
Abstract
SIRT6 is an NAD+ dependent deacetylase that belongs to the mammalian sirtuin family. SIRT6 is mainly located in the nucleus and regulates chromatin remodeling, genome stability, and gene transcription. SIRT6 extensively participates in various physiological activities such as DNA repair, energy metabolism, oxidative stress, inflammation, and fibrosis. In recent years, the role of epigenetics such as acetylation modification in renal disease has gradually received widespread attention. SIRT6 reduces oxidative stress, inflammation, and renal fibrosis, which is of great importance in maintaining cellular homeostasis and delaying the chronic progression of kidney disease. Here, we review the structure and biological function of SIRT6 and summarize the regulatory mechanisms of SIRT6 in kidney disease. Moreover, the role of SIRT6 as a potential therapeutic target for the progression of kidney disease will be discussed.
Graphical abstract
SIRT6 plays an important role in kidney disease. SIRT6 regulates mitochondrial dynamics and mitochondrial biogenesis, induces G2/M cycle arrest, and plays an antioxidant role in nephrotoxicity, IR, obstructive nephropathy, and sepsis-induced AKI. SIRT6 prevents and delays progressive CKD induced by hyperglycemia, kidney senescence, hypertension, and lipid accumulation by regulating mitochondrial biogenesis, and has antioxidant, anti-inflammatory, and antifibrosis effects. Additionally, hypoxia, inflammation, and fibrosis are the main mechanisms of the AKI-to-CKD transition. SIRT6 plays a critical role in the AKI-to-CKD transition and kidney repair through anti-inflammatory, antifibrotic, and mitochondrial quality control mechanisms. AKI Acute kidney injury, CKD Chronic kidney disease.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The kidney is one of the main energy-consuming organs in the human body [1]. Persistent chronic inflammation and incomplete recovery of kidney function after acute kidney injury (AKI) accelerate the progression to chronic kidney disease (CKD), which ultimately leads to an increased incidence of end-stage renal disease (ESRD) [2]. Under conditions of oxidative stress and aging, the physiological stress response capacity is reduced. The decreased antioxidant capacity exacerbates renal tubular epithelial cell (TEC) damage, vascular endothelial cell activation, and the inflammatory response, promoting abnormal kidney repair and ultimately resulting in irreversible kidney damage [3]. Identifying the key regulatory molecules and effective therapeutic targets is of vital importance in the prevention and treatment of kidney disease.
Mammalian sirtuins (SIRT1-7) are Class III histone deacetylases, which are a highly conserved protein family and are closely related to the development of diseases, including metabolic syndrome, diabetes, cancers, and aging [4, 5]. Among sirtuins, SIRT1, SIRT2, and SIRT3 belong to Class I based on sequence-based phylogenetic analysis, while SIRT4 belongs to Class II, SIRT5 is in Class III, and SIRT6 and SIRT7 are in Class IV [6]. Sirtuins localize in different subcellular compartments and perform different functions. SIRT1, the most well-studied sirtuin, is mainly located in the nucleus. SIRT1 shows strong histone deacetylation activity and shuttles between the nucleus and the cytoplasm, playing a vital role in DNA repair and the stress response [7]. SIRT2 is located in the nucleoplasm and exhibits robust deacetylase activity. SIRT2 mainly regulates the cell cycle and tumorigenesis [8]. SIRT3 is located in mitochondrial matrix, and is a mitochondrial protein deacetylase that regulates mitochondrial dynamics and metabolism [9]. SIRT4 is located in mitochondria, mainly functions as an ADP-ribosyltransferase and has weak substrate-specific deacetylase activity. SIRT4 plays a vital role in metabolism regulation [10]. SIRT5 primarily resides in mitochondria and exerts demalonylase, lysine desuccinylase, and deglutarylase activity, participating in the urea cycle and regulating metabolism [11]. SIRT6 is mainly located in the nucleus, is involved in DNA repair and energy metabolism, and plays a regulatory role in lifespan [12]. SIRT7 localizes in the nucleus and interacts with RNA polymerase I to regulate DNA repair and the aging process [13, 14]. Recently, SIRT6, which is a potential therapeutic target, has gradually drawn attention in maintaining kidney function and has been proven to participate in renal disease by regulating oxidative stress, inflammation, fibrosis, and mitochondrial biosynthesis [15,16,17]. In this review, we first summarize the structure and main biological functions of SIRT6 and then describe the regulatory mechanisms and potential role of SIRT6 as a target in kidney disease.
Structural features and biological functions of SIRT6
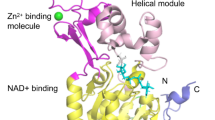
SIRT6 is composed of an N-terminal, a C-terminal, and a conserved central domain. The C terminus is a structural region related to nuclear localization that regulates the positioning of SIRT6. Central domain maintains catalytic activity. The N terminus of SIRT6 binds to the chromosome and contributes to intrinsic catalytic activity, such as regulating H3K9 and H3K56 deacetylation [18]. In contrast to other sirtuin family members, SIRT6 lacks a cofactor-binding loop and has a single helix instead. The crystal structure of SIRT6 contains two globular domains: a splayed zinc-binding domain and a stable single helix for NAD+ binding. Although SIRT6 lacks a conserved and highly flexible NAD+-binding loop, it can bind tightly to NAD+ in the absence of acetylated substrates [19]. SIRT6 possesses both NAD+-dependent protein deacetylase activity and ADP ribosyl transferase activity, participating in gene transcription, metabolism, telomere integrity, and DNA repair [20]. SIRT6 also exerts defatty acylase activity and plays an important role in protein secretion [21]. The unique structure and enzymatic activity of SIRT6 exerts a variety of unique biological effects. SIRT6 is actively recruited to target gene promoters and deacetylates H3K9, H3K18, and H3K56 to maintain cellular homeostasis [22,23,24]. The ADP ribosylation activity of SIRT6 is involved in DNA double-strand break (DSB) repair [25] (Fig. 1).

Structural features and biological functions of SIRT6. A Biological functions of SIRT6. SIRT6 can regulate telomere maintenance, DNA repair, energy metabolism, oxidative stress, the inflammatory response, and fibrosis to maintain cellular homeostasis. B Structural features of SIRT6. SIRT6 is composed of an N terminus (1–24), a C terminus (269–355), and a conserved central domain (25–274) and has a total length of 355 amino acids (aa). The conserved central domain is the main catalytic core, which includes the NAD+-binding Rossmann fold domain (RFD) (25–132 and 195–268) and a zinc-binding domain. Cysteine residues that bind to Zn2+ ions are located at positions 141, 144, 166, and 177. The C terminus is a disordered region that is proline-rich. The main phosphorylation and ubiquitylation sites are highlighted
SIRT6 in cellular homeostasis
As an upstream nucleoprotein, SIRT6 can regulate cell function and survival. SIRT6 plays a critical role in maintaining cellular homeostasis in multiple ways, including telomere maintenance, DNA repair, energy metabolism, oxidative stress, the inflammatory response, and fibrosis [26] (Fig. 1).
Telomere maintenance and DNA repair
Telomeres are specialized DNA protein structures at the end of chromosomal DNA that protect chromosome ends from degradation and the DNA damage response (DDR). Telomeres exhibit structural abnormalities and random loss of telomere sequences when SIRT6 is deficient [27]. SIRT6-deficient cells show base excision repair defects and genomic instability. Upregulation of SIRT6 may improve the ability of base excision repair to combat DNA damage and rescue genome instability [28]. The main types of DNA damage include base deletion, mismatch, DNA cross-linking, and DNA strand breaks that consist of DNA single-strand breaks (SSBs) and DSBs. SSBs occur more frequently in cells, but DSBs are the most toxic form of DNA damage that lead to impaired gene integrity and the subsequent partial loss of the genome [29, 30]. The repair of both SSBs and DSBs is closely related to SIRT6 [31, 32]. SIRT6 directly binds to DSBs and recruits key factors associated with DDR [33]. Upon DSB damage, H2AX is rapidly phosphorylated by ataxia-telangiectasia mutated (ATM) kinase. The phosphorylated histone H2A variant γH2AX anchors to SIRT6 and binds with DNA DSB sites to initiate the cellular DDR [30, 34]. Oxidative stress results in severe DNA damage, especially DSBs [35]. Under oxidative stress conditions, SIRT6 mono-ADP-ribosylates poly (ADP-ribose) polymerase 1 (PARP1) on lysine 521 to stimulate PARP1 activity, which enhances DSB repair [36]. c-Jun N-terminal kinases (JNKs) phosphorylate SIRT6 on serine 10, stimulating SIRT6 mono-ADP-ribosylation of PARP1 and promoting PARP1 recruitment to DNA breaks [37]. SIRT6 can also facilitate the recruitment of DNA repair factors, including Rad51 and NBS1 [38]. Telomere repeat binding factor 2 (TRF2) is involved in telomere maintenance and DDR. SIRT6 interacts with TRF2 and deacetylates the TRFH domain of TRF2, which is then ubiquitylated, activating ubiquitin-dependent proteolysis to regulate its stability [39]. SIRT6 responds to damaged telomeres in the early stage and then recruits MutY homolog (MYH) and Rad9-Rad1-Hus1 (9-1-1) to form the MYH/SIRT6/9-1-1 complex, which is important in DNA repair and maintaining telomere integrity [40]. In response to UV irradiation, SIRT6 binds with and deacetylates damage-specific DNA-binding protein 2 (DDB2) at the lysine residues K35 and K77 and then promotes DDB2 ubiquitination and segregation from chromatin, thereby facilitating nucleotide excision repair signal transduction [41]. SIRT6 mono-ADP ribosylates the lysine demethylase JHDM1A/histone demethylase 2A (KDM2A), which results in rapid displacement of KDM2A from chromatin and increased levels of H3K36me2, which recruits heterochromatin protein 1-alpha (HP1α) and promotes deposition of the H3K9me3 mark, leading to local chromatin compaction [42]. Chromodomain helicase DNA-binding protein 4 (CHD4) is a core subunit of mammalian nucleosome remodeling and the histone deacetylase (NuRD) complex, and is recruited to DNA damage sites. SIRT6 interacts with CHD4 and is required for the recruitment of CHD4 to mediate the DDR [28]. We also verified that SIRT6 alleviates DNA DSBs through the nuclear factor erythroid-related factor 2 (Nrf2)/heme oxygenase 1 (HO-1) pathway [43]. This evidence suggests that SIRT6 plays an important role in telomere maintenance and DNA repair.
Mitochondrial homeostasis
SIRT3 is highly expressed in mitochondria and is the most thoroughly studied mitochondrial sirtuin [44]. SIRT3 regulates mitochondrial energy metabolism by adjusting mitochondrial dynamics through fusion and fission, clearing damaged mitochondria through autophagy and generating new mitochondria through biosynthesis. SIRT3 overexpression activates peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α)-related mitochondrial protection mechanisms and blocks caspase 9-related apoptosis pathways, thus alleviating high glucose-induced endothelial cell injury [45]. In addition, renal ischemia–reperfusion (IR) leads to SIRT3 deficiency. SIRT3 enhances mitochondrial fusion triggered by optical atrophy 1 (OPA1) and thus maintains mitochondrial homeostasis and protects renal TECs from IR injury [46]. This evidence shows that SIRT3 maintains cellular mitochondrial homeostasis. Interestingly, SIRT6 overexpression may regulate mitochondrial homeostasis by cooperating with SIRT3. Under stress conditions, the lack of SIRT3 causes mitochondrial dysfunction and promotes mitochondrial reactive oxygen species (ROS) overproduction accompanied by the downregulation of SIRT6. SIRT3 overexpression significantly upregulates SIRT6 and reverses oxidative stress damage [47]. SIRT3 is an Nrf2-dependent gene, and it has also been proven that SIRT6 can activate Nrf2 to regulate downstream gene expression [48, 49]. SIRT6 promotes the recruitment and activation of RNA polymerase II to Nrf2-regulated antioxidant genes and then exerts antioxidant effects. In addition, SIRT6 inhibits the binding of Kelch-like ECH-associated protein 1 (Keap1) and Nrf2, stabilizes Nrf2 and activates the transcription of the Nrf2-dependent gene SIRT3, further maintaining mitochondrial homeostasis [50]. In addition, SIRT6 plasmid transfection significantly alleviated high glucose-induced mitochondrial defects by activating the AMP-activated protein kinase (AMPK) pathway [51]. In summary, SIRT6 participates in the regulation of mitochondrial function and is critical to mitochondrial homeostasis (Fig. 2).

The role of SIRT6 in the regulation of mitochondrial homeostasis. Under stress conditions, SIRT6 coordinates with SIRT3 to regulate mitochondrial dynamics through fission and fusion. In addition, SIRT6 activates AMPK signaling to promote autophagy and maintain mitochondrial homeostasis. AMPK AMP-activated protein kinase, LC3 Microtubule-associated protein 1 light chain 3, P62/SQSTM1 sequestosome-1, Mfn1/2 Mitofusin 1/2, Fis1 Mitochondrial fission protein 1, Drp1 Dynamin-related protein 1
Energy metabolism
As one of the most basic characteristics of life, energy metabolism has been examined in various research fields, including kidney disease research. Multiple studies have confirmed that SIRT6 acts as a regulator of glucose and lipid metabolism [52, 53]. SIRT6-knockout mice showed gradually increased blood glucose levels and fat mass, indicating the potential role of SIRT6 in regulating metabolism [54]. Next, we will summarize the role of SIRT6 in adjusting energy metabolism, including glucose metabolism and lipid metabolism.
Glucose metabolism
Glucose metabolism is crucial for tissue and organ energy supply. SIRT6 can directly regulate glycometabolism by inhibiting glucose metabolism genes. SIRT6 interacts with hypoxia inducible factor-1α (HIF-1α), deacetylates H3K9 at the HIF-1α promoter, regulates glucose metabolic genes such as phosphofructokinase-1 (PFK1), lactate dehydrogenase (LDH), and pyruvate dehydrogenase kinase (PDK), promotes glycolysis, and regulates mitochondrial respiration [55]. SIRT6 increases the liver gluconeogenic gene and NAD+ from de novo synthesis, and enhances glycerol release from adipose tissue to delay aging [56]. A lack of SIRT6 enhances the membrane association of glucose transporter 1 (GLUT1) and GLUT4, promoting glucose uptake. Simultaneously, SIRT6 regulates AKT signaling, which is negatively correlated with the effects of insulin [57]. Forkhead box protein O1 (FoxO1) and PGC-1α are transcriptional components of the insulin signaling pathway, and play crucial roles in gluconeogenesis. FoxO1, the first identified transcription factor for gluconeogenesis, is activated by PGC-1α. The interaction between FoxO1 and PGC-1α regulates gluconeogenesis [58]. Evidence suggests that statins increase the expression of the SIRT6 inhibitor microRNA (miR)-495 and then acetylate FoxO1, leading to increased gluconeogenesis and hyperglycemia [59]. In addition, SIRT6 controls gluconeogenesis by uniquely upregulating the acetylation of PGC-1α by activating and modifying general control nonrepressed 5 (GCN5) [60].
Lipid metabolism
SIRT6 is also a regulator of lipid metabolism [61]. SIRT6 deficiency results in the upregulation of triglycerides (TGs), cholesterol, and long-chain fatty acid uptake genes, but inhibits β-oxidation [62]. SIRT6 modulates lipid homeostasis by regulating peroxisome proliferator-activated receptor (PPAR) γ-related genes [63]. SIRT6 deficiency enhances the binding rate of the transcription factor PPARγ, thereby promoting fatty acid transporter expression, leading to lipid accumulation and fatty acid uptake [64]. Rosiglitazone (RGZ) is an agonist of PPARγ that can ameliorate hepatic lipid accumulation and increase SIRT6 expression [65, 66]. SIRT6 regulates cholesterol levels by repressing sterol regulatory element-binding proteins 1/2 (SREBP-1/2) and activating AMPK by increasing the AMP/ATP ratio [67]. The proprotein convertase subtilisin/kexin type 9 (PCSK9) gene plays a vital role in regulating LDL cholesterol metabolism. PCSK9 can be regulated by SIRT6 and FoxO3. FoxO3 recruits SIRT6 to the promoter of PCSK9 and deacetylates H3K9 and H3K56 to suppress PCSK9 expression, thus maintaining LDL cholesterol homeostasis [68]. Ubiquitin-specific peptidase 10 (USP10) deficiency induces metabolic dysfunction in high-fat diet (HFD)-treated mice, which can be ameliorated by SIRT6 [69]. SIRT6 can also modulate cholesterol efflux by regulating ATP-binding cassette transporter G1 (ABCG1) expression [70].
Oxidative stress
Under oxidative stress conditions stimulated by pathological factors, excessive ROS are generated. A disruption in the dynamic balance of oxidation and antioxidant capacity causes lipid, protein, and nucleic acid turbulences. Excessive ROS production directly damages tissues and organs, and acts as a second messenger to induce an immune inflammatory response [71]. Excessive ROS also lead to cellular phenotype transformation and induces apoptosis and necrosis [72,73,74]. Nrf2 is a crucial redox-sensitive transcription factor that belongs to the Cap-n-Collar (CNC) protein family, and is widely present in the liver, kidney, lung, and other organs. Nrf2 regulates redox homeostasis by interacting with antioxidant response elements (AREs) [75]. SIRT6 is known to attenuate apoptosis and oxidative stress by activating the Nrf2/ARE signaling pathway [76]. SIRT6 can act as a positive regulator of Nrf2. Studies have shown that SIRT6 activates the Nrf2 signaling pathway, mediates the expression of catalase, HO-1, and other downstream antioxidant proteins, and protects proximal renal TECs from oxidative stress [77]. Low SIRT6 expression is related to oxidative stress in diabetes, and patients with type 2 diabetes who are treated with sodium-dependent glucose transporter 2 (SGLT2) inhibitors have increased SIRT6 expression and reduced oxidative stress [78]. USP10 protects against a variety of environmental stresses, including oxidative stress [79]. USP10 promotes activation of the Nrf2/HO-1 pathway through SIRT6 to reduce oxidative stress and attenuate cell injury [80]. SIRT6 directly interacts with and deacetylates PGC‑1α through the AMPK pathway to maintain mitochondrial homeostasis and oxidative stress [81]. SIRT6 can also reduce oxidative stress by regulating the AKT signaling pathway. SIRT6 directly controls AKT signaling at the chromatin level through H3K9 and H3K56 deacetylation, negatively regulates the level of phosphorylated AKT, and induces autophagy, thus exerting an antioxidant effect [82,83,84]. Moreover, SIRT6 overexpression activates the phosphorylation of AMPKα, increases the levels of FoxO3α, decreases the phosphorylation activity of the protein kinase AKT, and further increases the expression of the downstream antioxidant proteins MnSOD and Catalase [85]. This evidence indicates that SIRT6 can inhibit oxidative stress through multiple pathways and exert a protective effect (Fig. 3).

The role of SIRT6 in the regulation of oxidative stress. ROS overproduction activates JNK, phosphorylates Ser10 on SIRT6, recruits SIRT6 to DNA damage sites, and regulates DNA repair. USP10 promotes activation of the Nrf2/HO-1 pathway through SIRT6, and further alleviates oxidative stress. SIRT6 inhibits AKT signaling and activates AMPK to regulate mitochondrial biogenesis and has an antioxidative effect. JNK Jun-N-terminal kinase, USP10 Ubiquitin-specific peptidase 10, AMPK AMP-activated protein kinase, Nrf2 Nuclear factor erythroid-related factor 2, HO-1 heme oxygenase 1, ROS Reactive oxygen species, FoxO3α Forkhead box O3α, PGC1α Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
Inflammation
SIRT6 also plays a unique regulatory role in the immune inflammatory response. SIRT6 deficiency increases proinflammatory cytokine production and adhesion molecule expression, leading to chronic inflammation and fibrosis [86]. SIRT6 deficiency promotes the expression of nuclear factor kappa-B (NF-κB) which is a well-known inflammatory factor that is widely expressed in cells [87]. The NF-κB transcription factor family can regulate inflammation-related gene expression and affect cellular activities that are closely related to various biological processes [88]. NF-κB activation induces an inflammatory response by promoting the release of inflammatory mediators from monocytes. NF-κB enables downstream pyrin domain containing protein 3 (NLRP3) signaling and tumor necrosis factor alpha (TNF-α) expression, further promoting the release of pro-interleukin-18 (pro-IL-18) and pro-IL-1β and the downstream inflammatory factors IL-1β and IL-18. SIRT6 can act as a negative regulator of NF-κB to regulate inflammation through histone H3K9 deacetylation [89, 90]. Mechanistically, SIRT6 binds to p65/RelA in the NF-κB promoter region, deacetylates histone H3K9, stabilizes RelA on chromatin, and inhibits transcriptional activity to terminate NF-κB signaling [91]. Additionally, SIRT6 plays an anti-inflammatory role by inhibiting the expression of c-JUN-dependent proinflammatory genes monocyte chemotactic protein 1 (MCP1) and IL-6 [92]. Notch signaling exacerbates cell damage by mediating the inflammatory response, apoptosis, and autophagy inhibition, which can be regulated by epigenetic modifications and is a potential target of SIRT6 [93]. A reduction in SIRT6 leads to increased H3K9 acetylation in the Notch1 and Notch4 promoters, and the activation of Notch signaling further exacerbates cell damage [94]. Multiple studies have shown the protective role of SIRT6 in alleviating inflammation, as mentioned previously, and SIRT6 increases TNF-α secretion by removing the fatty acyl modification on K19 and K20 of TNFα [95]. The precise modulatory effect of SIRT6 on inflammation needs further elucidation (Fig. 4).

The role of SIRT6 in the regulation of inflammation and fibrosis. SIRT6 plays a unique role in regulating inflammation and fibrosis. SIRT6 can act as a negative regulator of NF-κB to inhibit NLRP3 and TNF-α expression, and further reduce the release of pro-interleukin-18 (pro-IL-18) and pro-IL-1β and the downstream inflammatory factors IL-1β and IL-18. Additionally, SIRT6 plays an anti-inflammatory role by inhibiting the expression of the c-JUN-dependent proinflammatory genes MCP1 and IL-6. Notch signaling also promotes inflammation and can be negatively regulated by SIRT6. Fibrosis is the main pathological process of various chronic diseases at the end stage and can be driven by inflammation. SIRT6 negatively regulates TGF-β and reduces the activation of downstream Smad signaling. SIRT6 can also negatively regulate Wnt/β-catenin, thus alleviating fibrosis. c-JUN c-Jun-N-terminal kinase, TNF-α Tumor necrosis factor alpha, NF-κB Nuclear factor kappa-B, ECM Extracellular matrix, MCP1 Monocyte chemotactic protein 1
Fibrosis
Fibrosis is driven by inflammation and is the main pathological process of various chronic diseases that progress to an end stage [96]. TGF-β is the main factor involved in the phenotypic transformation of fibroblasts. While participating in cell growth and differentiation, TGF-β also plays an important role in regulating intercellular substance production, apoptosis, and the inflammatory response [97, 98]. TGF-β type II receptor (TGF-βR II) is activated through the autophosphorylation of Ser213 and Ser409, and then activated TGF-βR II interacts with TGF-βR I and enhances the enzymatic activity of TGF-βR I. Activated TGF-βR I further recruits and activates downstream Smad proteins to accumulate in the nucleus and acts as a transcription factor to regulate transcription [99]. In addition, TGF-β can also activate MAPK, PI3K–AKT, and PAK2 signaling molecules through nonclassical pathways to regulate fibrosis [100,101,102]. Wnt is a crucial pathway for regulating epithelial–mesenchymal transition (EMT). Wnt/β-catenin signaling regulates cell differentiation and regeneration and cross-linking with TGF-β and Notch signals [103]. TGF-β can activate the classic Wnt signaling pathway by downregulating the expression of the Wnt signaling pathway antagonist Dickkopf 1 (DKK1). On one hand, SIRT6 negatively regulates TGF-β, reduces the activation of downstream Smad signaling, and alleviates fibrosis [104]. On the other hand, SIRT6 interacts with β-catenin to regulate TGF-β, binds to the β-catenin promoter, and causes the deacetylation of histone H3K56, thereby preventing the transcription of genes related to fibrosis [16]. Some studies have proven that SIRT6 can directly or indirectly influence Notch signaling factor expression [105, 106]. The activation of Notch signaling induces TEC dedifferentiation and renal fibroblast proliferation, thus promoting renal fibrosis [107]. The interaction between Wnt and Notch signaling is critical for maintaining cellular function. Inhibiting the Wnt signaling pathway can restore the phenotype induced by the blockade of Notch signaling [108]. Collectively, SIRT6 can serve as a therapeutic target for fibrosis (Fig. 4).
SIRT6 in kidney disease
The high incidence of kidney disease remains a challenge worldwide in public health management. Exploring the pathogenesis and preventive mechanisms will help us to find better ways to treat kidney disease. SIRT6 has been proven to be involved in the progression of kidney disease. The potential role of SIRT6 in kidney disease needs to be further studied.
CKD
Diabetic kidney disease (DKD)
DKD is a common microvascular disease and one of the most serious chronic complications of diabetes [109]. With the increasing incidence of diabetes, DKD has become the main cause of ESRD [110]. The pathogenesis of DKD is complex. It is currently believed that genetic factors, hemodynamic changes, oxidative stress, inflammation, and mitochondrial damage jointly participate in the occurrence and development of DKD [111,112,113]. High glucose induces mitochondrial superoxide production in podocytes, further promoting mitochondrial dysfunction associated with mitochondrial morphological alterations and decreased mitochondrial membrane potential [114]. SIRT6 upregulation by plasmid transfection can protect mitochondrial function and alleviate oxidative stress by increasing AMPK phosphorylation, indicating that SIRT6 protects mitochondria and exerts antiapoptotic effects by activating the AMPK pathway [51]. Inflammation is also one of the main pathological features of DKD [115]. The mRNA levels of the inflammation-related factors IL-1β, IL-6, and TNF-α in podocytes were reduced by SIRT6. High glucose can activate the Notch signaling pathway in podocytes, and podocyte-specific overexpression of the intracellular domain of Notch1 (ICN1) induces proteinuria and glomerulosclerosis [116]. Activation of the Notch signaling pathway leads to endocytosis of nephrin and podocin, thereby destroying the structure of the podocyte split membrane and inducing proteinuria [117, 118]. SIRT6 protects against podocyte inflammation through epigenetic regulation of the Notch signaling pathway, suggesting that SIRT6 is a potential therapeutic target to protect podocytes from high glucose-induced injury. In brief, SIRT6 regulates the action of H3K9 deacetylation and binds to the promoter regions of Notch1 and Notch4 to inhibit transcription and activation of the downstream PTEN signaling pathway, further increasing autophagic flux and alleviating apoptosis and inflammation in podocytes [93]. In addition, SIRT6 protects podocytes against DKD by activating M2 macrophage transformation and acts as an immune regulator in inflammatory injury [119]. In addition, the NMN-producing enzyme nicotinamide phosphoribosyl-transferase (Nampt) has been proven to have a protective role in DKD. Proximal tubule Nampt-specific knockout mice showed SIRT6 downregulation, resulting in collagen deposition and a fibrotic phenotype, suggesting the protective role of Nampt-SIRT6 signaling in DKD [120]. This evidence shows that SIRT6 plays a protective role in high glucose-induced renal injury by reducing oxidative stress, mitochondrial damage, and inflammation, suggesting that SIRT6 could be a potential therapeutic target for preventing and delaying the progression of DKD.
Hypertensive kidney lesion
Hypertension is a chronic disease characterized by elevated systemic blood pressure and is considered to be a vital risk factor for coronary heart disease and CKD [121]. Endothelial dysfunction is associated with the occurrence of hypertension and is the main cause of hypertension-induced injury [122]. Although multiple pathogenic factors in hypertension have been revealed, the precise pathogenesis remains unclear. Previous studies have showed that hypertension reduces the level of the endothelium-dependent vasodilator nitric oxide, changes the permeability of endothelial cells, promotes the production of endothelin 1 (ET-1) and angiotensin II (Ang II), and then exacerbates target organ damage, including renal and cardiovascular injury [123]. The sirtuin family is involved in pathological processes associated with regulating blood pressure, fibrotic remodeling, and cell apoptosis [70, 124, 125]. As recently revealed, SIRT6 delays vascular aging and prevents hypertension by maintaining endothelial homeostatic functions [126]. MiR-122, acting as a risk biomarker of vascular fibrosis, has been confirmed to participate in the development of hypertension by inducing endothelial dysfunction [127]. SIRT6 is directly targeted by miR-122. Activation of the SIRT6-ELA pathway inhibits miR-122 and alleviates vascular oxidative injury and subsequent inflammation, negatively regulating Ang II-induced hypertension [128]. Additionally, SIRT6 overexpression weakens Ang II-induced apoptosis and oxidative stress in vascular cells by promoting Nrf2 signaling pathway activation [76]. SIRT6 also induces the expression of the blood pressure-related gene GATA5 by inhibiting the transcription of Nkx3.2, which is essential for endothelial homeostasis and protects vascular endothelial cells against hypertension and related organ injury [124]. Ang II induces cholesterol accumulation in podocytes and promotes CKD progression. Specific deletion of SIRT6 in podocytes exacerbates Ang II-induced kidney injury and affects cholesterol efflux by regulating the expression of ABCG1, suggesting that SIRT6 plays a protective role in the regulation of cholesterol metabolism in podocytes [70]. These studies indicate the unique role of SIRT6 signaling in the regulation of blood pressure and kidney injury.
Kidney senescence
Aging is an irreversible process and an important risk factor for multiple diseases [129]. Cellular senescence occurs in all renal cells, including TECs, mesangial cells, and podocytes. Age-related disruptions in kidney disease are associated with cellular senescence [130]. In kidney disease, age-related disruptions induce renal fibrosis and diminish glomerular filtration, decreasing kidney function. The reduction in renoprotective factors exacerbates cellular senescence. Mitochondrial ROS (mtROS) production drives stress-induced senescence and further leads to chronic inflammation and renal dysfunction, particularly in renal tubular cells [131]. Among the sirtuin members, SIRT6 has been implicated in regulating cellular senescence by attenuating inflammation, maintaining telomere integrity, and participating in DNA repair [132, 133]. SIRT6 knockdown results in activation of the NF-κB signaling pathway and accelerates cellular senescence. Caloric restriction (CR) significantly enhances SIRT6 expression and reverses age-dependent renal insufficiency [134]. In addition, SIRT6 maintains podocyte homeostasis and plays an important role in aging-associated glomerular function. SIRT6-deficient aging mice exhibit chronic inflammation and fibrosis and the loss of glomerular function [135]. This evidence supports the protective role of SIRT6 in renal cellular senescence.
Renal cancer
As a specific type of longevity gene, SIRT6 is considered as a tumor suppressor in renal cell carcinoma (RCC) [136]. SIRT6 is upregulated in RCC. Silencing SIRT6 expression promotes G1/S phase arrest and suppresses tumor growth [137]. The expression level of SIRT6 depends on tumor stage and histological grade and further corelates with prognosis in clear cell renal cell carcinoma (ccRCC) [138]. SIRT6 inhibition increases the sensitivity of ccRCC to cisplatin, which is strongly associated with Bcl-2 and BAX expression, and further initiates apoptosis-related processes [139].
AKI
Unilateral ureteral obstruction (UUO)
Obstructive nephropathy is a severe health problem and a critical factor in the development of CKD worldwide [140]. Persistent obstruction leads to renal pelvic effusion and irreversible kidney damage. UUO is characterized by progressive fibrosis [141, 142]. After UUO, tubular and interstitial cells release damage-associated molecular patterns (DAMPs), which are recognized by pattern recognition receptors (PRRs) and mediate the immune response, leading to inflammatory cell infiltration, increased levels of profibrotic factors, and matrix deposition [143]. SIRT6 deficiency specifically increases H3K56 acetylation at the promoter region of β-catenin, amplifies fibrosis-related protein expression, and exacerbates UUO-induced renal injury and fibrosis. The interaction between SIRT6 and TGF-β weakens the expression of β-catenin target genes and plays an antifibrotic role [16]. In addition, SIRT6 reduces inflammation by negatively regulating the NF-κB signaling pathway and synergistically regulates chronic renal fibrosis in ureteral obstruction [144]. In addition, accumulating evidence has confirmed that inflammation and fibrosis induced by UUO are closely related to mitochondrial dysfunction [145]. However, there is still no clear evidence indicating that SIRT6 alleviates UUO-induced AKI by regulating mitochondrial function. Based on this evidence, we conclude that SIRT6 ameliorates inflammation and fibrosis, and may be a therapeutic target for UUO.
Cisplatin-induced kidney damage
Cisplatin is an effective chemotherapeutic drug and is widely used to treat solid tumors, but severe side effects limit its applications [146]. Consecutive cisplatin administration results in irreversible nephrotoxicity [147]. Tubulointerstitial injury is the main pathological characteristic of cisplatin-induced renal injury. The accumulation of cisplatin in proximal tubular cells results in apoptosis and necrosis [148]. Nrf2 has been proven to play a significant role in cisplatin-induced renal injury by eliminating oxidative stress and subsequent inflammation mediated by NF-κB signaling [149]. SIRT6 is a positive regulator of the Nrf2 signaling pathway and acts as an antioxidative agent in cisplatin-induced renal injury [77]. Daphnetin, a natural coumarin, acts as an antioxidant in cisplatin-induced kidney damage through SIRT1/SIRT6-Nrf2 activation [77]. SIRT6 deficiency worsens cisplatin-mediated proximal tubular cell apoptosis, while SIRT6 overexpression inhibits extracellular signal-regulated kinase 1 (ERK1) and ERK2 expression, further inhibiting NF-κB signaling, suggesting that SIRT6 acts as an inhibitor of cisplatin-induced nephrotoxicity [151]. Taken together, this evidence hints at the protective role of SIRT6 in cisplatin-induced kidney damage, and SIRT6 may be a therapeutic target for nephrotoxic drug-induced kidney injury.
Other AKI models
In addition to kidney injury models, as mentioned previously, SIRT6 also plays a vital role in sepsis-induced AKI and cadmium-induced kidney toxicity. Sepsis caused by infection is a life-threatening condition and a crucial cause of AKI [152]. In the lipopolysaccharide (LPS)-induced sepsis AKI model, SIRT6 overexpression alleviated the LPS-induced inflammatory response and apoptosis in epithelial cells by promoting autophagy [153]. Cadmium exposure is a high-risk factor for kidney disease. Continuous exposure to cadmium increases the incidence of apoptosis and necrosis in proximal tubular cells [154]. SIRT6 has been shown to regulate cell apoptosis induced by cadmium through the polyubiquitinated (polyUb)-p62/SIRT6 signaling pathway [155]. These findings confirm that SIRT6 promotes autophagy and alleviates apoptosis and inflammation under stress conditions [156, 157].
SIRT6 in the AKI-to-CKD transition and kidney repair
AKI is a common complication among hospitalized patients and has become a growing public health problem associated with a high risk of developing CKD. Although the kidney has a strong compensatory ability, studies have shown that AKI causes irreversible microvasculature damage and impairs kidney structure and function [158, 159]. The incomplete recovery of renal function after AKI causes persistent chronic inflammation and fibrosis, which eventually progresses to ESRD [160]. The molecular mechanisms of the conversion of AKI to CKD are complicated. It is currently recognized that proximal tubular cell injury is the main pathological feature of chronic progression of AKI due to high energy demands [161, 162]. Proximal tubular cells are rich in mitochondria, which are sensitive to hypoxia and easily perceive changes in energy metabolism [163]. Persistent renal tubular injury caused by ischemia and hypoxia, mitochondrial dysfunction, and inflammation contribute to metabolic constraints and induce cytoskeletal rearrangement, extracellular matrix (ECM), and EMT in TECs. ECM accumulation and EMT account for tubulointerstitial fibrosis (TIF), leading to the development of CKD, and are considered to be the common pathway to ESRD [95, 164, 165]. SIRT6 has been shown to have a regulatory role in the progression of kidney disease. However, the current understanding of the pathological process, mechanisms of action, and clinical applications of SIRT6 in kidney disease, especially chronic progression, is still limited. SIRT6 may serve as a therapeutic target, and finding suitable treatments to prevent the chronic progression of kidney disease is critical. Here, we summarize the potential role of SIRT6 in the AKI-to-CKD transition and kidney repair.
Hypoperfusion after AKI causes persistent tissue ischemia and hypoxia, further leading to vascular endothelial cell damage, microvascular reductions, and proximal tubular cell injury [166]. Hypoxia leads to insufficient energy supply and inflammatory factor production in renal tubule epithelial cells. After renal tubule injury, the remaining epithelial cells enter the cell cycle and participate in regeneration through dedifferentiation and proliferation under the action of growth factors and chemokines [167]. However, in severe ischemia, hypoxia, persistent exposure to nephrotoxic drugs and inflammation, cell cycle arrest, and TGF-β activate profibrotic factor release, resulting in the accumulation of ECM [168, 169]. In addition, inflammatory factors initiate EMT, which promotes renal interstitial fibrosis [170, 171]. Hypoxia reduces the expression of SIRT6 in renal TECs. SIRT6 deficiency exacerbates hypoxia-induced inflammation and G2/M cycle arrest in renal TECs, which can be reversed by upregulating SIRT6, suggesting that SIRT6 protects renal TECs from hypoxia-induced tubular interstitial injury [172].
Mitochondria are central hubs that maintain cellular and redox homeostasis. The loss of mitochondrial quality control is the main mediator of incomplete repair after AKI [173]. ROS overproduction induces renal tubular cell injury and nephron dropout, further impairing mitochondrial structural integrity. Nrf2, acting as an antioxidant, has been proven to regulate mitochondrial quality control by binding to the promoter region of PINK1 [174]. SIRT6 activates Nrf2 and prevents Keap1 proteasomal degradation, increasing mitochondrial biogenesis, mitophagy, and the mitochondrial antioxidant response [49]. Additionally, the activation of AMPK stimulates downstream AKT signaling, phosphorylates FoxO3α, and further attenuates mitochondrial dysfunction [175]. SIRT6 promotes autophagy-related protein expression and maintains mitochondrial function by AMPK signaling pathway activation under oxidative stress conditions [176]. Collectively, mitochondrial quality control shows a protective role in kidney repair after AKI. SIRT6 increases the ROS-scavenging capacity and maintains mitochondrial quality, which is critical in AKI and kidney repair.
Lipid accumulation is involved in the progression of kidney disease [177]. The enzymes associated with fatty acid oxidation (FAO) are reduced in kidney fibrosis models, and restoring FAO through genetics may prevent the progression of CKD [178]. CK2 activity is regulated by SIRT6 and plays an important role in adipogenesis [179]. SIRT6 expression is decreased in obese pre-DM subjects, while the expression of NF-κB, PPAR-γ and SREBP-1 is increased. Of note, these effects can be reversed by metformin treatment [180].
Fibrosis is the common pathway associated with irreversible and progressive processes causing chronic development of kidney disease. TIF is mainly triggered by persistent chronic inflammation and fibrillary collagen accumulation. Increased proinflammatory cytokines interfere with intrarenal microcirculation and perfusion, contributing to EMT [181]. Activation of the inflammatory response-associated transcription factors NF-κB and STAT promotes tubulointerstitial inflammation and kidney fibrosis, and inhibiting these factors can ameliorate kidney fibrosis [182]. The TGF-β signaling pathway is the central mediator of renal fibrosis in progressive CKD. TGF-β promotes EMT and inhibits ECM degradation in renal tubular cells during the progression of CKD [183]. Wnt/β-catenin signaling regulates fibrosis and participates in the progression of CKD. β-Catenin overexpression in tubular cells induces epithelial dedifferentiation and EMT in mice [184]. The Notch signaling pathway also plays an important role in orchestrating the development of kidney disease. Notch expression in podocytes promotes glomerulosclerosis and albuminuria. Additionally, Notch expression in TECs promotes EMT-related snail1 and snail2 expression, thus contributing to TIF [185]. It is worth noting that the inflammatory response is also regulated by epigenetics. SIRT6 depletion induces chronic inflammation and fibrosis in the kidney and eventually leads to podocyte depletion, proteinuria, and the loss of kidney function. Studies have confirmed that SIRT6 negatively regulates the TGF-β and Wnt/β-catenin signaling pathways and plays an antifibrotic role [16]. In addition, high mobility group box 1 (HMGB1) exacerbates CKD progression by promoting vascular calcification. Bone marrow mesenchymal stem cell (BMSC)-derived exosomes downregulate HMGB1 expression through the SIRT6–HMGB1 pathway and ameliorate CKD-related fibrosis [17].
Overall, in-depth investigation of energy metabolism, inflammatory response, and fibrosis inhibition in kidney disease is essential in identifying specific and efficacious approaches for disease transition.
Conclusion and prospects
As a member of the sirtuin family, SIRT6 maintains intracellular homeostasis and is considered to be a powerful regulator of disease occurrence and development. In this review, we summarize the structure and biological function of SIRT6. We further summarize the regulatory mechanisms and potential roles of SIRT6 in multiple kidney diseases (Table 1). SIRT6 has shown a powerful regulatory effect on DNA repair, energy metabolism, oxidative stress, mitochondrial homeostasis, inflammation, fibrosis, and aging. Mechanistically, SIRT6 regulates transcription factors by deacetylating of histone H3K9, H3K56, and H3K18 on target gene promoters and controls downstream gene expression, thus maintaining cellular homeostasis. In kidney disease, SIRT6 regulates oxidative stress under hypoxia and stress conditions by regulating the Nrf2, AMPK, and AKT signaling pathways. SIRT6 participates in the pathogenesis of chronic inflammation and renal fibrosis by regulating the TGF-β1/Smad3, Wnt/β-catenin, NF-κB, β-catenin, and Notch signaling pathways. Moreover, SIRT6 synergistically maintains the content and integrity of mitochondria. These regulatory mechanisms are closely related to renal repair and survival. In-depth study of the regulatory mechanism of SIRT6 will help to identify new targets for kidney disease. Further exploration of the characteristics of SIRT6 has potential value and provides new ideas for the treatment of the chronic progression of kidney disease. In summary, focusing on SIRT6 as a target has important clinical significance for the prevention and treatment of kidney disease.
Availability of data and materials
Not applicable.
References
Bhargava P, Schnellmann RG (2017) Mitochondrial energetics in the kidney. Nat Rev Nephrol 13(10):629–646. https://doi.org/10.1038/nrneph.2017.107
Ferenbach DA, Bonventre JV (2015) Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11(5):264–276. https://doi.org/10.1038/nrneph.2015.3
Kim MG, Yang J, Ko YS, Lee HY, Oh SW, Cho WY, Jo SK (2019) Impact of aging on transition of acute kidney injury to chronic kidney disease. Sci Rep 9(1):18445. https://doi.org/10.1038/s41598-019-54585-1
Morigi M, Perico L, Benigni A (2018) Sirtuins in renal health and disease. J Am Soc Nephrol 29(7):1799–1809. https://doi.org/10.1681/ASN.2017111218
Hershberger KA, Martin AS, Hirschey MD (2017) Role of NAD and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol 13(4):213–225. https://doi.org/10.1038/nrneph.2017.5
Frye RA (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273(2):793–798. https://doi.org/10.1006/bbrc.2000.3000
Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y (2007) Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J Biol Chem 282(9):6823–6832. https://doi.org/10.1074/jbc.M609554200
Hamaidi I, Zhang L, Kim N, Wang MH, Iclozan C, Fang B, Liu M, Koomen JM, Berglund AE, Yoder SJ, Yao J, Engelman RW, Creelan BC, Conejo-Garcia JR, Antonia SJ, Mulé JJ, Kim S (2020) Sirt2 inhibition enhances metabolic fitness and effector functions of tumor-reactive T cells. Cell Metab 32(3):420–436. https://doi.org/10.1016/j.cmet.2020.07.008
Samant SA, Zhang HJ, Hong Z, Pillai VB, Sundaresan NR, Wolfgeher D, Archer SL, Chan DC, Gupta MP (2014) SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol Cell Biol 34(5):807–819. https://doi.org/10.1128/MCB.01483-13
Wang YS, Du L, Liang X, Meng P, Bi L, Wang YL, Wang C, Tang B (2019) Sirtuin 4 depletion promotes hepatocellular carcinoma tumorigenesis through regulating adenosine-monophosphate-activated protein kinase alpha/mammalian target of rapamycin axis in mice. Hepatology 69(4):1614–1631. https://doi.org/10.1002/hep.30421
Zhang Y, Bharathi SS, Rardin MJ, Lu J, Maringer KV, Sims-Lucas S, Prochownik EV, Gibson BW, Goetzman ES (2017) Lysine desuccinylase SIRT5 binds to cardiolipin and regulates the electron transport chain. J Biol Chem 292(24):10239–10249. https://doi.org/10.1074/jbc.M117.785022
Gertler AA, Cohen HY (2013) SIRT6, a protein with many faces. Biogerontology 14(6):629–639. https://doi.org/10.1007/s10522-013-9478-8
Yu AQ, Wang J, Jiang ST, Yuan LQ, Ma HY, Hu YM, Han XM, Tan LM, Wang ZX (2021) SIRT7-induced PHF5A decrotonylation regulates aging progress through alternative splicing-mediated downregulation of CDK2. Front Cell Dev Biol 9:710479. https://doi.org/10.3389/fcell.2021.710479
Ford E, Voit R, Liszt G, Magin C, Grummt I, Guarente L (2006) Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev 20(9):1075–1080. https://doi.org/10.1101/gad.1399706
Gewin LS (2020) Sirtuin 6 and renal injury: another link in the β-catenin chain? Kidney Int 97(1):24–27. https://doi.org/10.1016/j.kint.2019.09.022
Cai J, Liu Z, Huang X, Shu S, Hu X, Zheng M, Tang C, Liu Y, Chen G, Sun L, Liu H, Liu F, Cheng J, Dong Z (2020) The deacetylase sirtuin 6 protects against kidney fibrosis by epigenetically blocking β-catenin target gene expression. Kidney Int 97(1):106–118. https://doi.org/10.1016/j.kint.2019.08.028
Wei W, Guo X, Gu L, Jia J, Yang M, Yuan W, Rong S (2021) Bone marrow mesenchymal stem cell exosomes suppress phosphate-induced aortic calcification via SIRT6-HMGB1 deacetylation. Stem Cell Res Ther 12(1):235. https://doi.org/10.1186/s13287-021-02307-8
Tennen RI, Berber E, Chua KF (2010) Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech Ageing Dev 131(3):185–192. https://doi.org/10.1016/j.mad.2010.01.006
Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM (2011) Structure and biochemical functions of SIRT6. J Biol Chem 286(16):14575–14587. https://doi.org/10.1074/jbc.M111.218990
Chang AR, Ferrer CM, Mostoslavsky R (2020) SIRT6, a mammalian deacylase with multitasking abilities. Physiol Rev 100(1):145–169. https://doi.org/10.1152/physrev.00030.2018
Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H (2013) SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496(7443):110–113. https://doi.org/10.1038/nature12038
Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, Chang HY, Bohr VA, Ried T, Gozani O, Chua KF (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452(7186):492–496. https://doi.org/10.1038/nature06736
Michishita E, McCord RA, Boxer LD, Barber MF, Hong T, Gozani O, Chua KF (2009) Cell cycle-dependent deacetylation of telomeric histone H3 lysine K56 by human SIRT6. Cell Cycle 8(16):2664–2666. https://doi.org/10.4161/cc.8.16.9367
Tasselli L, Xi Y, Zheng W, Tennen RI, Odrowaz Z, Simeoni F, Li W, Chua KF (2016) SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat Struct Mol Biol 23(5):434–440. https://doi.org/10.1038/nsmb.3202
Liu G, Chen H, Liu H, Zhang W, Zhou J (2021) Emerging roles of SIRT6 in human diseases and its modulators. Med Res Rev 41(2):1089–1137. https://doi.org/10.1002/med.21753
Kugel S, Mostoslavsky R (2014) Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem Sci 39(2):72–81. https://doi.org/10.1016/j.tibs.2013.12.002
Tennen RI, Bua DJ, Wright WE, Chua KF (2011) SIRT6 is required for maintenance of telomere position effect in human cells. Nat Commun 2:433. https://doi.org/10.1038/ncomms1443
Hou T, Cao Z, Zhang J, Tang M, Tian Y, Li Y, Lu X, Chen Y, Wang H, Wei FZ, Wang L, Yang Y, Zhao Y, Wang Z, Wang H, Zhu WG (2020) SIRT6 coordinates with CHD4 to promote chromatin relaxation and DNA repair. Nucleic Acids Res 48(6):2982–3000. https://doi.org/10.1093/nar/gkaa006
Aleksandrov R, Hristova R, Stoynov S, Gospodinov A (2020) The chromatin response to double-strand DNA breaks and their repair. Cells 9(8):1853. https://doi.org/10.3390/cells9081853
Onn L, Portillo M, Ilic S, Cleitman G, Stein D, Kaluski S, Shirat I, Slobodnik Z, Einav M, Erdel F, Akabayov B, Toiber D (2020) SIRT6 is a DNA double-strand break sensor. Elife 9:e51636. https://doi.org/10.7554/eLife.51636
Koczor CA, Saville KM, Andrews JF, Clark J, Fang Q, Li J, Al-Rahahleh RQ, Ibrahim M, McClellan S, Makarov MV, Migaud ME, Sobol RW (2021) Temporal dynamics of base excision/single-strand break repair protein complex assembly/disassembly are modulated by the PARP/NAD+/SIRT6 axis. Cell Rep 37(5):109917. https://doi.org/10.1016/j.celrep.2021.109917
Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, Alt FW (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124(2):315–329. https://doi.org/10.1016/j.cell.2005.11.044
Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, Sebastian C, Cosentino C, Martinez-Pastor B, Giacosa S, D’Urso A, Näär AM, Kingston R, Rippe K, Mostoslavsky R (2013) SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell 51(4):454–468. https://doi.org/10.1016/j.molcel.2013.06.018
Meng F, Qian M, Peng B, Peng L, Wang X, Zheng K, Liu Z, Tang X, Zhang S, Sun S, Cao X, Pang Q, Zhao B, Ma W, Songyang Z, Xu B, Zhu WG, Xu X, Liu B (2020) Synergy between SIRT1 and SIRT6 helps recognize DNA breaks and potentiates the DNA damage response and repair in humans and mice. Elife 9:e55828. https://doi.org/10.7554/eLife.55828
Bakewell S, Conde I, Fallah Y, McCoy M, Jin L, Shajahan-Haq AN (2020) Inhibition of DNA repair pathways and induction of ROS are potential mechanisms of action of the small molecule inhibitor BOLD-100 in breast cancer. Cancers (Basel) 12(9):2647. https://doi.org/10.3390/cancers12092647
Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science 332(6036):1443–1446. https://doi.org/10.1126/science.1202723
Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, Bredbenner K, Park R, Sinclair DA, Bohr VA, Gorbunova V, Seluanov A (2016) JNK phosphorylates SIRT6 to stimulate DNA double-strand break repair in response to oxidative stress by recruiting PARP1 to DNA breaks. Cell Rep 16(10):2641–2650. https://doi.org/10.1016/j.celrep.2016.08.006
Mao Z, Tian X, Van Meter M, Ke Z, Gorbunova V, Seluanov A (2012) Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci USA 109(29):11800–11805. https://doi.org/10.1073/pnas.1200583109
Rizzo A, Iachettini S, Salvati E, Zizza P, Maresca C, D’Angelo C, Benarroch-Popivker D, Capolupo A, Del Gaudio F, Cosconati S, Di Maro S, Merlino F, Novellino E, Amoreo CA, Mottolese M, Sperduti I, Gilson E, Biroccio A (2017) SIRT6 interacts with TRF2 and promotes its degradation in response to DNA damage. Nucleic Acids Res 45(4):1820–1834. https://doi.org/10.1093/nar/gkw1202
Tan J, Wang X, Hwang BJ, Gonzales R, Konen O, Lan L, Lu AL (2020) An ordered assembly of MYH glycosylase, SIRT6 protein deacetylase, and Rad9-Rad1-Hus1 checkpoint clamp at oxidatively damaged telomeres. Aging (Albany NY) 12(18):17761–17785. https://doi.org/10.18632/aging.103934
Geng A, Tang H, Huang J, Qian Z, Qin N, Yao Y, Xu Z, Chen H, Lan L, Xie H, Zhang J, Jiang Y, Mao Z (2020) The deacetylase SIRT6 promotes the repair of UV-induced DNA damage by targeting DDB2. Nucleic Acids Res 48(16):9181–9194. https://doi.org/10.1093/nar/gkaa661
Rezazadeh S, Yang D, Biashad SA, Firsanov D, Takasugi M, Gilbert M, Tombline G, Bhanu NV, Garcia BA, Seluanov A, Gorbunova V (2020) SIRT6 mono-ADP ribosylates KDM2A to locally increase H3K36me2 at DNA damage sites to inhibit transcription and promote repair. Aging (Albany NY) 12(12):11165–11184. https://doi.org/10.18632/aging.103567
Fan Y, Cheng J, Yang Q, Feng J, Hu J, Ren Z, Yang H, Yang D, Ding G (2021) Sirt6-mediated Nrf2/HO-1 activation alleviates angiotensin II-induced DNA DSBs and apoptosis in podocytes. Food Funct 12(17):7867–7882. https://doi.org/10.1039/d0fo03467c
Wang S, Zhang J, Deng X, Zhao Y, Xu K (2020) Advances in characterization of SIRT3 deacetylation targets in mitochondrial function. Biochimie 179:1–13. https://doi.org/10.1016/j.biochi.2020.08.021
Wang Y, Zhang X, Wang P, Shen Y, Yuan K, Li M, Liang W, Que H (2019) Sirt3 overexpression alleviates hyperglycemia-induced vascular inflammation through regulating redox balance, cell survival, and AMPK-mediated mitochondrial homeostasis. J Recept Signal Transduct Res 39(4):341–349. https://doi.org/10.1080/10799893.2019.1684521
Wang Q, Xu J, Li X, Liu Z, Han Y, Xu X, Li X, Tang Y, Liu Y, Yu T, Li X (2019) Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J Cell Physiol 234(12):23495–23506. https://doi.org/10.1002/jcp.28918
Kanwal A, Pillai VB, Samant S, Gupta M, Gupta MP (2019) The nuclear and mitochondrial sirtuins, Sirt6 and Sirt3, regulate each other’s activity and protect the heart from developing obesity-mediated diabetic cardiomyopathy. FASEB J 33(10):10872–10888. https://doi.org/10.1096/fj.201900767R
Pan H, Guan D, Liu X, Li J, Wang L, Wu J, Zhou J, Zhang W, Ren R, Zhang W, Li Y, Yang J, Hao Y, Yuan T, Yuan G, Wang H, Ju Z, Mao Z, Li J, Qu J, Tang F, Liu GH et al (2016) SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell res 26(2):190–205. https://doi.org/10.1038/cr.2016.4
Zhang W, Wei R, Zhang L, Tan Y, Qian C (2017) Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience 366:95–104. https://doi.org/10.1016/j.neuroscience.2017.09.035
Kasai S, Shimizu S, Tatara Y, Mimura J, Itoh K (2020) Regulation of Nrf2 by mitochondrial reactive oxygen species in physiology and pathology. Biomolecules 10(2):320. https://doi.org/10.3390/biom10020320
Fan Y, Yang Q, Yang Y, Gao Z, Ma Y, Zhang L, Liang W, Ding G (2019) Sirt6 suppresses high glucose-induced mitochondrial dysfunction and apoptosis in podocytes through AMPK activation. Int J Biol Sci 15(3):701–713. https://doi.org/10.7150/ijbs.29323
Li Z, Xu K, Guo Y, Ping L, Gao Y, Qiu Y, Ni J, Liu Q, Wang Z (2020) A high-fat diet reverses metabolic disorders and premature aging by modulating insulin and IGF1 signaling in SIRT6 knockout mice. Aging Cell 19(3):e13104. https://doi.org/10.1111/acel.13104
Kuang J, Chen L, Tang Q, Zhang J, Li Y, He J (2018) The role of Sirt6 in obesity and diabetes. Front Physiol 9:135. https://doi.org/10.3389/fphys.2018.00135
Song MY, Kim SH, Ryoo GH, Kim MK, Cha HN, Park SY, Hwang HP, Yu HC, Bae EJ, Park BH (2019) Adipose sirtuin 6 drives macrophage polarization toward M2 through IL-4 production and maintains systemic insulin sensitivity in mice and humans. Exp Mol Med 51(5):1–10. https://doi.org/10.1038/s12276-019-0256-9
Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, Kurland I, Dor Y, Weissleder R, Shirihai OS, Ellisen LW, Espinosa JM, Mostoslavsky R (2010) The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140(2):280–293. https://doi.org/10.1016/j.cell.2009.12.041
Roichman A, Elhanati S, Aon MA, Abramovich I, Di Francesco A, Shahar Y, Avivi MY, Shurgi M, Rubinstein A, Wiesner Y, Shuchami A, Petrover Z, Lebenthal-Loinger I, Yaron O, Lyashkov A, Ubaida-Mohien C, Kanfi Y, Lerrer B, Fernández-Marcos PJ, Serrano M, Gottlieb E, de Cabo R, Cohen HY et al (2021) Restoration of energy homeostasis by SIRT6 extends healthy lifespan. Nat Commun 12(1):3208. https://doi.org/10.1038/s41467-021-23545-7
Xiao C, Kim HS, Lahusen T, Wang RH, Xu X, Gavrilova O, Jou W, Gius D, Deng CX (2010) SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. J Biol Chem 285(47):36776–36784. https://doi.org/10.1074/jbc.M110.168039
Gu L, Ding X, Wang Y, Gu M, Zhang J, Yan S, Li N, Song Z, Yin J, Lu L, Peng Y (2019) Spexin alleviates insulin resistance and inhibits hepatic gluconeogenesis via the FoxO1/PGC-1α pathway in high-fat-diet-induced rats and insulin resistant cells. Int J Biol Sci 15(13):2815–2829. https://doi.org/10.7150/ijbs.31781
Shi MY, Bang IH, Han CY, Lee DH, Park BH, Bae EJ (2020) Statin suppresses sirtuin 6 through miR-495, increasing FoxO1-dependent hepatic gluconeogenesis. Theranostics 10(25):11416–11427. https://doi.org/10.7150/thno.49770
Dominy JE Jr, Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, Ruan HB, Feldman J, Pierce K, Mostoslavsky R, Denu JM, Clish CB, Yang X, Shulman GI, Gygi SP, Puigserver P (2012) The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell 48(6):900–913. https://doi.org/10.1016/j.molcel.2012.09.030
Kuang J, Zhang Y, Liu Q, Shen J, Pu S, Cheng S, Chen L, Li H, Wu T, Li R, Li Y, Zou M, Zhang Z, Jiang W, Xu G, Qu A, Xie W, He J (2017) Fat-specific Sirt6 ablation sensitizes mice to high-fat diet-induced obesity and insulin resistance by inhibiting lipolysis. Diabetes 66(5):1159–1171. https://doi.org/10.2337/db16-1225
Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, Vazquez-Ortiz G, Jeong WI, Park O, Ki SH, Gao B, Deng CX (2010) Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab 12(3):224–236. https://doi.org/10.1016/j.cmet.2010.06.009
Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, Kronfeld-Schor N, Cohen HY (2010) SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 9(2):162–173. https://doi.org/10.1111/j.1474-9726.2009.00544.x
Khan D, Ara T, Ravi V, Rajagopal R, Tandon H, Parvathy J, Gonzalez EA, Asirvatham-Jeyaraj N, Krishna S, Mishra S, Raghu S, Bhati AS, Tamta AK, Dasgupta S, Kolthur-Seetharam U, Etchegaray JP, Mostoslavsky R, Rao PSM, Srinivasan N, Sundaresan NR (2021) SIRT6 transcriptionally regulates fatty acid transport by suppressing PPARγ. Cell Rep 35(9):109190. https://doi.org/10.1016/j.celrep.2021.109190
Ren S, Hou Y, Zuo Z, Liu Z, Wang H, Xu Y, Yamamoto M, Zhang Q, Fu J, Pi J (2020) Protracted rosiglitazone treatment exacerbates inflammation in white adipose tissues of adipocyte-specific Nfe2l1 knockout mice. Food Chem Toxicol 146:111836. https://doi.org/10.1016/j.fct.2020.111836
Yang SJ, Choi JM, Chae SW, Kim WJ, Park SE, Rhee EJ, Lee WY, Oh KW, Park SW, Kim SW, Park CY (2011) Activation of peroxisome proliferator-activated receptor gamma by rosiglitazone increases sirt6 expression and ameliorates hepatic steatosis in rats. PLoS ONE 6(2):e17057. https://doi.org/10.1371/journal.pone.0017057
Elhanati S, Kanfi Y, Varvak A, Roichman A, Carmel-Gross I, Barth S, Gibor G, Cohen HY (2013) Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep 4(5):905–912. https://doi.org/10.1016/j.celrep.2013.08.006
Tao R, Xiong X, DePinho RA, Deng CX, Dong XC (2013) FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J Biol Chem 288(41):29252–29259. https://doi.org/10.1074/jbc.M113.481473
Luo P, Qin C, Zhu L, Fang C, Zhang Y, Zhang H, Pei F, Tian S, Zhu XY, Gong J, Mao Q, Xiao C, Su Y, Zheng H, Xu T, Lu J, Zhang J (2018) Ubiquitin-specific peptidase 10 (USP10) inhibits hepatic steatosis, insulin resistance, and inflammation through Sirt6. Hepatology 68(5):1786–1803. https://doi.org/10.1002/hep.30062
Yang Q, Hu J, Yang Y, Chen Z, Feng J, Zhu Z, Wang H, Yang D, Liang W, Ding G (2020) Sirt6 deficiency aggravates angiotensin II-induced cholesterol accumulation and injury in podocytes. Theranostics 10(16):7465–7479. https://doi.org/10.7150/thno.45003
Zhuang J, Pan ZJ, Mengqiu-Li HFS, Zhu CK, Wu N, Chang G, Wang H, Zhao XX (2020) BDE-47 induced apoptosis in zebrafish embryos through mitochondrial ROS-mediated JNK signaling. Chemosphere 258:127385. https://doi.org/10.1016/j.chemosphere.2020.127385
Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, Zhang Z, Shen J, Zhou Y, Zhou W, Gu L, Lu R, Ni Z (2019) PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol 26:101254. https://doi.org/10.1016/j.redox.2019.101254
Chen CC, Chang ZY, Tsai FJ, Chen SY (2020) Resveratrol pretreatment ameliorates concanavalin A-induced advanced renal glomerulosclerosis in aged mice through upregulation of sirtuin 1-mediated Klotho expression. Int J Mol Sci 21(18):6766. https://doi.org/10.3390/ijms21186766
Salama RM, Nasr MM, Abdelhakeem JI, Roshdy OK, ElGamal MA (2020) Alogliptin attenuates cyclophosphamide-induced nephrotoxicity: a novel therapeutic approach through modulating MAP3K/JNK/SMAD3 signaling cascade. Drug Chem Toxicol. https://doi.org/10.1080/01480545.2020.1814319
Suzuki T, Yamamoto M (2015) Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med 88(Pt B):93–100. https://doi.org/10.1016/j.freeradbiomed.2015.06.006
Yang Y, Tian T, Wang Y, Li Z, Xing K, Tian G (2019) SIRT6 protects vascular endothelial cells from angiotensin II-induced apoptosis and oxidative stress by promoting the activation of Nrf2/ARE signaling. Eur J Pharmacol 859:172516. https://doi.org/10.1016/j.ejphar.2019.172516
Fan X, Wei W, Huang J, Liu X, Ci X (2020) Isoorientin attenuates cisplatin-induced nephrotoxicity through the inhibition of oxidative stress and apoptosis via activating the SIRT1/SIRT6/Nrf-2 pathway. Front Pharmacol 11:264. https://doi.org/10.3389/fphar.2020.00264
D’Onofrio N, Sardu C, Trotta MC, Scisciola L, Turriziani F, Ferraraccio F, Panarese I, Petrella L, Fanelli M, Modugno P, Massetti M, Marfella LV, Sasso FC, Rizzo MR, Barbieri M, Furbatto F, Minicucci F, Mauro C, Federici M, Balestrieri ML, Paolisso G, Marfella R (2021) Sodium-glucose co-transporter 2 expression and inflammatory activity in diabetic atherosclerotic plaques: Effects of sodium-glucose co-transporter 2 inhibitor treatment. Mol Metab 54:101337. https://doi.org/10.1016/j.molmet.2021.101337
Takahashi M, Higuchi M, Makokha GN, Matsuki H, Yoshita M, Tanaka Y, Fujii M (2013) HTLV-1 tax oncoprotein stimulates ROS production and apoptosis in T cells by interacting with USP10. Blood 122(5):715–725. https://doi.org/10.1182/blood-2013-03-493718
Gao F, Qian M, Liu G, Ao W, Dai D, Yin C (2021) USP10 alleviates sepsis-induced acute kidney injury by regulating Sirt6-mediated Nrf2/ARE signaling pathway. J Inflamm (Lond) 18(1):25. https://doi.org/10.1186/s12950-021-00291-7
Li J, Yu D, Chen S, Liu Y, Shi J, Zhang J, Wen P, Wang Z, Li J, Guo W, Zhang S (2020) Sirt6 opposes glycochenodeoxycholate-induced apoptosis of biliary epithelial cells through the AMPK/PGC-1alpha pathway. Cell Biosci 10:43. https://doi.org/10.1186/s13578-020-00402-6
He Y, Xiao Y, Yang X, Li Y, Wang B, Yao F, Shang C, Jin Z, Wang W, Lin R (2017) SIRT6 inhibits TNF-alpha-induced inflammation of vascular adventitial fibroblasts through ROS and Akt signaling pathway. Exp Cell Res 357(1):88–97. https://doi.org/10.1016/j.yexcr.2017.05.001
Shao J, Yang X, Liu T, Zhang T, Xie QR, Xia W (2016) Autophagy induction by SIRT6 is involved in oxidative stress-induced neuronal damage. Protein Cell 7(4):281–290. https://doi.org/10.1007/s13238-016-0257-6
Li W, Liu X, Qiao H (2020) Downregulation of hippocampal SIRT6 activates AKT/CRMP2 signaling and ameliorates chronic stress-induced depression-like behavior in mice. Acta Pharmacol Sin 41(12):1557–1567. https://doi.org/10.1038/s41401-020-0387-5
Wang XX, Wang XL, Tong MM, Gan L, Chen H, Wu SS, Chen JX, Li RL, Wu Y, Zhang HY, Zhu Y, Li YX, He JH, Wang M, Jiang W (2016) SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3α-dependent antioxidant defense mechanisms. Basic Res Cardiol 111(2):13. https://doi.org/10.1007/s00395-016-0531-z
Vitiello M, Zullo A, Servillo L, Mancini FP, Borriello A, Giovane A, Della Ragione F, D’Onofrio N, Balestrieri ML (2017) Multiple pathways of SIRT6 at the crossroads in the control of longevity, cancer, and cardiovascular diseases. Ageing Res Rev 35:301–311. https://doi.org/10.1016/j.arr.2016.10.008
Santos-Barriopedro I, Bosch-Presegué L, Marazuela-Duque A, de la Torre C, Colomer C, Vazquez BN, Fuhrmann T, Martínez-Pastor B, Lu W, Braun T, Bober E, Jenuwein T, Serrano L, Esteller M, Chen Z, Barceló-Batllori S, Mostoslavsky R, Espinosa L, Vaquero A (2018) SIRT6-dependent cysteine monoubiquitination in the PRE-SET domain of Suv39h1 regulates the NF-κB pathway. Nat Commun 9(1):101. https://doi.org/10.1038/s41467-017-02586-x
Tang SCW, Yiu WH (2020) Innate immunity in diabetic kidney disease. Nat Rev Nephrol 16(4):206–222. https://doi.org/10.1038/s41581-019-0234-4
Peng L, Wen L, Shi QF, Gao F, Huang B, Meng J, Hu CP, Wang CM (2020) Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis 11(11):978. https://doi.org/10.1038/s41419-020-03178-2
Li P, Jin Y, Qi F, Wu F, Luo S, Cheng Y, Montgomery RR, Qian F (2018) SIRT6 acts as a negative regulator in dengue virus-induced inflammatory response by targeting the DNA binding domain of NF-κB p65. Front Cell Infect Microbiol 8:113. https://doi.org/10.3389/fcimb.2018.00113
Mendes KL, Lelis DF, Santos SHS (2017) Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev 38:98–105. https://doi.org/10.1016/j.cytogfr.2017.11.001
Xiao C, Wang RH, Lahusen TJ, Park O, Bertola A, Maruyama T, Reynolds D, Chen Q, Xu X, Young HA, Chen WJ, Gao B, Deng CX (2012) Progression of chronic liver inflammation and fibrosis driven by activation of c-JUN signaling in Sirt6 mutant mice. J Biol Chem 287(50):41903–41913. https://doi.org/10.1074/jbc.M112.415182
Hu X, Zhu S, Liu R, Miller JD, Merkley K, Tilton RG, Liu H (2018) Sirt6 deficiency impairs corneal epithelial wound healing. Aging (Albany NY) 10(8):1932–1946. https://doi.org/10.18632/aging.101513
Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z, Wei X, Zhang Y, Sun Y, Zhou Z, Su H, Zhang C, Li N, Gao C, Peng J, Yi F (2017) Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat Commun 8(1):413. https://doi.org/10.1038/s41467-017-00498-4
Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H (2013) SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496(7443):110–113. https://doi.org/10.1038/nature12038
Sato Y, Yanagita M (2019) Functional heterogeneity of resident fibroblasts in the kidney. Proc Jpn Acad Ser B Phys Biol Sci 95(8):468–478. https://doi.org/10.2183/pjab.95.033
Ma J, Sanchez-Duffhues G, Goumans MJ, Ten Dijke P (2020) TGF-β-induced endothelial to mesenchymal transition in disease and tissue engineering. Front Cell Dev Biol 8:260. https://doi.org/10.3389/fcell.2020.00260
Alyaseer AAA, de Lima MHS, Braga TT (2020) The role of NLRP3 inflammasome activation in the epithelial to mesenchymal transition process during the fibrosis. Front Immunol 11:883. https://doi.org/10.3389/fimmu.2020.00883
Xu X, Xu C, Saud SM, Lu X, Liu L, Fang L, Zhang X, Hu J, Li W (2016) Effect of Kuijie granule on the expression of TGF-β/Smads signaling pathway in patients with ulcerative colitis. Evid Based Complement Alternat Med 2016:2601830. https://doi.org/10.1155/2016/2601830
Zhang H, Chen X, Xue P, Ma X, Li J, Zhang J (2021) FN1 promotes chondrocyte differentiation and collagen production via TGF-β/PI3K/Akt pathway in mice with femoral fracture. Gene 769:145253. https://doi.org/10.1016/j.gene.2020.145253
Deng B, Yang W, Wang D, Cheng L, Bu L, Rao J, Zhang J, Xie J, Zhang B (2020) Peptide DR8 suppresses epithelial-to-mesenchymal transition via the TGF-β/MAPK signaling pathway in renal fibrosis. Life Sci 261:118465. https://doi.org/10.1016/j.lfs.2020.11846
Wang S, Wilkes MC, Leof EB, Hirschberg R (2010) Noncanonical TGF-beta pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am J Physiol Renal Physiol 298(1):F142–F149. https://doi.org/10.1152/ajprenal.00320.2009
Meng P, Zhu M, Ling X, Zhou L (2020) Wnt signaling in kidney: the initiator or terminator? J Mol Med (Berl) 98(11):1511–1523. https://doi.org/10.1007/s00109-020-01978-9
Zhong X, Huang M, Kim HG, Zhang Y, Chowdhury K, Cai W, Saxena R, Schwabe RF, Liangpunsakul S, Dong XC (2020) SIRT6 protects against liver fibrosis by deacetylation and suppression of SMAD3 in hepatic stellate cells. Cell Mol Gastroenterol Hepatol 10(2):341–364. https://doi.org/10.1016/j.jcmgh.2020.04.005
Han Q, Xie QR, Li F, Cheng Y, Wu T, Zhang Y, Lu X, Wong AST, Sha J, Xia W (2021) Targeted inhibition of SIRT6 via engineered exosomes impairs tumorigenesis and metastasis in prostate cancer. Theranostics 11(13):6526–6541. https://doi.org/10.7150/thno.53886
Carraway HE, Malkaram SA, Cen Y, Shatnawi A, Fan J, Ali HEA, Abd Elmageed ZY, Buttolph T, Denvir J, Primerano DA, Fandy TE (2020) Activation of SIRT6 by DNA hypomethylating agents and clinical consequences on combination therapy in leukemia. Sci Rep 10(1):10325. https://doi.org/10.1038/s41598-020-67170-8
Yu C, Xiong C, Tang J, Hou X, Liu N, Bayliss G, Zhuang S (2021) Histone demethylase JMJD3 protects against renal fibrosis by suppressing TGFβ and Notch signaling and preserving PTEN expression. Theranostics 11(6):2706–2721. https://doi.org/10.7150/thno.48679
Nasser F, Moussa N, Helmy MW, Haroun M (2021) Dual targeting of Notch and Wnt/β-catenin pathways: Potential approach in triple-negative breast cancer treatment. Naunyn Schmiedebergs Arch Pharmacol 394(3):481–490. https://doi.org/10.1007/s00210-020-01988-x
Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KA, Zoungas S, Rossing P, Groop PH, Cooper ME (2015) Diabetic kidney disease. Nat Rev Dis Primers 1:15018. https://doi.org/10.1038/nrdp.2015.18
Ayinde KS, Olaoba OT, Ibrahim B, Lei D, Lu Q, Yin X, Adelusi TI (2020) AMPK allostery: a therapeutic target for the management/treatment of diabetic nephropathy. Life Sci 261:118455. https://doi.org/10.1016/j.lfs.2020.118455
Barrera-Chimal J, Jaisser F (2020) Pathophysiologic mechanisms in diabetic kidney disease: a focus on current and future therapeutic targets. Diabetes Obes Metab 22(Suppl 1):16–31. https://doi.org/10.1111/dom.13969
Matoba K, Takeda Y, Nagai Y, Yokota T, Utsunomiya K, Nishimura R (2020) Targeting redox imbalance as an approach for diabetic kidney disease. Biomedicines 8(2):40. https://doi.org/10.3390/biomedicines8020040
Matoba K, Takeda Y, Nagai Y, Kawanami D, Utsunomiya K, Nishimura R (2019) Unraveling the role of inflammation in the pathogenesis of diabetic kidney disease. Int J Mol Sci 20(14):3393. https://doi.org/10.3390/ijms20143393
Feng J, Ma Y, Chen Z, Hu J, Yang Q, Ding G (2019) Mitochondrial pyruvate carrier 2 mediates mitochondrial dysfunction and apoptosis in high glucose-treated podocytes. Life Sci 237:116941. https://doi.org/10.1016/j.lfs.2019.116941
Niewczas MA, Pavkov ME, Skupien J, Smiles A, Md Dom ZI, Wilson JM, Park J, Nair V, Schlafly A, Saulnier PJ, Satake E, Simeone CA, Shah H, Qiu C, Looker HC, Fiorina P, Ware CF, Sun JK, Doria A, Kretzler M, Susztak K, Duffin KL, Nelson RG, Krolewski AS (2019) A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat Med 25(5):805–813. https://doi.org/10.1038/s41591-019-0415-5
Niranjan T, Bielesz B, Gruenwald A, Ponda MP, Kopp JB, Thomas DB, Susztak K (2008) The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med 14(3):290–298. https://doi.org/10.1038/nm1731
El Machhour F, Keuylian Z, Kavvadas P, Dussaule JC, Chatziantoniou C (2015) Activation of Notch3 in glomeruli promotes the development of rapidly progressive renal disease. J Am Soc Nephrol 26(7):1561–1575. https://doi.org/10.1681/ASN.2013090968
Waters AM, Wu MY, Huang YW, Liu GY, Holmyard D, Onay T, Jones N, Egan SE, Robinson LA, Piscione TD (2012) Notch promotes dynamin-dependent endocytosis of nephrin. J Am Soc Nephrol 23(1):27–35. https://doi.org/10.1681/ASN.2011010027
Ji L, Chen Y, Wang H, Zhang W, He L, Wu J, Liu Y (2019) Overexpression of Sirt6 promotes M2 macrophage transformation, alleviating renal injury in diabetic nephropathy. Int J Oncol 55(1):103–115. https://doi.org/10.3892/ijo.2019.4800
Muraoka H, Hasegawa K, Sakamaki Y, Minakuchi H, Kawaguchi T, Yasuda I, Kanda T, Tokuyama H, Wakino S, Itoh H (2019) Role of Nampt-Sirt6 axis in renal proximal tubules in extracellular matrix deposition in diabetic nephropathy. Cell Rep 27(1):199–212. https://doi.org/10.1016/j.celrep.2019.03.024
Govender S, Pfister C, Rayner B, Dyer R, Flint M, Roodt F, Davids J, Nejthardt MB, Swanevelder JL, Chiu E, Cloete E, Koller V, Pretorius T, Fullerton Z, Roos J, Van Zyl R, Biccard BM (2020) A multicentre cross-sectional descriptive study evaluating the cardiovascular risk profile of preoperatively identified patients with hypertension. S Afr Med J 111(11):74–79. https://doi.org/10.7196/SAMJ.2020.v111i1.14640
Mennuni S, Rubattu S, Pierelli G, Tocci G, Fofi C, Volpe M (2014) Hypertension and kidneys: unraveling complex molecular mechanisms underlying hypertensive renal damage. J Hum Hypertens 28(2):74–79. https://doi.org/10.1038/jhh.2013.55
Zhang W, Cheng B, Lu Q, Sheng Y, Sun X, Chen X (2019) Analysis of the correlations of hypertension complicated with or without hypertensive nephropathy with glucose and lipid metabolism, vascular endothelial function, inflammation, oxidative stress and course of disease. Panminerva Med 62(3):180–183. https://doi.org/10.23736/S0031-0808.19.03649-8
Guo J, Wang Z, Wu J, Liu M, Li M, Sun Y, Huang W, Li Y, Zhang Y, Tang W, Li X, Zhang C, Hong F, Li N, Nie J, Yi F (2019) Endothelial SIRT6 is vital to prevent hypertension and associated cardiorenal injury through targeting Nkx3.2-GATA5 signaling. Circ Res 124(10):1448–1461. https://doi.org/10.1161/CIRCRESAHA.118.314032
D’Onofrio N, Servillo L, Balestrieri ML (2018) SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal 28(8):711–732. https://doi.org/10.1089/ars.2017.7178
Cardus A, Uryga AK, Walters G, Erusalimsky JD (2013) SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence. Cardiovasc Res 97(3):571–579. https://doi.org/10.1093/cvr/cvs352
Liu Y, Song JW, Lin JY, Miao R, Zhong JC (2020) Roles of MicroRNA-122 in cardiovascular fibrosis and related diseases. Cardiovasc Toxicol 20(5):463–473. https://doi.org/10.1007/s12012-020-09603-4
Song JJ, Yang M, Liu Y, Song JW, Wang J, Chi HJ, Liu XY, Zuo K, Yang XC, Zhong JC (2020) MicroRNA-122 aggravates angiotensin II-mediated apoptosis and autophagy imbalance in rat aortic adventitial fibroblasts via the modulation of SIRT6-elabela-ACE2 signaling. Eur J Pharmacol 883:173374. https://doi.org/10.1016/j.ejphar.2020.173374
Schroth J, Thiemermann C, Henson SM (2020) Senescence and the aging immune system as major drivers of chronic kidney disease. Front Cell Dev Biol 8:564461. https://doi.org/10.3389/fcell.2020.564461
Kitada M, Kume S, Takeda-Watanabe A, Kanasaki K, Koya D (2013) Sirtuins and renal diseases: relationship with aging and diabetic nephropathy. Clin Sci (Lond) 124(3):153–164. https://doi.org/10.1042/CS20120190
Zhang J, Hansen KM, Pippin JW, Chang AM, Taniguchi Y, Krofft RD, Pickering SG, Liu ZH, Abrass CK, Shankland SJ (2012) De novo expression of podocyte proteins in parietal epithelial cells in experimental aging nephropathy. Am J Physiol Renal Physiol 302(5):F571–F580. https://doi.org/10.1152/ajprenal.00516.2011
Grootaert MOJ, Finigan A, Figg NL, Uryga AK, Bennett MR (2021) SIRT6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res 128(4):474–491. https://doi.org/10.1161/CIRCRESAHA.120.318353
Pillai VB, Samant S, Hund S, Gupta M, Gupta MP (2021) The nuclear sirtuin SIRT6 protects the heart from developing aging-associated myocyte senescence and cardiac hypertrophy. Aging (Albany NY) 13(9):12334–12358. https://doi.org/10.18632/aging.203027
Zhang N, Li Z, Mu W, Li L, Liang Y, Lu M, Wang Z, Qiu Y, Wang Z (2016) Calorie restriction-induced SIRT6 activation delays aging by suppressing NF-κB signaling. Cell Cycle 15(7):1009–1018. https://doi.org/10.1080/15384101.2016.1152427
Huang W, Liu H, Zhu S, Woodson M, Liu R, Tilton RG, Miller JD, Zhang W (2017) Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging (Albany NY) 9(3):1069–1083. https://doi.org/10.18632/aging.101214
Jeh SU, Park JJ, Lee JS, Kim DC, Do J, Lee SW, Choi SM, Hyun JS, Seo DH, Lee C, Kam SC, Chung KH, Hwa JS (2017) Differential expression of the sirtuin family in renal cell carcinoma: Aspects of carcinogenesis and prognostic significance. Urol Oncol 35(12):675–679. https://doi.org/10.1016/j.urolonc.2017.08.016
Ding Y, Wu S, Huo Y, Chen X, Chai L, Wang Y, Wang X, Zhu G, Jiang W (2019) Inhibition of Sirt6 suppresses tumor growth by inducing G1/S phase arrest in renal cancer cells. Int J Clin Exp Pathol 12(7):2526–2535
Tan Y, Li B, Peng F, Gong G, Li N (2020) Integrative analysis of sirtuins and their prognostic significance in clear cell renal cell carcinoma. Front Oncol 10:218. https://doi.org/10.3389/fonc.2020.00218
An J, Yang J, Yao Y, Lu K, Zhao Z, Yu M, Zhu Y (2021) Sirtuin 6 regulates the proliferation and survival of clear cell renal cell carcinoma cells via B-cell lymphoma 2. Oncol Lett 21(4):293. https://doi.org/10.3892/ol.2021.12554
Ren H, Zuo S, Hou Y, Shang W, Liu N, Yin Z (2020) Inhibition of α1-adrenoceptor reduces TGF-β1-induced epithelial-to-mesenchymal transition and attenuates UUO-induced renal fibrosis in mice. FASEB J 34(11):14892–14904. https://doi.org/10.1096/fj.202000737RRR
Wetzel MD, Stanley K, Wang WW, Maity S, Awad AS (2020) Selective inhibition of arginase-2 in endothelial cells but not proximal tubules reduces renal fibrosis. JCI Insight 5(19):e142187. https://doi.org/10.1172/jci.insight.142187
Li X, Pan J, Li H, Li G, Liu X, Liu B, He Z, Peng Z, Zhang H, Li Y, Xiang X, Chai X, Yuan Y, Zheng P, Liu F, Zhang D (2020) DsbA-L mediated renal tubulointerstitial fibrosis in UUO mice. Nat Commun 11(1):4467. https://doi.org/10.1038/s41467-020-18304-z
Wyczanska M, Lange-Sperandio B (2020) DAMPs in unilateral ureteral obstruction. Front Immunol 11:581300. https://doi.org/10.3389/fimmu.2020.581300
You YK, Luo Q, Wu WF, Zhang JJ, Zhu HJ, Lao L, Lan HY, Chen HY, Cheng YX (2019) Petchiether A attenuates obstructive nephropathy by suppressing TGF-β/Smad3 and NF-κB signalling. J Cell Mol Med 23(8):5576–5587. https://doi.org/10.1111/jcmm.14454
Martínez-Klimova E, Aparicio-Trejo OE, Gómez-Sierra T, Jiménez-Uribe AP, Bellido B, Pedraza-Chaverri J (2020) Mitochondrial dysfunction and endoplasmic reticulum stress in the promotion of fibrosis in obstructive nephropathy induced by unilateral ureteral obstruction. BioFactors 46(5):716–733. https://doi.org/10.1002/biof.1673
Duan Z, Cai G, Li J, Chen X (2020) Cisplatin-induced renal toxicity in elderly people. Ther Adv Med Oncol 12:1758835920923430. https://doi.org/10.1177/1758835920923430
Crona DJ, Faso A, Nishijima TF, McGraw KA, Galsky MD, Milowsky MI (2017) A systematic review of strategies to prevent cisplatin-Induced nephrotoxicity. Oncologist 22(5):609–619. https://doi.org/10.1634/theoncologist.2016-0319
Wang H, Xia W, Long G, Pei Z, Li Y, Wu M, Wang Q, Zhang Y, Jia Z, Chen H (2020) Isoquercitrin ameliorates cisplatin-induced nephrotoxicity the inhibition of apoptosis, inflammation, and oxidative stress. Front Pharmacol 11:599416. https://doi.org/10.3389/fphar.2020.599416
Zhang Y, Chen Y, Li B, Ding P, Jin D, Hou S, Cai X, Sheng X (2020) The effect of monotropein on alleviating cisplatin-induced acute kidney injury by inhibiting oxidative damage, inflammation and apoptosis. Biomed Pharmacother 129:110408. https://doi.org/10.1016/j.biopha.2020.110408
Fan X, Wei W, Huang J, Peng L, Ci X (2020) Daphnetin attenuated cisplatin-induced acute nephrotoxicity with enhancing antitumor activity of cisplatin by upregulating SIRT1/SIRT6-Nrf2 pathway. Front Pharmacol 11:579178. https://doi.org/10.3389/fphar.2020.579178
Li Z, Xu K, Zhang N, Amador G, Wang Y, Zhao S, Li L, Qiu Y, Wang Z (2018) Overexpressed SIRT6 attenuates cisplatin-induced acute kidney injury by inhibiting ERK1/2 signaling. Kidney Int 93(4):881–892. https://doi.org/10.1016/j.kint.2017.10.021
Nadeem A, Ahmad SF, Al-Harbi NO, Ibrahim KE, Alqahtani F, Alanazi WA, Mahmood HM, Alsanea S, Attia SM (2021) Bruton’s tyrosine kinase inhibition attenuates oxidative stress in systemic immune cells and renal compartment during sepsis-induced acute kidney injury in mice. Int Immunopharmacol 90:107123. https://doi.org/10.1016/j.intimp.2020.107123
Zhang Y, Wang L, Meng L, Cao G, Wu Y (2019) Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell Cycle 18(4):425–436. https://doi.org/10.1080/15384101.2019.1568746
Luo T, Yuan Y, Yu Q, Liu G, Long M, Zhang K, Bian J, Gu J, Zou H, Wang Y, Zhu J, Liu X, Liu Z (2017) PARP-1 overexpression contributes to Cadmium-induced death in rat proximal tubular cells via parthanatos and the MAPK signalling pathway. Sci Rep 7(1):4331. https://doi.org/10.1038/s41598-017-04555-2
So KY, Park BH, Oh SH (2020) Cytoplasmic sirtuin 6 translocation mediated by p62 polyubiquitination plays a critical role in cadmium-induced kidney toxicity. Cell Biol Toxicol 37(2):193–207. https://doi.org/10.1007/s10565-020-09528-2
Yuan X, Chen G, Guo D, Xu L, Gu Y (2020) Polydatin alleviates septic myocardial injury by promoting SIRT6-mediated autophagy. Inflammation 43(3):785–795. https://doi.org/10.1007/s10753-019-01153-4
Li Y, Liu M, Song X, Zheng X, Yi J, Liu D, Wang S, Chu C, Yang J (2020) Exogenous hydrogen sulfide ameliorates diabetic myocardial fibrosis by inhibiting cell aging through SIRT6/AMPK autophagy. Front Pharmacol 11:1150. https://doi.org/10.3389/fphar.2020.01150
Basile DP, Donohoe D, Roethe K, Osborn JL (2001) Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol 281(5):F887–F899. https://doi.org/10.1152/ajprenal.2001.281.5.F887
Tammaro A, Kers J, Scantlebery AML, Florquin S (2020) Metabolic flexibility and innate immunity in renal ischemia reperfusion injury: the fine balance between adaptive repair and tissue degeneration. Front Immunol 11:1346. https://doi.org/10.3389/fimmu.2020.01346
Arshad A, Ayaz A, Rehman S, Ukrani RD, Akbar I, Jamil B (2020) Progression of acute kidney injury to chronic kidney disease in sepsis survivors: 1-year follow-up study. J Intensive Care Med 36(11):1366–1370. https://doi.org/10.1177/0885066620956621
Kruger C, Nguyen TT, Breaux C, Guillory A, Mangelli M, Fridianto KT, Kovalik JP, Burk DH, Noland RC, Mynatt R, Stadler K (2019) Proximal tubular cell-specific ablation of carnitine acetyltransferase causes tubular disease and secondary glomerulosclerosis. Diabetes 68(4):819–831. https://doi.org/10.2337/db18-0090
Console L, Scalise M, Giangregorio N, Tonazzi A, Barile M, Indiveri C (2020) The link between the mitochondrial fatty acid oxidation derangement and kidney injury. Front Physiol 11:794. https://doi.org/10.3389/fphys.2020.00794
Liu R, Wang SC, Li M, Ma XH, Jia XN, Bu Y, Sun L, Yu KJ (2020) An inhibitor of DRP1 (Mdivi-1) alleviates LPS-induced septic AKI by inhibiting NLRP3 inflammasome activation. Biomed Res Int 2020:2398420. https://doi.org/10.1155/2020/2398420
Ullah MM, Basile DP (2019) Role of renal hypoxia in the progression from acute kidney injury to chronic kidney disease. Semin Nephrol 39(6):567–580. https://doi.org/10.1016/j.semnephrol.2019.10.006
Liao W, Liang P, Liu B, Xu Z, Zhang L, Feng M, Tang Y, Xu A (2020) MicroRNA-140-5p mediates renal fibrosis through TGF-β1/Smad signaling pathway by directly targeting TGFBR1. Front Physiol 11:1093. https://doi.org/10.3389/fphys.2020.01093
Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR (2009) Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 53(6):961–973. https://doi.org/10.1053/j.ajkd.2008.11.034
Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J (2009) A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA 106(29):11960–11965. https://doi.org/10.1073/pnas.0904875106
Lok SWY, Yiu WH, Li H, Xue R, Zou Y, Li B, Chan KW, Chan LYY, Leung JCK, Lai KN, Tang SCW (2020) The PAR-1 antagonist vorapaxar ameliorates kidney injury and tubulointerstitial fibrosis. Clin Sci (Lond) 134(21):2873–2891. https://doi.org/10.1042/CS20200923
Liang Y, Liang L, Liu Z, Wang Y, Dong X, Qu L, Gou R, Wang Y, Wang Q, Liu Z, Tang L (2020) Inhibition of IRE1/JNK pathway in HK-2 cells subjected to hypoxia-reoxygenation attenuates mesangial cells-derived extracellular matrix production. J Cell Mol Med 24(22):13408–13420. https://doi.org/10.1111/jcmm.15964
Ke Q, Yuan Q, Qin N, Shi C, Luo J, Fang Y, Xu L, Sun Q, Zen K, Jiang L, Zhou Y, Yang J (2020) UCP2-induced hypoxia promotes lipid accumulation and tubulointerstitial fibrosis during ischemic kidney injury. Cell Death Dis 11(1):26. https://doi.org/10.1038/s41419-019-2219-4
Zhang L, Liu L, Bai M, Liu M, Wei L, Yang Z, Qian Q, Ning X, Sun S (2020) Hypoxia-induced HE4 in tubular epithelial cells promotes extracellular matrix accumulation and renal fibrosis via NF-κB. FASEB J 34(2):2554–2567. https://doi.org/10.1096/fj.201901950R
Gao Z, Chen X, Fan Y, Zhu K, Shi M, Ding G (2020) Sirt6 attenuates hypoxia-induced tubular epithelial cell injury via targeting G2/M phase arrest. J Cell Physiol 235(4):3463–3473. https://doi.org/10.1002/jcp.29235
Tang C, Cai J, Yin XM, Weinberg JM, Venkatachalam MA, Dong Z (2021) Mitochondrial quality control in kidney injury and repair. Nat Rev Nephrol 17(5):299–318. https://doi.org/10.1038/s41581-020-00369-0
Murata H, Takamatsu H, Liu S, Kataoka K, Huh NH, Sakaguchi M (2015) NRF2 regulates PINK1 expression under oxidative stress conditions. PLoS ONE 10(11):e142438. https://doi.org/10.1371/journal.pone.0142438
Yu LM, Dong X, Xue XD, Xu S, Zhang X, Xu YL, Wang ZS, Wang Y, Gao H, Liang YX, Yang Y, Wang HS (2021) Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: role of SIRT6. J Pineal Res 70(1):e12698. https://doi.org/10.1111/jpi.12698
Iachettini S, Trisciuoglio D, Rotili D, Lucidi A, Salvati E, Zizza P, Di Leo L, Del Bufalo D, Ciriolo MR, Leonetti C, Steegborn C, Mai A, Rizzo A, Biroccio A (2018) Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis 9(10):996. https://doi.org/10.1038/s41419-018-1065-0
Cases A, Coll E (2005) Dyslipidemia and the progression of renal disease in chronic renal failure patients. Kidney Int Suppl. https://doi.org/10.1111/j.1523-1755.2005.09916.x
Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ, Susztak K (2015) Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21(1):37–46. https://doi.org/10.1038/nm.3762
Chen Q, Hao W, Xiao C, Wang R, Xu X, Lu H, Chen W, Deng CX (2017) SIRT6 is essential for adipocyte differentiation by regulating mitotic clonal expansion. Cell Rep 18(13):3155–3166. https://doi.org/10.1016/j.celrep.2017.03.006
D’Onofrio N, Pieretti G, Ciccarelli F, Gambardella A, Passariello N, Rizzo MR, Barbieri M, Marfella R, Nicoletti G, Balestrieri ML, Sardu C (2019) Abdominal fat SIRT6 expression and Its relationship with inflammatory and metabolic pathways in pre-diabetic overweight patients. Int J Mol Sci 20(5):1153. https://doi.org/10.3390/ijms20051153
Yang F, Huang XR, Chung AC, Hou CC, Lai KN, Lan HY (2010) Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J Pathol 221(4):390–401. https://doi.org/10.1002/path.2721
Hassan NME, Said E, Shehatou GSG (2021) Nifuroxazide suppresses UUO-induced renal fibrosis in rats via inhibiting STAT-3/NF-κB signaling, oxidative stress and inflammation. Life Sci 272:119241. https://doi.org/10.1016/j.lfs.2021.119241
Lu A, Pallero MA, Owusu BY, Borovjagin AV, Lei W, Sanders PW, Murphy-Ullrich JE (2020) Calreticulin is important for the development of renal fibrosis and dysfunction in diabetic nephropathy. Matrix Biol Plus 8:100034. https://doi.org/10.1016/j.mbplus.2020.100034
Zhou D, Li Y, Lin L, Zhou L, Igarashi P, Liu Y (2012) Tubule-specific ablation of endogenous β-catenin aggravates acute kidney injury in mice. Kidney Int 82(5):537–547. https://doi.org/10.1038/ki.2012.173
Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH, Susztak K (2010) Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest 120(11):4040–4054. https://doi.org/10.1172/JCI430254
Acknowledgements
This study was supported by the Grants from National Natural Science Foundation of China (No. 81770687 and 82070713 to G.D., 81970631 to W.L., and 81670631 to D.Y.).
Author information
Authors and Affiliations
Contributions
XY, JF, WL, DY, and GD put forward the concept. XY and JF performed a review of article and wrote the manuscript. ZZ, ZC, and JH contributed to language modification and content adjustment. XY, JF, WL, and GD participated in the revision of this manuscript. All authors have read the article and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, X., Feng, J., Liang, W. et al. Roles of SIRT6 in kidney disease: a novel therapeutic target. Cell. Mol. Life Sci. 79, 53 (2022). https://doi.org/10.1007/s00018-021-04061-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-021-04061-9