
Abstract
The α7 nicotinic acetylcholine receptor is involved in neurological, neurodegenerative, and inflammatory disorders. It operates both as a ligand-gated cationic channel and as a metabotropic receptor in neuronal and non-neuronal cells. As protein phosphorylation is an important cell function regulatory mechanism, deciphering how tyrosine phosphorylation modulates α7 dual ionotropic/metabotropic molecular function is required for understanding its integral role in physiological and pathological processes. α7 single-channel activity elicited by ACh appears as brief isolated openings and less often as episodes of few openings in quick succession. The reduction of phosphorylation by tyrosine kinase inhibition increases the duration and frequency of activation episodes, whereas the inhibition of phosphatases has the opposite effect. Removal of two tyrosine residues at the α7 intracellular domain recapitulates the effects mediated by tyrosine kinase inhibition. The tyrosine-free mutant receptor shows longer duration-activation episodes, reduced desensitization rate and significantly faster recovery from desensitization, indicating that phosphorylation decreases α7 channel activity by favoring the desensitized state. However, the mutant receptor is incapable of triggering ERK1/2 phosphorylation in response to the α7-agonist. Thus, while tyrosine phosphorylation is absolutely required for α7-triggered ERK pathway, it negatively modulates α7 ionotropic activity. Overall, phosphorylation/dephosphorylation events fine-tune the integrated cell response mediated by α7 activation, thus having a broad impact on α7 cholinergic signaling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The α7 nicotinic acetylcholine receptor (nAChR) is highly expressed in the brain, mainly in the cortex, hippocampus, and subcortical limbic regions, where it contributes to cognition, attention, and working memory. In neurons, α7 receptors are found at presynaptic locations where they facilitate the release of neurotransmitters; at postsynaptic locations where they mediate fast synaptic transmission, and at perisynaptic locations where they modulate neuronal excitability and activate a variety of signaling pathways through volume transmission [1,2,3]. α7 reduced activity has been associated with several neurological and neurodegenerative disorders, particularly schizophrenia and Alzheimer’s disease [4, 5]. α7 is also expressed in non-neuronal cells, such as astrocytes, microglia, oligodendrocytes, endothelial cells, and immune cells, where it has anti-inflammatory and neuroprotective activities [6, 7]. Potentiation of α7 has therefore emerged as a therapeutic strategy for neurological, neurodegenerative and inflammatory disorders. Moreover, α7 has been recently proposed to be involved in some clinical manifestations of COVID-19 [8]. On the other hand, increased α7 activity in certain cells may contribute to cancer progression via the promotion of cell proliferation, apoptosis inhibition and tumor angiogenesis stimulation [9, 10].
α7 belongs to the family of pentameric ligand-gated ion channels that contain an extracellular domain (ECD), which carries the agonist binding sites; a transmembrane domain (TMD), which forms the ion pore and the gate; and an intracellular domain (ICD), which contains sites for modulation and intracellular signaling [11].
α7 responds to acetylcholine (ACh) by opening an intrinsic cationic channel which triggers rapid membrane depolarization and calcium influx [12]. The hallmarks of its ionotropic activity are rapid kinetics, rapid desensitization, and high calcium permeability. In addition to its ionotropic actions, α7 shows important metabotropic activity. Its activation induces the release of calcium from intracellular stores and triggers several signal transduction cascades, including JAK2/STAT3 and PI3K/Akt pathways, activation of Ca2+-calmodulin-dependent protein kinase and MAPK/ERK pathway [4, 6, 12,13,14,15,16]. In some non-neuronal cells, signaling pathways elicited by activation of α7 have been shown to be independent of its activity as an ion channel [17, 18].
The metabotropic responses triggered upon receptor activation depend on cell-specific protein-α7 receptor interactions that are mainly mediated through the α7-ICD [19, 20]. The α7 ICD is a loop of 145 amino acids starting from the C-terminal of the transmembrane segment M3 to the N-terminal of M4. This region carries determinants of ion conductance [21] and sites for tyrosine and serine/threonine phosphorylation [22,23,24]. A special sequence of α7-ICD has been proposed to be involved in the interaction with Gα and Gβγ proteins [14, 25]. Also, direct interactions of α7 with several proteins involved in signal transduction, including phosphatases, kinases and G proteins, have been suggested by proteomic and bioinformatic studies [19, 20, 26].
Protein phosphorylation occurs in a reversible manner responding to the cellular balance of kinases, phosphatases, ATP and ADP. It is an important cellular regulatory mechanism involved in physiological processes, including metabolism, proliferation, apoptosis, subcellular trafficking, synaptic plasticity, membrane excitability, and pathological processes, such as inflammation and cancer [27]. Phosphorylation has been shown to modulate several types of nAChRs, thus impacting on neuromuscular transmission, synaptic plasticity, and neurotransmitter secretion [28,29,30]. At the molecular level, it was shown that tyrosine dephosphorylation of α7 increased its macroscopic responses to the agonist, an effect attributed either to a direct channel modulation [22] or to changes in the number of surface α7 receptors [23]. In addition, it was reported that α7 tyrosine phosphorylation induced by T-cell receptor activation led to decreased number of α7 surface receptors and decreased channel conductance [31]. Thus, although these few studies showed α7 modulation by phosphorylation, the conclusions are discrepant, and the underlying molecular mechanisms remain largely unknown. The fact that α7 activates several downstream signaling pathways and that activation of kinases leads to receptor phosphorylation suggest that the receptor might be physically associated with components of these pathways and receptor phosphorylation may influence signal transduction.
We here deciphered the molecular basis of the modulation of human α7 by tyrosine phosphorylation using high-resolution single-channel recordings, which provide unequivocal evidence of the microscopic functional changes, and we revealed how tyrosine phosphorylation impacts on α7-triggered signaling pathways. Our study unveils molecular mechanisms through which the integrated response of α7, including ionotropic and metabotropic responses, is coupled to phosphorylation events. It also enhances our understanding of how phosphorylation associated with normal or pathological situations may fine-tune α7 signaling and contributes to the design of novel therapeutic strategies targeting α7.
Materials and methods
Drugs
Acetylcholine (ACh), 5-Hydroxyindole (5-HI), 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2), RIPA buffer drugs, phenylmethylsulfonyl fluoride and protease inhibitor mixture, luminol, p-cumaric acid, dimethylsulfoxide (DMSO), pervanadate, SU6656, were purchased from Merck (USA). PNU-282987 (N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride) was obtained from Tocris Biosciences (Bristol, UK).
Stock solutions were prepared in water (ACh, pervanadate) or in DMSO (PNU-282987, PP2, SU6656).
Cell culture
BOSC-23 cells, derived from HEK-293 cells (kindly provided by Dr. Sine, Mayo Clinic, USA) were cultured with HEPES-buffered DMEM culture medium (GIBCO, USA) supplemented with 100 µg/mL streptomycin − 100 IU/mL penicillin (Invitrogen, USA), 10% Fetal Bovine Serum (Internegocios, Argentina).
Receptor expression, mutations and cell treatments
Human α7 cDNA subunit was subcloned into the pRBG4 expression vector [32]. Single and double mutations introduced at phosphorylation sites in the intracellular domain were α7-Y386F, α7-Y442F and α7-Y386F/Y442F. The mutations were carried out using the Quick-Change kit (Stratagene, USA) and were confirmed by sequencing.
BOSC-23 cells were transfected by the calcium phosphate procedure with wild type or mutant α7 subunit cDNAs together with the α7 chaperone Ric-3 cDNA (cDNA ratio 1:4 and total amount of cDNA 4 μg/35 mm dish) and GFP cDNA plasmid to allow identification of transfected cells as previously described [21, 33]. For some experiments, the Src-K297R cDNA was used for transfection together with α7 and Ric-3 cDNAs.
All transfections were carried out for about 8–12 h in DMEM with 10% FBS. Cells were used for experiments two to three days after transfection at which time maximum functional expression levels are usually achieved [32,33,34].
Exposure of cells to modulators of protein phosphorylation was carried out following different protocols. For some experiments, drugs were added to the dish and cells in DMEM were incubated during 30–60 min at 37 °C before use. In other experiments, drugs were added to the dish during the course of the electrophysiological recording. PP2 and SU6656 were used as Src kinase inhibitors. Both inhibitors have been shown to exert similar actions in different cell contexts and similar effects on α7 macroscopic currents [22, 35,36,37].
Single-channel recordings
Single channels were recorded in the cell-attached patch configuration [32, 38]. The bath and pipette solutions contained 142 mM KCl, 5.4 mM NaCl, 1.8 mM CaCl2, 1.7 mM MgCl2 and 10 mM HEPES (pH 7.4). ACh was solubilized directly in the pipette solution. Single-channel currents were digitized at 5–10 μs intervals and low-pass filtered at a cut-off frequency of 10 kHz using an Axopatch 200B patch-clamp amplifier (Molecular Devices, CA, USA). Analysis was performed with the program TAC (Bruxton Corporation, Seattle, WA, USA) with the Gaussian digital filter at 9 kHz (Final cut-off frequency 6.7 kHz). Events were detected by the half-amplitude threshold criterion [32]. To determine channel amplitude, events were tracked regardless of current amplitude and amplitude histograms were then constructed.
Open-time histograms were fitted by the sum of exponential functions by maximum likelihood using the program TACFit (Bruxton Corporation, Seattle, WA, USA). Bursts of channel openings were identified as a series of closely separated openings preceded and followed by closings longer than a critical duration, which was taken as the point of intersection between closed components as previously described [32, 34, 38]. Typically, critical durations were defined by the intersection between the first and second briefest components in the closed-time histogram for bursts of α7 (~ 200–400 µs).
Probability of channel opening (Po) was determined only for experiments in which the drug was added during the course of the recording. It corresponded to the fraction of time that the channel was in the open state and was determined in the same recording for the same period before and after the addition of the drug.
Whole-cell recordings
Macroscopic currents were recorded in the whole-cell configuration at − 50 mV as described previously [32, 39]. The pipette was filled with intracellular solution (ICS) containing 134 mM KCl, 5 mM EGTA, 1 mM MgCl2, and 10 mM HEPES (pH 7.3). The extracellular solution (ECS) contained 150 mM NaCl, 1.8 mM CaCl2, 1 mM MgCl2, and 10 mM HEPES (pH 7.3). The solution exchange time was estimated by the open pipette method as described in Corradi et al. [39]. This method consists of applying a pulse of 50% diluted ECS to an open patch pipette, which produces a sudden change in the current measured by the patch-clamp amplifier. After proper adjustment of the electrode position, the current jump in our system varied between 0.1 and 1 ms. After the whole-cell formation, ECS containing 1 mM ACh was rapidly applied during 300 ms using a three-tube perfusion system with elevated solution reservoirs for gravity-driven flow and switching valves controlled by a VC3 controller (ALA Scientific). Currents were filtered at 5 kHz and digitized at 20 kHz using an Axopatch 200B patch-clamp amplifier (Molecular Devices, CA, USA) and acquired using WinWCP software (Strathclyde Electrophysiology Software, University of Strathclyde, Glasgow, UK). The recordings were analysed using the ClampFit software (Molecular Devices, CA, USA). Current decays were fitted by a single exponential function according to the equation:
in which t is time, I is the peak current, I∞ is the steady-state current value, and τd is the decay time constant.
Recovery from desensitization was determined by using a twin pulse procedure as described before [32]. Briefly, whole-cell currents at -50 mV were elicited by a 300 ms step pulse of 1 mM ACh. After removal of ACh, a second pulse of 1 mM ACh was applied at intervals between 100 and 6000 ms after the end of the first ACh pulse. The percentage of recovery of the peak current elicited by the second pulse with respect to the first was plotted against the interpulse interval. The points were fitted using SigmaPlot 12 (Systat Software Inc) by the exponential rise equation:
in which t is the interpulse interval and τr is the recovery time constant.
Western blot
Cells seeded into 6-well plates (4 × 105 cells/well) were transfected and cultured for an additional 48 h in DMEM with 10% FBS. Then cells were growth factor/serum starved for 24 h. On the experimental day, cells were pretreated or not for 30 min with the indicated inhibitor. For activation, the specific α7 agonist PNU-282987 was added to the dish. Cells were then rapidly washed with ice-cold PBS and lysed in RIPA buffer (10 mM Tris, pH 7.5; 150 mM NaCl; 2 mM Na ortho-vanadate; 0.1% SDS; 1% Igepal and 1% Na deoxycholate) in the presence of protease inhibitors (phenylmethylsulfonyl fluoride and protease inhibitor cocktail). Equal amounts of protein (20 μg) were separated on SDS-PAGE and transferred to nitrocellulose membranes (Amersham Biosciences GE Healthcare, England). Phosphorylated ERK1/2 (p-ERK1/2) was detected using anti-p-ERK1/2 monoclonal antibody 1:100 (sc-7383, Santa Cruz Biotechnology, Dallas, TX, USA). Immunocomplexes were revealed by chemiluminescence using horseradish peroxidase-conjugated secondary antibody (Amersham), used at 1:2000 dilution in the same solution as primary antibodies. Immunostaining for ERK1/2 (sc-94, Santa Cruz Biotechnology) was used for protein loading normalization. Chemiluminescence detection was performed using an enhanced detection solution [1.25 mM luminol, 0.2 mM p-coumaric acid, 0.06% (v/v) hydrogen peroxide, 100 mM Tris–HCl pH 8.8]. Immunoblots were exposed to autoradiographic film (Santa Cruz Biotechnology).
Statistical analysis
Data are presented as mean ± SD. Data sets were analyzed using Student´s t-test or Mann–Whitney rank sum test with SigmaPlot 12.0 (Sysat Software, Inc.). Statistically significant differences between two groups of data were established at p values < 0.05. For each condition, n indicates the number of independent experiments, each from different cell patches, and N, the number of cell transfections, each from different days and cell batches.
Results
Inhibition of tyrosine phosphorylation increases the duration and frequency of activation episodes of human α7
To explore if tyrosine phosphorylation modulates α7 ionotropic activity, we first incubated cells with the Src-family protein tyrosine-kinase inhibitor, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2, 10 µM), or with the vehicle alone (0.1% DMSO, control) for 1 h in DMEM. Then, single-channel currents of α7 activated by 100 μM ACh were recorded in the presence of PP2 in the bath solution. In non-treated cells and in the absence of PP2, channel activity appeared mainly as brief and isolated openings or as several openings in quick succession, known as bursts, which correspond to the activation episode of a single receptor molecule (Fig. 1a) [32, 38]. Open and burst duration histograms were described by two exponential components, with mean durations of the slowest components of 0.32 ± 0.07 ms and 0.48 ± 0.13 ms (n = 17), respectively (Fig. 1a, c). For the PP2-treated cells, open time histograms were also fitted by two components with no significant differences in the duration of the slowest component with respect to the control condition (0.35 ± 0.04 ms, n = 11, p = 0.15, Fig. 1b, c). However, burst duration histograms from treated cells revealed an additional longer duration component of 2.96 ± 1.30 ms (n = 11), indicating that PP2 produced a 6.2-fold, statistically significant, increase in the duration of the activation episodes (p < 0.001, Fig. 1b,c).

Effects of cell treatment with PP2 on α7 single-channel activity. Single-channel currents of α7 activated by 100 µM ACh from cells pre-treated for 1 h with 0.1% DMSO (a, control) or 10 µM PP2 in 0.1% DMSO (b). Channel openings are shown as upward deflections. Representative open and burst duration histograms are shown for each condition. Membrane potential: − 70 mV. Filter: 9 kHz. c Bar charts showing the mean open (τopen) and burst (τburst) durations for each condition. The durations were obtained from the slowest components of the corresponding histograms. Data are plotted as mean ± SD. The n (number of independent experiments, each from different cell patches) were 17 and 11 for (a, b), respectively, and the N (number of cell transfections) were 4 and 3 for (a, b), respectively. Statistical comparisons were performed by two-tailed Student-t test for τopen [ns not significant (p = 0.15)] and Mann–Whitney rank-sum test for τburst (***p < 0.001)
To further explore the molecular effects of tyrosine phosphorylation on α7, we followed a different application protocol. Single-channel currents were recorded during 4–25 min to allow detection of more than 1000 opening events. Then, 200 μl ECS solution with 0.1% DMSO containing or not 10 μM PP2 were added to the dish, and channels were recorded again for a similar period (Fig. 2). The advantage of this protocol is that it allows recording both the control and treated conditions in the same patch, thus overcoming the variability of receptor expression among different patches. As shown in Fig. 2a, b, PP2 induced a marked increase in the frequency of channel opening in real time. To quantify this effect, we compared for each seal the number of bursts/min and the fraction of time that the channel can be found in the open state (Open probability, Po) before and after the treatment. The changes determined for each patch were averaged for different experiments. Control recordings showed slightly reduced frequency and Po after the addition of DMSO (0.1%), probably due to the expected desensitization of α7 as a function of time (Fig. 2, n = 9). In contrast, a profound increase in the frequency of bursts (~ 3-fold, n = 6, p = 0.002) and Po (~ 4.2-fold, n = 6, p = 0.002) took place after the addition of PP2 (Fig. 2c).

Application of PP2 during the course of the single-channel recording. Single-channel currents of α7 activated by 100 µM ACh were first recorded in the cell-attached patch configuration (Before), and DMSO or PP2 was added to the dish during the course of the recording to a final concentration of 0.1% or 10 µM, respectively (After). a Single-channel traces of a typical recording before and after addition of DMSO (left) or PP2 (right). Channel openings are shown as upward deflections. Membrane potential: − 70 mV. b The plot shows the number of bursts of openings at each minute of a typical recording before and after the addition of DMSO (left) or PP2 in DMSO (right). c Bar charts showing the change in a number of bursts (left) and in the probability of channel opening (Po, right) after the addition of DMSO or PP2. For each recording, the number of bursts and Po were measured during the same period of time before and after the addition of the drug. The ratio of the number of bursts and Po after addition of the drug/before the addition of the drug were calculated for each patch. The figure corresponds to the mean ± SD of the ratios for 9 (DMSO) and 5 (PP2) patches. d Bar chart showing the change in mean open (left, τopen) and mean burst durations (right, τburst) after addition of DMSO or PP2. The τopen and τburst after addition of the drug were normalized to those before the drug exposure in the same patch. Data are plotted as mean ± SD. The n (number of independent experiments, each from different cell patches) and the N (number of cell transfections) for each condition were 9 and 5 for DMSO, 6 and 3 for PP2, respectively. Statistical comparisons were performed by Mann–Whitney rank sum test, **p < 0.010 (p = 0.002 for After/Before coefficient for the four measured parameters: Nº of bursts, Po, τopen and τburst)
For each patch, we also determined the mean open and burst durations before and after the addition of DMSO (control) or PP2. We found that both parameters remained constant in the control condition (Open: before = 0.29 ± 0.04 ms, after = 0.28 ± 0.05 ms, n = 9, p = 0.771; Burst: before = 0.42 ± 0.08 ms, after = 0.42 ± 0.08 ms, n = 9, p = 0.724). In contrast, after the addition of PP2, the mean open and burst durations showed a statistically significant increase of ~ 1.4-fold (before = 0.24 ± 0.07 ms, after = 0.33 ± 0.07 ms, n = 6, p = 0.003) and ~ 5-fold (before = 0.38 ± 0.06 ms, after = 1.88 ± 0.40 ms, n = 6, p = 0.002), respectively (Fig. 2d).
Taken together this set of experiments confirmed that reduced phosphorylation increases burst duration and revealed that it also increases the frequency and the probability of finding the receptor in an open state.
Receptor phosphorylation is mediated by endogenous Src kinases.
To evaluate endogenous players involved in the phosphorylation effects, we co-expressed α7 with an inactive Src kinase (Src-K297R), following the hypothesis that its competition with endogenous cell kinases would lead to reduced α7 phosphorylation. Single-channel recordings of α7 activated by 100 µM ACh from cells transfected with α7 and Src-K297R cDNAs showed non statistically significant changes in the mean open duration compared to cells not expressing the inactive kinase (0.30 ± 0.06 ms, n = 38, and 0.35 ± 0.10 ms, n = 4, for control and Src-K297R-expressing cells, respectively, p = 0.21). However, they showed a statistically significant increase in the mean burst duration (0.46 ± 0.12 ms, n = 38, and 0.65 ± 0.10 ms, n = 4, for control and Src-K297R-expressing cells, respectively, p = 0.005).
Inhibition of tyrosine phosphatases by pervanadate decreases open and burst durations of α7 without affecting channel amplitude.
Given that the inhibition of phosphorylation increases the duration of channel opening episodes, we hypothesized that the inhibition of tyrosine phosphatases by pervanadate should produce the opposite effect. However, the mean open and burst durations of α7 are in the sub-millisecond range, close to the limit of the temporal resolution of our system, and, therefore, a reduction in these durations cannot be detected. To overcome this limitation, we used a positive allosteric modulator (PAM) as a tool to increase open and burst durations. We chose 5-hydroxyindole (5-HI), which is a type I PAM, whose effects at the single-channel level have been well described [34]. Cells were preincubated for 0.5–1 h in the absence or presence of 100 μM pervanadate before performing single-channel recordings. In non-treated cells, single-channel activity in the presence of 100 μM ACh and 2 mM 5-HI appeared mainly as longer bursts composed of successive openings of prolonged durations with respect to those recorded in the sole presence of 100 μM ACh. The mean open and burst durations were 1.40 ± 0.37 ms and 3.27 ± 0.91 ms, respectively (n = 12, Fig. 3a, c). Recordings from pervanadate-treated cells showed evident kinetic changes. The mean open and burst durations under these conditions were 0.67 ± 0.31 ms and 1.36 ± 0.58 ms (n = 19), respectively, indicating a statistically significant reduction with respect to the control condition (p = 0.000002 and p = 0.00000007, respectively) (Fig. 3b, c).

Effects of inhibition of phosphatases on channel activity. Single-channel currents of α7 activated by 100 µM ACh and potentiated by 2 mM 5-HI were recorded from cells that were preincubated for 0.5–1 h in the absence (a, control) or presence of 100 µM pervanadate (b). Channel openings are shown as upward deflections. Representative open and burst duration histograms are shown for each condition. Membrane potential: − 70 mV. Filter: 9 kHz. c Bar charts showing the mean open (τopen) and burst (τburst) durations for each condition. The durations were obtained from the slowest components of the corresponding histograms. Data are plotted as mean ± SD. The n (number of independent experiments, each from different cell patches) and the N (number of cell transfections) for each condition were 12 and 4 for A, 19 and 4 for B; respectively. Statistical comparisons were performed by two-tailed Student-t test, ***p < 0.001 (p = 0.000002 for τopen, p = 0.00000007 for τburst). d Representative amplitude histograms constructed with events longer than 0.2 ms are shown for control (left) and pervanadate (right). Bar chart shows the mean amplitude for each condition. Data are plotted as mean ± SD. The n (number of independent experiments, each from different cell patches) and the N (number of cell transfections) for each condition were 4 and 4 for control, 4 and 3 for pervanadate treatment, respectively. Statistical comparisons were performed by two-tailed Student-t test, ns not significant (p = 0.21)
We also evaluated if enhanced phosphorylation affected single-channel amplitude. For α7 openings, there is a wide range of channel amplitudes because their brief open channel lifetimes do not allow full amplitude resolution. However, in the presence of 5-HI and considering only events longer than 0.2 ms, it is possible to resolve the full channel amplitude [32, 38]. There were no statistically significant differences in single-channel amplitudes of openings elicited by 100 µM ACh and 2 mM 5-HI between cells treated or not with pervanadate (10.18 ± 0.31 pA, n = 4, and 9.90 ± 0.25 pA, n = 4, respectively; p = 0.21, Fig. 3d). Thus, the use of a PAM allowed us to unequivocally demonstrate that an increased phosphorylated state reduces α7 open channel lifetime and burst duration but does not affect channel amplitude.
Mutant receptors lacking tyrosine residues at the intracellular domain exhibit longer burst durations than α7 wild type
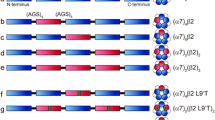
The intracellular domain contains two tyrosine residues with the potential of being phosphorylated. We generated a tyrosine-free α7-ICD by introducing the conservative mutation of tyrosine to phenylalanine at both positions (α7-Y386F/Y442F) (Fig. 4). The mean amplitude of single-channel currents (considering only events longer than 0.2 ms) elicited by 100 μM ACh from cells expressing this mutant receptor was not statistically different from the wild type (9.92 ± 0.29 pA, n = 4, and 9.91 ± 0.19 pA, n = 4, respectively; p = 0.951) (Fig. 4d).

Single-channel characterization of mutant α7 receptors lacking tyrosine residues at ICD. Single-channel currents elicited by 100 µM ACh from cells expressing α7 (a) or the mutant α7-Y386F/Y442F (b). Traces are shown at two different time scales. Channel openings are shown as upward deflections. Representative open and burst duration histograms are shown for each receptor. Membrane potential: − 70 mV. Filter: 9 kHz. c Bar charts showing the mean open (τopen) and burst (τburst) durations for each receptor. The durations were obtained from the slowest components of the corresponding histograms. Data are plotted as mean ± SD. The n (number of independent experiments, each from different cell patches) and the N (number of cell transfections) were 38 and 13 for wild-type, 12 and 5 for the mutant, respectively. Statistical comparisons were performed by two-tailed Student-t test; ns: not significant (p = 0.38 for τopen), ***p < 0.001 (p = 0.0003 for τburst). d Representative amplitude histograms constructed with events longer than 0.2 ms are shown for wild-type (left) and mutant (right) receptors. Bar chart shows the mean amplitude for each receptor. Data are plotted as mean ± SD. The n (number of independent experiments, each from different cell patches) and the N (number of cell transfections) for each receptor were 4 and 4 for wild-type, 4 and 3 for mutant; respectively. Statistical comparisons were performed by two-tailed Student-t test, ns not significant (p = 0.95)
The open channel lifetime did not show statistically significant changes (0.32 ± 0.05 ms, p = 0.38, n = 12) whereas the mean burst duration showed a statistically significant increase with respect to α7 wild-type (0.63 ± 0.14 ms, n = 12, p = 0.0003, Fig. 4a–c). Moreover, the increased burst duration was similar to that observed for α7 wild-type receptors expressed together with Src-K297R, a condition in which the dephosphorylation state is enhanced (p = 0.756). These results confirmed that tyrosine residues 386 and/or 442 at the ICD are involved in the modulation of α7 channel by phosphorylation.
To identify which of the two tyrosine residues is mediating the effect on channel properties, we analysed the burst durations of the two single mutant receptors, α7-Y386F and α7-Y442F. For both single mutant receptors, we detected slightly increased mean burst durations with respect to wild-type receptors, indicating a trend to longer durations (0.52 ± 0.17 ms, n = 5, for α7-Y386F and 0.52 ± 0.05 ms, n = 5, for α7-Y442F). However, for neither single mutant the increase was statistically significant with respect to the wild-type receptor (p = 0.322 for α7-Y386F and p = 0.352 for α7-Y442F). Thus, the effect of phosphorylation on channel kinetics appears to increase with the number of tyrosine residues capable of being phosphorylated.
To further confirm that Y386 and Y442 residues are involved in the modulatory effects of phosphorylation, we expressed the α7-Y386F/Y442F receptor in cells, recorded single-channels activated by 100 µM ACh and then added 10 µM PP2 (final concentration) to the dish during the course of the recording as in the assays shown in Fig. 2. We determined the changes in mean open and burst durations, frequency of openings and Po induced by PP2. In contrast to the results shown for α7 wild type (Fig. 2), the analysis showed no changes in the mean duration and frequency of bursts and in Po after the addition of PP2 (Fig. 5, n = 5). In addition, the changes in mean open duration, mean burst duration, frequency of channels and Po induced by PP2 were statistically significantly different between wild-type and mutant receptors (p = 0.0005, 0.000005, 0.009 and 0.009, respectively). As a control, we measured the changes in the frequency of bursts without the addition of PP2. The results showed a slight reduction in channel activity of the mutant α7-Y386/Y442F in the absence of PP2 probably due to desensitization during a long time of recording (0.77 ± 0.12).

Effect of PP2 on mutant α7-Y386F/Y442F receptors. Single-channel currents of α7-Y386F/Y442F activated by 100 µM ACh were first recorded in the cell-attached patch configuration (before), and PP2 was added to the dish during the course of the recording to a final concentration of 10 µM (after). a Single-channel traces of a typical recording before and after addition of PP2. Channel openings are shown as upward deflections. Membrane potential: − 70 mV. Filter: 6 kHz. b The plot shows the number of bursts of openings at each minute of a typical recording before and after the addition of PP2. c Bar chart showing the change in a number of bursts, probability of channel opening (Po), mean open durations (τopen) and mean burst durations (τburst) after addition of PP2 for α7 and α7-Y386F/Y442F. For each recording, the total number of bursts, Po, τopen and τburst were measured during the same period of time before and after the addition of the drug. The number of bursts, Po, τopen and τburst after addition of the drug were normalized to those before drug exposure in the same patch. Data are plotted as mean ± SD. The n (number of independent experiments, each from different cell patches) and the N (number of cell transfections) for each receptor were 6 and 3 for α7, 5 and 3 for α7-Y386F/Y442F; respectively. Statistical comparisons were performed by two-tailed Student-t test, ***p < 0.001 (p: 0.009, 0.009, 0.0005 and 0.000005 for After/Before PP2 ratio for the four measured parameters: Nº of bursts, Po, τopen and τburst, respectively)
Thus, the lack of tyrosine residues in α7 ICD abolishes the increase in the frequency of openings and Po mediated by PP2 which takes place in wild-type receptors.
Tyrosine phosphorylation of α7 ICD favors the desensitized state of the receptor
To get further insights into the mechanism by which phosphorylation affects single-channel kinetics, we recorded macroscopic currents from wild-type and α7-Y386F/Y442F mutant receptors to explore effects on desensitization.
Rapid application of a 300 ms-pulse of ACh to BOSC-23 cells expressing human α7 or α7-Y386/Y442F elicited macroscopic currents that reached the peak in about 5–10 ms and decayed in the presence of the agonist due to desensitization (Fig. 6a). Current decays were fitted by a single component, and the decay time constants were 28.9 ± 6.5 ms (n = 7) and 39.1 ± 7.1 ms (n = 7) for wild-type and mutant receptors, respectively. The analysis indicated that the decay rate is statistically significantly slower in the mutant receptor (p = 0.0165).

Macroscopic current responses and recovery from desensitization. Whole cell currents were elicited by a 300 ms-pulse of 1 mM ACh from cells expressing α7 or α7-Y386F/Y442F. To measure the time of recovery from desensitization, after the first ACh pulse (300 ms), the cell was exposed to ECS free of agonist for intervals between 100 and 6000 ms, and then a second pulse of ACh was applied. a The figure corresponds to representative experiments for wild-type and mutant α7, each from a single cell. For each receptor, the control current and the recovered currents after exposure for different times to ECS without ACh are shown. b For both receptors, recovery was measured by the ratio of the peak currents between the second and first pulse of agonist. The ratios (expressed in percentage) were plotted against the interval time in the absence of ACh and fitted by a single exponential function. The data correspond to the mean and SD of the percentage of recovery at each interval from eight independent experiments for each receptor
An increase in channel activity could be due to faster recovery from a desensitized to an activatable state. Thus, we measured the time course of recovery from desensitization using a twin pulse procedure. A 300 ms-pulse of 1 mM ACh was applied, then the agonist was removed, and, after increasing recovery times in ECS, a second 300 ms-pulse of 1 mM ACh was applied. The results showed that currents were detected after ~ 300–400 ms in ECS for the mutant receptor but not for the wild-type receptor, for which a ~ 600–800 ms interval was required to elicit detectable currents by the second ACh pulse (Fig. 6a).
The percentage of the peak current of the second pulse related to the first pulse of agonist (control pulse) was plotted as a function of the interval time without ACh (interpulse) for wild-type and mutant receptors and fitted by an exponential function. Figure 6b shows the mean and SD of the percentage of the recovery as a function of the interpulse duration for eight different experiments for wild-type and mutant receptors. The results showed that the mutant receptor recovers faster from the desensitized state than the wild-type receptor. The mean time constants for recovery (τr), obtained from the fit of each different experiment, were 1930 ± 550 ms (n = 8) for wild-type and 664 ± 200 ms (n = 8) for mutant receptors, thus indicating a statistically significant (p < 0.001), threefold faster recovery from desensitization for the mutant receptor.
Thus, the lack of tyrosine residues capable of being phosphorylated decreases the desensitization rate from the open state and accelerates the recovery from the desensitized state to an activatable state.
Tyrosine phosphorylation of ICD is involved in α7 metabotropic responses
Given that α7 has a dual ionotropic/metabotropic activity, we explored if phosphorylation affects signaling pathways elicited by α7 activation. We chose the ERK1/2 pathway that is associated to α7-induced neuroprotection [40].
A brief exposure of α7-expressing BOSC-23 cells to the specific α7 agonist PNU-282987 (PNU, 10 μM) for 2.5 or 5 min increased significantly ERK1/2 phosphorylation (p-ERK1/2) with respect to cells not exposed to the agonist, thus confirming that this pathway is induced by α7 activation in BOSC-23 cells (Fig. 7a). Pretreatment of cells (30 min) with the Src family kinase inhibitor SU6656 (10 μM) before PNU exposure abolished α7-triggered p-ERK1/2 (Fig. 7b, lanes 2 and 4). SU6656 did not significantly affect p-ERK1/2 content in cells not exposed to the agonist (lane 3) with respect to the basal condition (lane 1). To further confirm that the inhibitory effect of the Src inhibitor was specifically related to α7 phosphorylation, we also measured p-ERK1/2 after the PNU-pulse in cells expressing the double mutant α7-Y386F/Y442F receptor in the absence of the kinase inhibitor (Fig. 7b). p-ERK1/2 generated by PNU-exposure was not detected in α7-Y386F/Y442F expressing cells (lane 6), indicating the requirement of the tyrosine residues in the α7-triggered ERK1/2 pathway. As control of receptor expression, we verified by single-channel recordings that the mutant receptor was active in the cells.

Effects of phosphorylation on α7-triggered ERK1/2 pathway. a Western blot of pERK1/2 after exposure of α7-expressing BOSC-23 cells to the α7 specific agonist PNU-282987 (10 μM PNU) for 0, 2.5 or 5 min. b Actions of SFK inhibitor SU6656 (10 μM) on PNU-induced pERK1/2 in cells expressing α7 or α7-Y386F/Y442F receptors. The cells were incubated for 30 min with SU6656 before PNU-282987 exposure (5 min)
In summary, phosphorylation of two tyrosine residues in ICD involves an intrinsic mode of α7 regulation, coordinating the control of both the receptor ionotropic activity and the ERK metabotropic pathway.
Discussion
The role of protein phosphorylation/dephosphorylation as an important regulator of cell function is well established. We here deciphered molecular mechanisms and structures underlying the fine regulation of α7 ionotropic and metabotropic functions by phosphorylation. We found that phosphorylation/dephosphorylation events of two tyrosine residues at the ICD modulate the frequency and duration of α7 channel activation episodes elicited by ACh mainly by changing the rate of fast desensitization and the stability of the desensitized state. Although tyrosine phosphorylation exerts a negative modulation of α7 ionotropic activity, it is required for triggering the ERK signaling pathway, which takes place at a different temporal scale to the ion flux response. Thus, tyrosine phosphorylation has contrasting actions as well as the different degrees of participation in α7 ionotropic and metabotropic activities. Overall, α7 pleiotropic responses can be fine-tuned by the phosphorylation state of the cells.
The importance of understanding how phosphorylation alters Cys-loop receptor function is supported by its association with pathological states. Inappropriate phosphorylation of Cys-loop receptors has pathological consequences and it has, therefore, been postulated that modulation of receptor phosphorylation may be an avenue for novel therapeutic strategies [24, 41]. For example, phosphorylation of glycine receptors in spinal pain sensory neurons is associated with chronic pain [42, 43], and of GABAA receptors, with status epilepticus, alcoholism and spatial memory deficits [24]. In nAChRs, it has been reported that phosphorylation of the muscle-type, α4β2 and α7 receptors is relevant to muscle disorders, nicotine addiction, and fertilization, respectively [24, 44, 45]. α7 is involved in different pathological situations in which aberrant phosphorylation has been described, including Alzheimer’s disease, inflammatory and respiratory disorders, and cancer [46,47,48,49]. Enhancement of α7 activity improves cognition and memory, favors long-term potentiation, and has neuroprotective and anti-inflammatory effects [4, 50, 51], all processes in which tyrosine phosphorylation events are involved. Therefore, further exploration of the direct impact of α7 phosphorylation state on these physiological and pathological situations is highly relevant for therapeutic strategies.
Tyrosine phosphorylation of α7 takes place solely at the least conserved intracellular region, the ICD, whose length and sequence are highly variable among Cys-loop receptors. In line with the great spectrum of ICD sequences and phosphorylation sites, phosphorylation has been shown to elicit a wide variety of molecular effects among Cys-loop receptors, including changes in expression levels, synaptic targeting, receptor activity and desensitization [24].
Previous studies using whole-cell recordings have shown that tyrosine phosphorylation of α7 decreases the maximal macroscopic responses to the agonist whereas dephosphorylation produces the opposite effect [22, 23]. Indeed, the explanation for the increase in α7 currents upon dephosphorylation was discrepant as it was attributed to a higher number of receptors on the surface [23] or to direct changes in receptor function [22, 28]. Macroscopic currents cannot provide information about the underlying mechanism for such effect since their amplitudes depend on the amplitude of each individual channel, the number of channels in the cell and the probability of channel opening. In contrast, our approach that uses high-resolution single-channel recordings unequivocally determined that phosphorylation directly affects the receptor conformational states. Particularly, the increase in the mean burst duration cannot be explained by a change in the number of receptors, clearly indicating a direct effect on channel kinetics. Nevertheless, our study cannot discard small changes in the number of receptors in the membrane occurring at longer times, whose study was out of the scope of the work.
At the single-channel level, we revealed that the lack of tyrosine residues and the inhibition of kinases increase burst duration. Increased burst duration could be the resultant of a reduced rate of desensitization since desensitization governs the open channel lifetime of α7 [32]. In line with this finding, macroscopic currents from the mutant receptor decay slower than currents from wild-type receptors. Although in α7 the decay rates of whole-cell currents do not reflect exactly its desensitization rate due to the temporal resolution limitation of the system [32, 52], the comparison of the times constants under identical conditions suggests decreased desensitization rate from the open state in the non-phosphorylated receptor.
Our macroscopic current recordings also showed that the mutant receptor recovers faster from desensitization. This latter effect is clearly evidenced at the single-channel level as an increase in the frequency of opening events in real time after the addition of the kinase inhibitor in the wild-type receptor. Thus, the enhancement of burst duration and frequency of opening in the absence of tyrosine phosphorylation indicates that phosphorylation reduces the energetic barrier from the open to desensitized states and stabilizes the receptor in a desensitized state.
Changes in α7 channel amplitude due to tyrosine phosphorylation indirectly mediated by T-cell receptor activation were suggested by current fluctuation analysis [31]. This finding does not agree with our results as our high-resolution single-channel recordings showed no changes in single-channel amplitude. A possible explanation for this discrepancy may reside in the fact that, due to its very brief open channel lifetime, α7 channel amplitude cannot be fully resolved. Because of this, single channels of α7 typically show a broad spectrum of amplitudes [32, 52]. Only if channel openings longer than a given duration (which depends on the recording and filtering conditions) are selected for amplitude histograms or if a PAM that enhances duration is used, α7 amplitude can be accurately determined. Our recordings in the presence of 5-HI clearly revealed that tyrosine phosphorylation decreases open and burst durations but does not affect channel amplitude. In agreement, no changes in channel amplitude due to tyrosine phosphorylation were reported for the muscle nAChR [53].
There are only two tyrosine residues in α7 ICD, Y386, located at the middle of ICD and Y442, located before the beginning of the MA helix. Whereas the first is not conserved, the second one is conserved among α1, α4, and β1 nAChR subunits. Muscle nAChRs are phosphorylated by tyrosine kinases at β, γ and δ subunits [24, 54, 55]; and tyrosine phosphorylation is of great relevance in the formation of the neuromuscular junction and receptor clustering [28]. At the molecular functional level, tyrosine phosphorylation has been correlated with an increased rate of receptor desensitization [53, 56]. Respect to neuronal α4β2 nAChRs, which together with α7 are the most abundant nAChRs in the brain, only one report shows that tyrosine phosphorylation may negatively affect function and expression [44]. Interestingly, inhibition of phosphorylation by PP2 or expression of a kinase defective Src reduced the amplitude of macroscopic currents of α3β4α5 nAChRs, indicating that phosphorylation acts as a positive modulator, opposite to its actions in α7 [57].
In line with our conclusion that phosphorylation affects directly α7 function, it has been shown that phosphorylation can cause global allosteric conformational changes in glycine receptors [58]. Also, it has been proposed that phosphorylated conformations could in principle be targeted by drugs with therapeutic potential (Reviewed in [24]). As a proof of concept, it was shown that a non-anesthetic propofol derivative, 2,6-di-tert-butylphenol, was accessible to its α3β glycine receptor binding site only after PKA-dependent phosphorylation [43].
In addition to its ionotropic action, which leads to a rapid increase of intracellular calcium and depolarization, α7 triggers intracellular signaling pathways, some of which may be also triggered by the calcium influx. These actions are of great importance for α7 neuroprotective, antioxidant and anti-inflammatory activities and take place at a different temporal scale from the rapid ion flux. Thus, we here evaluated phosphorylation effects on α7 metabotropic function.
To determine how tyrosine phosphorylation is coupled to α7 metabotropic activity, we chose to explore the effects on the MAPK/ERK pathway, which is associated with nicotine-induced neuroprotection and cell proliferation [15, 40, 59, 60]. We found that a very brief exposure of α7-expressing BOSC-23 cells to the specific α7 agonist PNU-282987 markedly increased p-ERK levels, in good agreement with results from a variety of neuronal and non-neuronal cells expressing α7 [40, 61, 62]. The enhancement of p-ERK levels was fully abolished in the presence of a Src kinase inhibitor, indicating the involvement of tyrosine phosphorylation in this effect. More importantly, the removal of tyrosine residues in α7 ICD closely recapitulated the effects of the kinase inhibitor. Thus, phosphorylated tyrosine residues in α7 ICD are strictly required for triggering ERK1/2 signaling pathway. Future studies should be directed to analyze if other α7-activated signaling pathways also require tyrosine phosphorylation of α7 ICD.
It remains to be elucidated what governs the phosphorylated state of α7 and how SFKs regulate α7 signaling towards ERK phosphorylation. A direct association between Src and α7 may occur. This is supported by in silico studies showing SH2- and SH3-domain binding sites in the α7 ICD [19], which may be recognized by SFKs, and by studies showing the binding of Src to fusion proteins containing the N-terminal half of the ICD of the β1 nAChR subunit [63]. Moreover, a direct interaction between α7 and Fyn was suggested in primary cultures of rat cortex neurons [64]. Agonist activation of α7 may be required to produce a conformational change that exposes tyrosine residues for SFK-mediated phosphorylation. The involvement of adaptor proteins, as shown for tyrosine phosphorylation of the muscle nAChR δ-subunit [65], the existence of crosstalk between α7 and other receptors [31, 60, 66], and Src-activation by other mediators triggered by α7-induced calcium influx may also take place. Most likely, different actions might occur depending on the scaffolding proteins present in the receptor microdomain and the cellular context, which may result in the activation of different intracellular signaling pathways and calcium movement initiated by agonist binding to α7.
We found that while α7 tyrosine phosphorylation is absolutely required for α7-triggered ERK pathway, it acts as a negative modulator of α7 ionotropic activity (Fig. 8). Given that α7 is highly permeable to Ca2+, decreased phosphorylation may act, by increasing open channel probability, as a regulatory mechanism that enhances calcium entrance into the cell, which in turn, leads to most of α7 functions, including neurotransmitter release, postsynaptic depolarization, and triggering of cell signaling processes [67]. Also, this transient increase of calcium is converted into a sustained, wide-ranging phenomena by the release of calcium from intracellular stores through a calcium-induced calcium release (CICR) mechanism [4, 13]. On the other hand, decreased α7 phosphorylation decreases metabotropic responses requiring phosphorylated tyrosine residues, such as the ERK pathway here described, and therefore would redirect responses to those mainly governed by its ionotropic activity. Therefore, phosphorylation/dephosphorylation events of α7 fine-tune the integrated cell response initiated by α7 activation, thus having a broad impact on α7 cholinergic signaling and on its involvement in human disorders, such as neurodegenerative diseases, cancer and inflammatory disorders.

Scheme showing interrelationships between ionotropic and metabotropic responses and receptor phosphorylation
Availability of data and material
The data that support the findings of this study are available within the article.
Abbreviations
- nAChR:
-
Nicotinic acetylcholine receptor
- ACh:
-
Acetylcholine
- ICD:
-
Intracellular domain
- ECD:
-
Extracellular domain
- TMD:
-
Transmembrane domain
- 5-HI:
-
5-Hydroxyindole
- PP2:
-
4-Amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- PNU-282987:
-
N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride
- Src:
-
Proto-oncogene tyrosine-protein kinase
- SFKs :
-
Src-family kinases
- MAPK:
-
Mitogen-activated protein kinase
- ERK:
-
Extracellular signal-regulated kinase
- PAM:
-
Positive allosteric modulator
- Po :
-
Open probability
- ECS:
-
Extracellular solution
References
Dickinson JA, Kew JNC, Wonnacott S (2008) Presynaptic α7- and β2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol 74:348–359. https://doi.org/10.1124/mol.108.046623
Albuquerque EX, Pereira EFR, Alkondon M, Rogers SW (2009) Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol Rev 89:73–120. https://doi.org/10.1152/physrev.00015.2008
Zoli M, Pucci S, Vilella A, Gotti C (2018) Neuronal and extraneuronal nicotinic acetylcholine receptors. Curr Neuropharmacol 16:338–349. https://doi.org/10.2174/1570159x15666170912110450
Corradi J, Bouzat C (2016) Understanding the bases of function and modulation of α7 nicotinic receptors: implications for drug discovery. Mol Pharmacol 90:288–299. https://doi.org/10.1124/mol.116.104240
Buckingham SD, Jones AK, Brown LA, Sattelle DB (2009) Nicotinic acetylcholine receptor signalling: roles in Alzheimer’s disease and amyloid neuroprotection. Pharmacol Rev 61:39–61. https://doi.org/10.1124/pr.108.000562
Egea J, Buendia I, Parada E et al (2015) Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol 97:463–472. https://doi.org/10.1016/j.bcp.2015.07.032
de Jonge WJ, Ulloa L (2007) The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol 151:915–929. https://doi.org/10.1038/sj.bjp.0707264
Changeux JP, Amoura Z, Rey FA, Miyara M (2020) A nicotinic hypothesis for Covid-19 with preventive and therapeutic implications. Comptes Rendus - Biol 343:33–39. https://doi.org/10.5802/crbiol.8
Zhao Y (2016) The oncogenic functions of nicotinic acetylcholine receptors. J Oncol 2016:1–9. https://doi.org/10.1155/2016/9650481
Egleton RD, Brown KC, Dasgupta P (2008) Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol Sci 29:151–158. https://doi.org/10.1016/j.tips.2007.12.006
Noviello CM, Gharpure A, Mukhtasimova N et al (2021) Structure and gating mechanism of the α7 nicotinic acetylcholine receptor. Cell. https://doi.org/10.1016/j.cell.2021.02.049
Dajas-Bailador F, Wonnacott S (2004) Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci 25:317–324. https://doi.org/10.1016/j.tips.2004.04.006
Bouzat C, Lasala M, Nielsen BE et al (2018) Molecular function of α7 nicotinic receptors as drug targets. J Physiol 596:1847–1861. https://doi.org/10.1113/JP275101
Kabbani N, Nichols RA (2018) Beyond the channel: metabotropic signaling by nicotinic receptors. Trends Pharmacol Sci 39:354–366. https://doi.org/10.1016/j.tips.2018.01.002
Elnagar MR, Walls AB, Helal GK et al (2018) Functional characterization of α7 nicotinic acetylcholine and NMDA receptor signaling in SH-SY5Y neuroblastoma cells in an ERK phosphorylation assay. Eur J Pharmacol 826:106–113. https://doi.org/10.1016/j.ejphar.2018.02.047
Cheng Q, Yakel JL (2015) The effect of α7 nicotinic receptor activation on glutamatergic transmission in the hippocampus. Biochem Pharmacol 97:439–444. https://doi.org/10.1016/j.bcp.2015.07.015
Zanetti SR, Ziblat A, Torres NI et al (2016) Expression and functional role of α7 nicotinic receptor in human cytokine-stimulated natural killer (NK) cells. J Biol Chem 291:16541–16552. https://doi.org/10.1074/jbc.M115.710574
Razani-Boroujerdi S, Boyd RT, Dávila-García MI et al (2007) T cells express α7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol 179:2889–2898. https://doi.org/10.4049/jimmunol.179.5.2889
Stokes C, Treinin M, Papke RL (2015) Looking below the surface of nicotinic acetylcholine receptors. Trends Pharmacol Sci 36:514–523. https://doi.org/10.1016/j.tips.2015.05.002
Paulo JA, Brucker WJ, Hawrot E (2009) Proteomic analysis of an alpha7 nicotinic acetylcholine receptor interactome. J Proteome Res 8:1849–1858. https://doi.org/10.1021/pr800731z
Lasala M, Corradi J, Bruzzone A et al (2018) A human-specific, truncated α7 nicotinic receptor subunit assembles with full-length α7 and forms functional receptors with different stoichiometries. J Biol Chem 293:10707–10717. https://doi.org/10.1074/jbc.RA117.001698
Charpantier E, Wiesner A, Huh KH et al (2005) α7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J Neurosci 25:9836–9849. https://doi.org/10.1523/JNEUROSCI.3497-05.2005
Cho CH, Song W, Leitzell K et al (2005) Rapid upregulation of α7 nicotinic acetylcholine receptors by tyrosine dephosphorylation. J Neurosci 25:3712–3723. https://doi.org/10.1523/JNEUROSCI.5389-03.2005
Talwar S, Lynch JW (2014) Phosphorylation mediated structural and functional changes in pentameric ligand-gated ion channels: Implications for drug discovery. Int J Biochem Cell Biol 53:218–223. https://doi.org/10.1016/j.biocel.2014.05.028
Nordman JC, Kabbani N (2014) Microtubule dynamics at the growth cone are mediated by α7 nicotinic receptor activation of a Gαq and IP3 receptor pathway. FASEB J 28:2995–3006. https://doi.org/10.1096/fj.14-251439
Jones AK, Buckingham SD, Sattelle DB (2010) Proteins interacting with nicotinic acetylcholine receptors: expanding functional and therapeutic horizons. Trends Pharmacol Sci 31:455–462. https://doi.org/10.1016/j.tips.2010.07.001
Ardito F, Giuliani M, Perrone D et al (2017) The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int J Mol Med 40:271–280. https://doi.org/10.3892/ijmm.2017.3036
Wiesner A, Fuhrer C (2006) Regulation of nicotinic acetylcholine receptors by tyrosine kinases in the peripheral and central nervous system: Same players, different roles. Cell Mol Life Sci 63:2818–2828. https://doi.org/10.1007/s00018-006-6081-z
Bermudez I, Moroni M (2006) Phosphorylation and function of α4β2 receptor. J Mol Neurosci 30:97–98. https://doi.org/10.1385/JMN:30:1:97
Rudell JB, Ferns MJ (2013) Regulation of muscle acetylcholine receptor turnover by β subunit tyrosine phosphorylation. Dev Neurobiol 73:399–410. https://doi.org/10.1002/dneu.22070
Komal P, Gudavicius G, Nelson CJ, Nashmi R (2014) T-cell receptor activation decreases excitability of cortical interneurons by inhibiting α7 nicotinic receptors. J Neurosci 34:22–35. https://doi.org/10.1523/JNEUROSCI.2093-13.2014
Bouzat C, Bartos M, Corradi J, Sine SM (2008) The interface between extracellular and transmembrane domains of homomeric Cys-loop receptors governs open-channel lifetime and rate of desensitization. J Neurosci 28:7808–7819. https://doi.org/10.1523/JNEUROSCI.0448-08.2008
Nielsen BE, Minguez T, Bermudez I, Bouzat C (2018) Molecular function of the novel α7β2 nicotinic receptor. Cell Mol Life Sci 75:2457–2471. https://doi.org/10.1007/s00018-017-2741-4
Andersen ND, Nielsen BE, Corradi J et al (2016) Exploring the positive allosteric modulation of human α7 nicotinic receptors from a single-channel perspective. Neuropharmacology 107:189–200. https://doi.org/10.1016/j.neuropharm.2016.02.032
Knock GA, Snetkov VA, Shaifta Y et al (2008) Role of src-family kinases in hypoxic vasoconstriction of rat pulmonary artery. Cardiovasc Res 80:453–462. https://doi.org/10.1093/cvr/cvn209
Taniguchi K, Xia L, Goldberg HJ et al (2013) Inhibition of src kinase blocks high glucose-induced EGFR transactivation and collagen synthesis in mesangial cells and prevents diabetic nephropathy in mice. Diabetes 62:3874–3886. https://doi.org/10.2337/db12-1010
Angers-Loustau A, Hering R, Werbowetski TE et al (2004) Src regulates actin dynamics and invasion of malignant glial cells in three dimensions. Mol Cancer Res 2:595–605
Nielsen BE, Bermudez I, Bouzat C (2019) Flavonoids as positive allosteric modulators of α7 nicotinic receptors. Neuropharmacology 160:107794. https://doi.org/10.1016/j.neuropharm.2019.107794
Corradi J, Gumilar F, Bouzat C (2009) Single-channel kinetic analysis for activation and desensitization of homomeric 5-HT3A receptors. Biophys J 97:1335–1345. https://doi.org/10.1016/j.bpj.2009.06.018
Toborek M, Son KW, Pudelko A et al (2007) ERK 1/2 signaling pathway is involved in nicotine-mediated neuroprotection in spinal cord neurons. J Cell Biochem 100:279–292. https://doi.org/10.1002/jcb.21013
Lee AM, Wu DF, Dadgar J et al (2015) PKCε phosphorylates α4β2 nicotinic ACh receptors and promotes recovery from desensitization. Br J Pharmacol 172:4430–4441. https://doi.org/10.1111/bph.13228
Harvey RJ, Depner UB, Wässle H et al (2004) GlyR α3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science 304:884–887. https://doi.org/10.1126/science.1094925
Acuña MA, Yévenes GE, Ralvenius WT et al (2016) Phosphorylation state-dependent modulation of spinal glycine receptors alleviates inflammatory pain. J Clin Invest 126:2547–2560. https://doi.org/10.1172/JCI83817
Biaggi-Labiosa NM, Avilés-Pagán E, Caballero-Rivera D et al (2015) Engineering α4β2 nAChRs with reduced or increased nicotine sensitivity via selective disruption of consensus sites in the M3–M4 cytoplasmic loop of the α4 subunit. Neuropharmacology 99:273–284. https://doi.org/10.1016/j.neuropharm.2015.04.022
Kumar P, Meizel S (2005) Nicotinic acetylcholine receptor subunits and associated proteins in human sperm. J Biol Chem 280:25928–25935. https://doi.org/10.1074/jbc.M502435200
Hendriks WJAJ, Elson A, Harroch S et al (2013) Protein tyrosine phosphatases in health and disease. FEBS J 280:708–730. https://doi.org/10.1111/febs.12000
Perluigi M, Barone E, Di Domenico F, Butterfield DA (2016) Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim Biophys Acta Mol Basis Dis 1862:1871–1882. https://doi.org/10.1016/j.bbadis.2016.07.005
Aschner Y, Downey GP (2018) The importance of tyrosine phosphorylation control of cellular signaling pathways in respiratory disease: pY and pY not. Am J Respir Cell Mol Biol 59:535–547. https://doi.org/10.1165/rcmb.2018-0049TR
Mallozzi C, Crestini A, D’Amore C et al (2020) Activation of tyrosine phosphorylation signaling in erythrocytes of patients with Alzheimer’s disease. Neuroscience 433:36–41. https://doi.org/10.1016/j.neuroscience.2020.02.050
Dineley KT, Pandya AA, Yakel JL (2015) Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol Sci 36:96–108. https://doi.org/10.1016/j.tips.2014.12.002
Uteshev VV (2014) The therapeutic promise of positive allosteric modulation of nicotinic receptors. Eur J Pharmacol 727:181–185. https://doi.org/10.1016/j.ejphar.2014.01.072
Bouzat C, Sine SM (2018) Nicotinic acetylcholine receptors at the single-channel level. Br J Pharmacol 175:1789–1804. https://doi.org/10.1111/bph.13770
Hopfield JF, Tank DW, Greengard P, Huganir RL (1988) Functional modulation of the nicotinic acetylcholine receptor by tyrosine phosphorylation. Nature 336:677–680. https://doi.org/10.1038/336677a0
Huganir RL, Miles K, Greengard P (1984) Phosphorylation of the nicotinic acetylcholine receptor by an endogenous tyrosine-specific protein kinase. Proc Natl Acad Sci USA 81:6968–6972. https://doi.org/10.1073/pnas.81.22.6968
Wagner K, Edson K, Heginbotham L et al (1991) Determination of the tyrosine phosphorylation sites of the nicotinic acetylcholine receptor. J Biol Chem 266:23784–23789. https://doi.org/10.1016/S0021-9258(18)54351-9
Swope SL, Qu Z, Huganir RL (1995) Phosphorylation of the nicotinic acetylcholine receptor by protein tyrosine kinases. Ann N Y Acad Sci 757:197–214. https://doi.org/10.1111/j.1749-6632.1995.tb17476.x
Wang K, Hackett JT, Cox ME et al (2004) Regulation of the neuronal nicotinic acetylcholine receptor by Src family tyrosine kinases. J Biol Chem 279:8779–8786. https://doi.org/10.1074/jbc.M309652200
Han L, Talwar S, Wang Q et al (2013) Phosphorylation of α3 glycine receptors induces a conformational change in the glycine-binding site. ACS Chem Neurosci 4:1361–1370. https://doi.org/10.1021/cn400097j
Larsen HM, Hansen SK, Mikkelsen JD et al (2019) Alpha7 nicotinic acetylcholine receptors and neural network synaptic transmission in human induced pluripotent stem cell-derived neurons. Stem Cell Res 41:101642. https://doi.org/10.1016/j.scr.2019.101642
Nishioka T, Tada H, Ibaragi S et al (2019) Nicotine exposure induces the proliferation of oral cancer cells through the α7 subunit of the nicotinic acetylcholine receptor. Biochem Biophys Res Commun 509:514–520. https://doi.org/10.1016/j.bbrc.2018.12.154
Kouhen REL, Hu M, Anderson DJ et al (2009) Pharmacology of α7 nicotinic acetylcholine receptor mediated extracellular signal-regulated kinase signalling in PC12 cells. Br J Pharmacol 156:638–648. https://doi.org/10.1111/j.1476-5381.2008.00069.x
Gubbins EJ, Gopalakrishnan M, Li J (2010) α7 nAChR-mediated activation of MAP kinase pathways in PC12 cells. Brain Res 1328:1–11. https://doi.org/10.1016/j.brainres.2010.02.083
Fuhrer C, Hall ZW (1996) Functional interaction of Src family kinases with the acetylcholine receptor in C2 myotubes. J Biol Chem 271:32474–32481. https://doi.org/10.1074/jbc.271.50.32474
Kihara T, Shimohama S, Sawada H et al (2001) α7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A β-amyloid-induced neurotoxicity. J Biol Chem 276:13541–13546. https://doi.org/10.1074/jbc.M008035200
Colledge M, Froehner SC (1997) Tyrosine phosphorylation of nicotinic acetylcholine receptor mediates Grb2 binding. J Neurosci 17:5038–5045. https://doi.org/10.1523/jneurosci.17-13-05038.1997
Pestana IA, Vazquez-Padron RI, Aitouche A, Pham SM (2005) Nicotinic and PDGF-receptor function are essential for nicotine-stimulated mitogenesis in human vascular smooth muscle cells. J Cell Biochem 96:986–995. https://doi.org/10.1002/jcb.20564
Gotti C, Clementi F (2004) Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol 74:363–396. https://doi.org/10.1016/j.pneurobio.2004.09.006
Funding
This work was supported by grants from Universidad Nacional del Sur (PGI 24/B298 to CB and PGI 24/ZB79 to MCE), Agencia Nacional de Promoción Científica y Tecnológica (PICT 2015-0941-and PICT 2017-1170 to CB).
Author information
Authors and Affiliations
Contributions
CB, JFC and MCE designed the work; JFC, AB and MCE performed experiments; CB supervised the study and wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors report no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chrestia, J.F., Bruzzone, A., Esandi, M. et al. Tyrosine phosphorylation differentially fine-tunes ionotropic and metabotropic responses of human α7 nicotinic acetylcholine receptor. Cell. Mol. Life Sci. 78, 5381–5395 (2021). https://doi.org/10.1007/s00018-021-03853-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03853-3