Unifrac (Fig. 5e), but not Sorensen (Fig. 5d). When the standardized effect sizes of PD and Unifrac were considered, we found that biotic components were the main drivers, showing pure effects of 19.4% and 18.9%, respectively (Fig. 5c), while spatial variables could also explain 4.1% of the variation in ses.Unifrac (Fig. 5f).

The relative importance of biotic (B), abiotic (A) and spatial (S) variables in explaining alpha and beta diversity within taxonomic and phylogenetic contexts. The top-left panel is the general outline. The pure variation explained by each factor is represented by the edges of the triangle. The sides and middle of the triangles indicate the percentages of variation explained by interactions of two or all factors, respectively. a Species richness, b observed phylogenetic alpha diversity (PD), c standardized effect size of phylogenetic alpha diversity (ses.PD), d taxonomic beta diversity (Sorensen), e observed phylogenetic beta diversity (Unifrac), f standardized effect size of phylogenetic beta diversity (ses.Unifrac). Statistical significance was determined according to a Monte Carlo permutation test (9999, P < 0.01)
Discussion
Species richness and PD were generally highly correlated, which is consistent with previous reports (e.g., Wang et al. 2012). When the phylogenetic independency of species was considered, however, there was no correlation between species richness and ses.PD or between Sorensen and ses.Unifrac after the phylogenetic null models were applied to alpha and beta diversity. Abiotic variables showed a dominant influence on both observed alpha and beta diversity within taxonomic and phylogenetic contexts. Interestingly, biotic variables were important for both diversity and community composition at taxonomic and observed phylogenetic levels but showed dominant roles in explaining standardized effect size of phylogenetic diversity.
Taxonomic data indicated Proteobacteria was the most abundant phylum followed by Cyanobacteria and Bacteroidetes. Similar to our study, at the bacterial phylum level, Proteobacteria, Cyanobacteria, Bacteroidetes and Actinobacteria dominate epilithic bacterial communities in the streams located along the main chain of the Austrian Alps (Wilhelm et al. 2014). In Boulder Creek watershed, the stream bacterioplankton communities were, on average, dominated by a relatively limited number of bacterial phyla, including Actinobacteria, and Bacteroidetes (Portillo et al. 2012). In Miers Stream, bacterial assemblages are dominated by Cyanobacteria, which comprised 47–79% of OTU abundances (Stanish et al. 2013). While in Mina Stream, most bacterial OTUs were affiliated with two phyla: Proteobacteria and Bacteroidetes (Costa et al. 2015). These results collectively indicate that the above phyla typically dominate bacterial communities in stream environments.
We found high correlations between taxonomic and observed phylogenetic diversity at both alpha (species richness and PD) and beta (Sorensen and Unifrac) levels. Similar abiotic and biotic variables were observed to correlate with species richness and PD. This result is not surprising, because taxonomic and observed phylogenetic diversity are typically related (Morlon et al. 2011). Such high correlations may indicate that taxonomic and observed phylogenetic levels respond to similar gradients in the environment. This is supported by the fact that there were several common environmental variables that were identified to be correlated with alpha (pH, CPOM, total cover of periphyton and species richness of macroinvertebrates) and beta (pH, Pfankuch index and total cover of periphyton) diversity between taxonomic and phylogenetic metrics.
Abiotic variables related to both alpha and beta diversity at the taxonomic and phylogenetic levels included water chemistry and habitat factors. Total nitrogen and nitrogen oxide (NOx) were the most influential water chemistry variables for PD, while habitat factors, such as CPOM and Pfankuch index, were more important for both richness and PD, and pH showed a significant correlation for all three beta diversity metrics. This result supports the notion that stream ecosystems and their communities are under strong abiotic control (Heino et al. 2014). Similar to our findings, previous studies also showed that among the water chemistry variables, pH was the most influential variable for bacteria community structure, while habitat factors were of minor importance (Heino et al. 2014; Lear et al. 2009). Water temperature and conductivity were significantly correlated with the bacterial communities in plateau freshwater lakes (Liu et al. 2016; Wu et al. 2019). Nutrients such as TN could limit bacterial biomass, as TN showed a significantly negative correlation with bacterial diversity (Carr et al. 2010) whereas the increases in coarse particulate organic matter could enhance bacterial biomass (Artigas et al. 2009; Gulis et al. 2004; Gulis and Suberkropp 2003).
We found that biotic variables such as macroinvertebrate species richness and total cover of periphyton, were also important in accounting for the bacterial alpha and beta diversity within taxonomic and observed phylogenetic contexts. Previous studies found that PD of bacteria was positively related to algal biomass and macroinvertebrate richness (Langenheder et al. 2017), which is consistent with our findings. The effects of biotic variables on bacterial communities were further supported by the standardized effect size of phylogenetic metrics when we used null model to break down the association between species phylogeny and co-occurrence. Biotic interactions are clearly capable of influencing species distributions, as biotic variables have been demonstrated to influence species distributions and realized assemblages of species across all spatial extents (Alofs and Jackson 2015; Wang and Jackson 2011; Wisz et al. 2013b) For example, the organic substrates released by periphyton are often highly available and readily used by bacteria (Wagner et al. 2014), suggesting that the dependence of consumers on producers could be important in determining species diversity and community (Wisz et al. 2013a). However, the extent to which biotic interactions structure species distributions relative to abiotic drivers remains uncertain, and inferring interactions from co-occurrence data is highly problematic, so caution is required when interpreting such patterns (Blanchet et al. 2020).
Specifically, we would like to highlight that the species richness and community compositions of macroinvertebrates had significant correlations with bacterial diversity and community composition. Both macroinvertebrates and bacteria play a key role in the processing of detritus, and macroinvertebrates could enhance bacterial activity and detritus processing (Vandebund et al. 1994; Wieltschnig et al. 2008), thereby increasing the utilization of bacteria by the substrate. Detrital organic matter is a key energy source in stream ecosystems that are dominated by leaf litter (Marcarelli et al. 2011). Litter decomposition can affect bacteria richness and community composition due to the substrate preferences of bacteria and the availability of energy and nutrients (Moragomez et al. 2016). Moreover, bacterial community structure could be strongly affected by macroinvertebrate bioturbation and feeding traits. For example, up to 50% of the bacterial community originated from the macroinvertebrates, suggesting that bacteria introduced by macroinvertebrates strongly affected bacterial community structure (Hunting et al. 2012). Furthermore, abiotic and other biotic variables affect the bacterial community via an indirect pathway through macroinvertebrates. For example, CPOM could provide food for leaf shredding macroinvertebrates, and the taxonomic composition and abundance of macroinvertebrates are influenced by substrate type and food, such as periphyton biomass (Mieczan et al. 2018).
Conclusions
Taken together, both alpha and beta diversity could be substantially explained by abiotic and biotic variables within both taxonomic and phylogenetic contexts. Furthermore, we used null model to break down the association between species phylogeny and co-occurrence and found that biotic variables showed dominant effects on both ses.PD and ses.Unifrac. Such results add weight to the notion that biotic variables play important roles in influencing the spatial structuring of bacterial communities. Although the explained variation in phylogenetic diversity and community was still relatively limited, we found important biotic variables for bacterial communities, including macroinvertebrates, total cover of periphyton and submerged macrophytes. Our results contribute to the evidence highlighting the importance of the potential role of biological interactions in streams. Further studies are encouraged to explore the importance of biotic variables among different community assembly processes along environmental gradients.
References
Alfonso VB, Miguel V (2008) Temporal shifts from facilitation to competition occur between closely related taxa. J Ecol 96:489–494
Alofs KM, Jackson DA (2015) The abiotic and biotic factors limiting establishment of predatory fishes at their expanding northern range boundaries in Ontario. Canada Glob Change Biol 21:2227–2237. https://doi.org/10.1111/gcb.12853
Amin SA, Parker MS, Virginia EA (2012) Interactions between diatoms and bacteria. Microbiol Mol Biol Rev Membr 76:667–684
Anderson M, Cribble N (1998) Partitioning the variation among spatial, temporal and environmental components in a multivariate data set. Aust J Ecol 23:158–167
Artigas J, RomanÍ AM, Gaudes A, MuÑOz I, Sabater S (2009) Organic matter availability structures microbial biomass and activity in a Mediterranean stream. Freshw Biol 54:2025–2036. https://doi.org/10.1111/j.1365-2427.2008.02140.x
Association APH, Association AWW, Federation WPC, Federation WE (1915) Standard methods for the examination of water and wastewater, 2nd edn. American Public Health Association, Washington
Blanchet FG, Cazelles K, Gravel D (2020) Co-occurrence is not evidence of ecological interactions. Ecol Lett 23:1050–1063
Borcard D, Legendre P (2002) All-scale spatial analysis of ecological data by means of principal coordinates of neighbour matrices. Ecol Model 153:51–68
Borcard D, Legendre P, Drapeau P (1992) Partialling out the spatial component of ecological variation. Ecology 73:1045–1055
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R (2009) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267
Caporaso JG et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335
Carlson ML, Flagstad LA, Gillet F, Mitchell EAD (2010) Community development along a proglacial chronosequence: are above-ground and below-ground community structure controlled more by biotic than abiotic factors? J Ecol 98:1084–1095
Carr GM, Morin A, Chambers PA (2010) Bacteria and algae in stream periphyton along a nutrient gradient. Freshw Biol 50:1337–1350
Cavender-Bares J, Wilczek A (2003) Integrating micro-and macroevolutionary processes in community ecology. Ecology 84:592–597
Costa PS et al (2015) Metagenome of a microbial community inhabiting a metal-rich tropical stream sediment. PLoS ONE 10:e0119465
DeSantis TZ et al (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. https://doi.org/10.1128/aem.03006-05
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Faith DP (1992) Conservation evaluation and phylogenetic diversity. Biol Conserv 61:1–10
Fegel TS, Baron JS, Fountain AG, Johnson G, Hall EK (2016) The differing biogeochemical and microbial signatures of glaciers and rock glaciers. J Geophys Res 121:919–932
Fierer N, Morse JL, Berthrong ST, Bernhardt ES, Jackson RB (2007) Environmental controls on the landscape-scale biogeography of stream bacterial communities. Ecology 88:2162–2173
Fine PVA, Kembel SW (2011) Phylogenetic community structure and phylogenetic turnover across space and edaphic gradients in western Amazonian tree communities. Ecography 34:552–565. https://doi.org/10.1111/j.1600-0587.2010.06548.x
Gotelli NJ (2000) Null model analysis of species co-occurrence patterns. Ecology 81:2606–2621
Gothe E, Angeler DG, Gottschalk S, Lofgren S, Sandin L (2013) The influence of environmental, biotic and spatial factors on diatom metacommunity structure in Swedish headwater streams. PLoS ONE 8:e72237. https://doi.org/10.1371/journal.pone.0072237
Graham AA, Mccaughan DJ, Mckee FS (1988) Measurement of surface area of stones. Hydrobiologia 157:85–87
Gulis V, Rosemond AD, Suberkropp K, Weyers HS, Benstead JP (2004) Effects of nutrient enrichment on the decomposition of wood and associated microbial activity in streams. Freshw Biol 49:1437–1447
Gulis V, Suberkropp K (2003) Effect of inorganic nutrients on relative contributions of fungi and bacteria to carbon flow from submerged decomposing leaf litter. Microb Ecol 45:11–19
Hardy OJ (2008) Testing the spatial phylogenetic structure of local communities: statistical performances of different null models and test statistics on a locally neutral community. J Ecol 96:914–926
Heino J, Bini LM, Karjalainen SM, Mykrä H, Soininen J, Vieira LCG, Diniz-Filho JAF (2010) Geographical patterns of micro-organismal community structure: are diatoms ubiquitously distributed across boreal streams? Oikos 119:129–137
Heino J, Tolkkinen M, Pirttilä AM, Aisala H, Mykrä H, Maggs C (2014) Microbial diversity and community-environment relationships in boreal streams. J Biogeogr 41:2234–2244. https://doi.org/10.1111/jbi.12369
Hill BH, Stevenson RJ, Pan Y, Herlihy AT, Kaufmann PR, Johnson CB (2001) Comparison of correlations between environmental characteristics and stream diatom assemblages characterized at genus and species levels. J N Am Benthol Soc 20:299–310
Hotaling S, Finn DS, Giersch JJ, Weisrock DW, Jacobsen D (2017) Climate change and alpine stream biology: progress, challenges, and opportunities for the future. Biol Rev 92:2024–2045
Hotaling S et al (2019) Microbial assemblages reflect environmental heterogeneity in alpine streams. Glob Change Biol 25:2576–2590
Hunting ER, Whatley MH, Geest HGVD, Mulder C, Kraak MHS, Breure AM, Admiraal WJFS (2012) Invertebrate footprints on detritus processing, bacterial community structure, and spatiotemporal redox profiles. Freshw Sci 31:724–732
Johnson RK, Daniel H (2010) Spatial congruency of benthic diatom, invertebrate, macrophyte, and fish assemblages in European streams. Ecol Appl 20:978–992
Jones CM, Hallin S (2010) Ecological and evolutionary factors underlying global and local assembly of denitrifier communities. ISME J 4:633
Kembel SW (2009) Disentangling niche and neutral influences on community assembly: assessing the performance of community phylogenetic structure tests. Ecol Lett 12:949–960
Langenheder S, Wang J, Karjalainen SM, Laamanen TM, Tolonen KT, Vilmi A, Heino J (2017) Bacterial metacommunity organization in a highly connected aquatic system. FEMS Microbiol Ecol. https://doi.org/10.1093/femsec/fiw225
Lear G, Niyogi D, Harding J, Dong Y, Lewis G (2009) Biofilm bacterial community structure in streams affected by acid mine drainage. Appl Environ Microbiol 75:3455–3460. https://doi.org/10.1128/AEM.00274-09
Legendre P, Anderson MJ (1999) Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monogr 69:1–24
Lindström ES, Langenheder S (2012) Local and regional factors influencing bacterial community assembly. Environ Microbiol Rep 4:1–9. https://doi.org/10.1111/j.1758-2229.2011.00257.x
Liu K, Liu Y, Jiao N, Zhu L, Wang J, Hu A, Liu X (2016) Vertical variation of bacterial community in Nam Co, a large stratified lake in central Tibetan Plateau. Antonie Van Leeuwenhoek 109:1323–1335
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. https://doi.org/10.1128/AEM.71.12.8228-8235.2005
Marcarelli A, Baxter CV, Mineau MM, Hall RO (2011) Quantity and quality: unifying food web and ecosystem perspectives on the role of resource subsidies in freshwaters. Ecology 92:1215–1225
Martiny JBH et al (2006) Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4:102–112
Mieczan T, Tarkowskakukuryk M, Ȃrva D, Berzninagy L, Novak Z, Vajda C (2018) The effect of epiphytic macroinvertebrates on microbial communities in different types of macrophyte-dominated shallow lakes. Knowl Manag Aquat Ecosyst 419:13
Wolman MG (1954) A method of sampling coarse river-bed material. Eos Trans Am Geophys Union 35:951–956
Moragomez J, Elosegi A, Duarte SAF, Cassio F, Pascoal C, Romani AM (2016) Differences in the sensitivity of fungi and bacteria to season and invertebrates affect leaf litter decomposition in a Mediterranean stream. FEMS Microbiol Ecol 92:fiw121
Morlon H et al (2011) Spatial patterns of phylogenetic diversity. Ecol Lett 14:141–149. https://doi.org/10.1111/j.1461-0248.2010.01563.x
Morse JC, Yang L, Tian L (1994) Aquatic insects of China useful for monitoring water quality. Hohai University Press, Nanjing
Mykra H, Tolkkinen M, Markkola A, Pirttila AM, Muotka T (2016) Phylogenetic clustering of fungal communities in human-disturbed streams. Ecosphere 7:e01316
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H, Oksanen MJ (2013) Package ‘vegan’. Community ecology package, version, 2(9), pp 1–295. https://cran.r-project.org/web/packages/vegan/
Pigot AL, Etienne RS (2015) A new dynamic null model for phylogenetic community structure. Ecol Lett 18:153–163
Portillo MC, Anderson SP, Fierer N (2012) Temporal variability in the diversity and composition of stream bacterioplankton communities. Environ Microbiol 14:2417–2428
Quinn JM, Hickey CW (1990) Characterisation and classification of benthic invertebrate communities in 88 New Zealand rivers in relation to environmental factors. N Z J Mar Freshw Res 24:387–409
R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. R V3.6.2. https://www.R-project.org/.
Reeder J, Knight R (2010) Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods 7:668
Region USFSN, Pfankuch DJ (1975) Stream reach inventory and channel stability evaluation. United States Department of Agriculture Forest Service, Northern Region, Missoula
Roeder M, McLeish M, Beckschäfer P, de Blécourt M, Paudel E, Harrison RD, Slik F (2015) Phylogenetic clustering increases with succession for lianas in a Chinese tropical montane rain forest. Ecography 38:832–841
Schwendel AC, Death RG, Fuller IC, Tonkin JD (2012) A new approach to asses bed stability relevant for onvertebrate communities in upland streams. River Res Appl 28:1726–1739
Stanish LF et al (2013) Bacteria and diatom co-occurrence patterns in microbial mats from polar desert streams. Environ Microbiol 15:1115–1131
Steinman AD, Lamberti GA, Leavitt PR, Uzarski DG (2017) Biomass and pigments of benthic algae. Methods in stream ecology, 1st edn. Elsevier, New York, pp 223–241
Tonkin JD, Death RG, Barquín J (2014) Periphyton control on stream invertebrate diversity: Is periphyton architecture more important than biomass? Mar Freshw Res 65:818–829
Vandebund WJ, Goedkoop W, Johnson RK (1994) Effect of deposit-feeder activity on bacterial production and abundance in profundal lake sediment. J N Am Benthol Soc 13:532–539. https://doi.org/10.2307/1467849
Vandegehuchte ML, Peña EDL, Bonte D (2010) Relative importance of biotic and abiotic soil components to plant growth and insect herbivore population dynamics. PLoS ONE 5:e12937
Verleyen E et al (2009) The importance of dispersal related and local factors in shaping the taxonomic structure of diatom metacommunities. Oikos 118:1239–1249
Wagner K, Bengtsson MM, Besemer K, Sieczko A, Burns NR, Herberg ER, Battin TJ (2014) Functional and structural responses of hyporheic biofilms to varying sources of dissolved organic matter. Appl Environ Microbiol 80:6004–6012
Wang J et al (2017) Regional and global elevational patterns of microbial species richness and evenness. Ecography 40:393–402. https://doi.org/10.1111/ecog.02216
Wang J, Soininen J, He J, Shen J (2012) Phylogenetic clustering increases with elevation for microbes. Environ Microbiol Rep 4:217–226. https://doi.org/10.1111/j.1758-2229.2011.00324.x
Wang J, Soininen J, Zhang Y, Wang B, Yang X, Shen J (2011) Contrasting patterns in elevational diversity between microorganisms and macroorganisms. J Biogeogr 38:595–603. https://doi.org/10.1111/j.1365-2699.2010.02423.x
Wang LF, Jackson DA (2011) Modeling the establishment of invasive species: habitat and biotic interactions influencing the establishment of Bythotrephes longimanus. Biol Invasions 13:2499–2512. https://doi.org/10.1007/s10530-011-0071-2
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Webb CO (2000a) Exploring the phylogenetic structure of ecological communities: an example for rain forest trees. Am Nat 156:145–155
Webb CO (2000b) Exploring the phylogenetic structure of ecological communities: an example for rain forest trees. Am Nat 156:145–155
Webb CO, Ackerly DD, McPeek MA, Donoghue MJ (2002) Phylogenies and community ecology. Annu Rev Ecol Syst 33:475–505. https://doi.org/10.1146/annurev.ecolysis.33.010802.150448
Wentworth CK (1922) A scale of grade and class terms for clastic sediments. J Geol 30:377–392
Wieltschnig C, Fischer UR, Velimirov B, Kirschner AKT (2008) Effects of deposit-feeding macrofauna on benthic bacteria, viruses, and protozoa in a silty freshwater sediment. Microbial Ecol 56:1–12. https://doi.org/10.1007/s00248-007-9318-y
Wilhelm L, Besemer K, Fasching C, Urich T, Singer GA, Quince C, Battin TJ (2014) Rare but active taxa contribute to community dynamics of benthic biofilms in glacier-fed streams. Environ Microbiol 16:2514–2524
Wisz MS et al (2013a) The role of biotic interactions in shaping distributions and realised assemblages of species: implications for species distribution modelling. Biol Rev 88:15–30
Wisz MS et al (2013b) The role of biotic interactions in shaping distributions and realised assemblages of species: implications for species distribution modelling. Biol Rev 88:15–30. https://doi.org/10.1111/j.1469-185X.2012.00235.x
Wu K et al (2019) The relative abundance of benthic bacterial phyla along a water-depth gradient in a Plateau Lake: physical, chemical, and biotic drivers. Front Microbiol 10:1521
Acknowledgements
We are grateful to Esta Tonkin for assistance with field sampling. The work was financially supported by the Program of Global Change and Mitigation (2017YFA0605200), National Natural Science Foundation of China (41871048, 41830757, 41571058), CAS Key Research Program of Frontier Sciences (QYZDB-SSW-DQC043), and Xi’an Jiaotong-Liverpool University Research Development (RDF-12-01-24).
Author information
Authors and Affiliations
Contributions
JW and NX conceived the idea. JT took field samples and provided physiochemical and biological data. JW performed the bioinformatic analyses. ML led the statistical analyses with the contributions from JL and JW. ML wrote the first draft of the manuscript. ML and JW finished the manuscript with the contributions from JS, NX, JT and JL. All the authors contributed to development of the study.
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, M., Liu, J., Tonkin, J.D. et al. The effects of abiotic and biotic factors on taxonomic and phylogenetic diversity of stream epilithic bacteria around Qiandao Lake. Aquat Sci 82, 71 (2020). https://doi.org/10.1007/s00027-020-00746-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00027-020-00746-8