
Abstract
It is well-established that Lysine-specific demethylase 1 (LSD1, also known as KDM1A) roles as a lysine demethylase canonically acting on H3K4me1/2 and H3K9me1/2 for regulating gene expression. Though the discovery of non-histone substrates methylated by LSD1 has largely expanded the functions of LSD1 as a typical demethylase, recent groundbreaking studies unveiled its non-catalytic functions as a second life for this demethylase. We and others found that LSD1 is implicated in the interaction with a line of proteins to exhibit additional non-canonical functions in a demethylase-independent manner. Here, we present an integrated overview of these recent literatures charging LSD1 with unforeseen functions to re-evaluate and summarize its non-catalytic biological roles beyond the current understanding of its demethylase activity. Given LSD1 is reported to be ubiquitously overexpressed in a variety of tumors, it has been generally considered as an innovative target for cancer therapy. We anticipate that these non-canonical functions of LSD1 will arouse the consideration that extending the LSD1-based drug discovery to targeting LSD1 protein interactions non-catalytically, not only its demethylase activity, may be a novel strategy for cancer prevention.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
LSD1 (also known as KDM1A, BHC110 and AOF2), as one of the members of flavin adenine dinucleotide-dependent amine oxidase superfamily, is the first identified histone demethylase [1]. It was originally recognized as a required constituent of the BRAF-HDAC (HDAC1/2) transcriptional complex, enclosing the CoREST (REST (RE1-silencing transcription factor) corepressor) [2]. By coordinating with CoREST, LSD1 was revealed to specifically catalyze the demethylation of mono- and di-methylated, but not tri-methylated, histone 3 lysine 4 (H3K4me1, H3K4me2) for transcriptional repression [1, 3]. Subsequent works realized that LSD1 also interacts with AR (androgen receptor) in a ligand-dependent manner and demethylates mono- and di-methylated H3K9 (H3K9me1, H3K9me2) to trigger target gene activation programs [4,5,6]. In addition to histone substrates, studies demonstrate that LSD1 also regulates the methylation dynamics of a substantial number of cancer-relevant non-histone proteins, such as tumor suppressor p53 [7], transcription factor E2F1 [8], DNA methyltransferase 1 (Dnmt1) [9], hypoxia-inducible factor-1 (HIF-1α) [10] and Signal Transducer and Activator of Transcription 3 (STAT3) [11], resulting in different functioning modulations.
Based on its demethylase enzymatic activity targeting both histones and non-histone proteins, it has been increasingly described that LSD1 plays a pivotal role in vast range of cellular processes, such as cell proliferation [12], epithelial-to-mesenchymal transition (EMT) [13], chromosome segregation [14], metabolism [15] stem cell pluripotent regulation [16] and embryonic development etc.[16], and its dysregulation is closely associated with human cancer development [17]. While the demethylase enzymatic activity of LSD1 has been studied in detail and, until recent, most known LSD1 functions are owing to its demethylase activity, the understanding of its biochemical functions independent of demethylation is relatively limited. Reports of function diversity in the most recent years indicate that LSD1 might have additional biological functions beyond catalyzing demethylation. We and others found that, by interacting with FBXW7 (F-box and WD repeat-containing protein 7) [18] or p62 [19], LSD1 accelerates their protein degradation, leading to different biological consequences. And in collaboration with ZNF217, LSD1 promotes prostate cancer cell survival independently of its demethylase function and of AR [20]. In this review, we will focus on the these recent evidences that charging LSD1 with non-canonical functions by protein and protein interactions independent of its demethylase enzymatic activity and discussing its translational significance in LSD1-based drug discovery for cancer prevention.
Overview of the structure analysis into LSD1
The functional diversity of LSD1 is supported by its complex structure which allows it to interact with a variety of cellular proteins. LSD1 is composed of 852 amino acids coding 3 major domains: (1) SWIRM (swi3p/Rsc8p/Moira, small alpha-helical domain) in the N-terminal which is critical for the protein–protein interactions and contributes to the steadiness of the protein [21]; (2) The C-terminal contains an amine oxidase-like (AOL) domain, which shares 20% sequence similarity with FAD-dependent oxidase [22]; (3) The AOL domain is separated into two halves by a large insertion that forms a central protruding Tower domain [22] (Fig. 1). LSD1 has a homolog called LSD2 (also known as KDM1B, AOF1), representing the second member in the FAD-dependent AOL-domain-containing demethylase protein family. LSD1 and LSD2 share less than 31% sequence similarity and many structural properties [23]. Both of them contain a catalytic AOL domain and a SWIRM domain responsible for protein interactions, which are all required for the catalytic action of demethylation. However, the SWIRM domain of LSD2 shows a slight difference with that of LSD1, which is involved in interacting with glyoxylate reductase 1 (GLYR1) that positively regulates its demethylase activity [24]. Moreover, the key difference between LSD1 and LSD2 is that LSD2 does not contain a Tower domain but comprises an N-terminal zinc-finger (ZF-CW) domain. It was described that both of ZF-CW domain and SWIRM are crucial for the demethylase activity of LSD2 [25].

Structural domain overview of LSD1 and LSD2. Both LSD1 and LSD2 contain the catalytic amine oxidase-like domain (AOL), two subdomains of LSD1 AOL domain are shown in green (FAD-binding subdomain) and in reseda (substrate binding subdomain), and a SWIRM domain (shown in red). Tower domain (shown in yellow) and ZFs domain (shown in blue) are unique for LSD1 and LSD2, respectively. The N-terminal flexible region and the C-terminal tail are colored in gray
We mentioned that the AOL domain of LSD1 shares a high similarity in sequence and catalytic mechanism with that of other typical amine oxidases, however, it was buried by a different structural architectures. Specifically, LSD1 contains a flexible region in the N-terminal, which is a non-catalytic domain involving in protein interactions and subject to post-translational modifications (PTMs) [26, 27]. Moreover, the SWIRM domain is located far away from both the FAD-binding site and the catalytic center and it packs together with AOL domain to form a functional structure interface which may contribute to its involvement of divers protein interactions [22]. Furthermore, AOL, was defined as the catalytic center regulating the enzymatic activity and responsible for targeting substrate proteins. Particularly, it is divided by Tower domain into two lobes: (1) One forms a noncovalent FAD-binding site, contains three fragments (residues 271–356, 559–657, and 770–833), which is similar with other amine oxidases; (2) The other forms a more open funnel-shaped active site for substrate binding and recognition, comprises three fragments (residues 357–417, 523–558, and 658–769), allowing LSD1 to pocket more surrounding residues near the target lysine, such as accommodate a long basic histone tails by specifically interacting with the first 20 amino acids of histone 3 (H3) (Fig. 1) [22, 28]. This structural feature not only enhances the exquisite specificity of this enzyme, but also provides the potential that LSD1 may have other non-catalytic functions by specifically binding with various proteins.
LSD1 roles as a demethylase targeting both histones and non-histone proteins
Roles of LSD1 in histones demethylation
The primary substrates of LSD1 are mono- and di-methylated H3K4, which are known as important marks for the activation of gene transcription. Together with several co-factor proteins, LSD1 is assembled into different complexes to demethylate H3K4me1/2 and shape chromatin into a repressive conformation, thus negatively regulating gene expression [1, 3] (Fig. 2). Laterally, H3K9 was also found to be the substrate of LSD1, which leading to an opposite effect that transcriptionally activate gene expression [4,5,6] (Fig. 2). In most of the cases, the target specificity of LSD1 is depend on its association with different partners. Specifically, when it interacts with CoREST [2], CtBP (C-terminal-binding protein 1) [29] and NuRD (nucleosome remodeling and deacetylase) [30] complexes, LSD1 demethylates H3K4me1/2 to act as transcriptional co-repressor. On the contrary, when its coactivators are AR and ER (estrogen receptor) nuclear hormone receptors, LSD1 switches to demethylate H3K9me1/2, thus facilitating gene transcription [5, 31, 32].

Substrate specificity and regulation of gene expression by LSD1. a The binding of LSD1 to the CoREST, CtBP or NuRD complex allows the demethylation of H3K4me1/2 and leads to the transcriptional repression of target gene. b LSD1 catalyzes the demethylation of the repressive mark H3K9me2 when it interacts with the AR or ERα, thus activating the target gene expression. c LSD1n, a neuronal-specific isoform, demethylates the repressive mark H4K20me2, by interacting with CREB (cAMP-response element-binding protein) and MEF2 (myocyte enhancer factor 2)
Moreover, a neuron-specific isoform of LSD1, LSD1n, was described to acquire a new substrate specificity targeting H4K20me2 methylation for transcription activation of neuronal-regulated genes [33]. Through regulating the methylation status of H3K4, H3K9 or H4K20me2 in a context-dependent manner, LSD1 serves as a master epigenetic governor of transcription to control gene expression [34] (Fig. 2).
LSD1 interacts with its cofactors to regulate gene transcription.
In addition to interacting with corepressor CoREST mentioned above, it was found that LSD1 could also bind with SNAG domain family transcription repressors, such as growth factor independence (GFI) proteins [35], SCRT1, SNAI2 (or known as Slug) [36] and Snail, via its substrate binding and recognition cleft [37,38,39]. Given the SNAG domain is a histone H3-mimicking motif exhibiting potent binding affinity with chromatin-modifying enzymes like LSD1, studies found that SNAG is required and sufficient for functioning as a molecular hook in the interactions of LSD1 and its cofactors [35,36,37,38]. When the binding cleft of is occupied by a SNAG domain, H3 tails will be outcompeted to the catalytic activity of LSD1, leading various biological consequences. In hematopoiesis, LSD1–CoREST was hooked by SNAG to coordinate with GFI proteins, which mediates transcriptional repression for controlling hematopoietic differentiation [35]; and disruption of the LSD1–GFI interaction by pharmacologic agents, such as OG86, largely inhibits the transcriptional activity of GFI1, resulting in promoting blast cell differentiation in acute myeloid leukemia (AML) with MLL translocations [40]. Similarly, the SNAG domain of Snail1 resembles a histone H3-like structure for recruiting LSD1–CoREST to its target gene promoters and resulting in suppression of cell migration and invasion in metastasis [37]. Also, it is worth highlighting that LSD1 also associated with a plethora of lncRNAs, such as HOTAIR, TERRA, SRA and other oncogenic lncRNAs, for transcriptional regulation and leading to various phenotypes [41].
Roles of LSD1 in demethylating non-histone proteins
Despite it is beyond the scope of this review to discuss the demethylase activity of LSD1 in detail which has been described in a line of review articles [34, 42, 43], it is worth noting that the discovery of non-histone proteins as substrates of this enzyme is of great significance. Beyond histones, a large amount of non-histone proteins have been identified as the substrates of LSD1. For example, the tumor suppressor p53 is the first non-histone protein identified as the substrate of LSD1 [7]. In this case, LSD1 was reported to preferentially demethylate the demethylated K370 residue of p53 in cancer cells, resulting in repressing p53 transcriptional activity [7]. In addition to affecting the protein function, LSD1 also demethylates several non-histone proteins to regulate their protein stability, such as destabilizes MYPT1 [44] and stabilizes E2F1 [45], and HIF-1α [46]. Most recently, it was reported that Ago2 is demethylated by LSD1 upon the site of K726me1, which promotes its protein stability and accumulates dsRNA expression to regulate tumor T cell response [47]. With the development of proteomic analysis using mass spectrometry, more and more methylation sites on various proteins is to be identified. In particular, we summarize the non-histone substrates of LSD1 identified so far as presented in Table 1. Similar to other PTMs such as phosphorylation and acetylation, lysine methylation dynamic has emerged as another important homeostasis in the regulation of cellular signal transduction pathways.
Collectively, by regulating the methylation dynamics of both histones and non-histone proteins as mentioned above, LSD1 is involved in the control of various cellular processes, including cell proliferation [12], EMT [13], stemness, differentiation, cell motility, autophagy, senescence, etc.[43], and its dysregulation is closely associated with embryonic development [16], human cancer development and other diseases [17].
Non-canonical functions of LSD1 beyond its demethylase activity
The best-characterized and fully validated function of LSD1 is serving as a transcriptional governor by targeting histones for demethylation. The discovery of non-histone proteins as demethylation substrates unveils a wider range of functions on LSD1 and largely associates its relevant to human cancer. Beyond its demethylase activity, recently, LSD1 was reported to interact with a couple of cellular proteins, in a non-canonical manner independent of its demethylation activity, leading to different biological consequence. We will discuss these researches in the following contents.
LSD1 destabilizes FBXW7 and abrogates FBXW7 functions by promoting FBXW7 self-ubiquitylation
As mentioned above, the demethylation of non-histone proteins driven by LSD1 enzymatic activity can either positively (e.g. Dnmt1, E2F1) or negatively (e.g. Mypt1) regulate protein stability. Most recently, we found that LSD1 also negatively regulates the protein stability of FBXW7 in a demethylase-independent manner [48]. FBXW7 is an F-box protein acting as substrate recognition subunit of SCF (SKP1-CUL1-F-box protein) E3 ubiquitin ligase complex. By targeting a line of critical human oncoproteins, such as Cyclin E, c-JUN, c-MYC, NOTCH-1 and MCL-1, for ubiquitylation and proteasome degradation, FBXW7 functions as a typical tumor suppressor [49]. In most of the cases, FBXW7 recognizes and binds with its substrates upon the phosphorylation of an evolutionarily conserved motif termed as CPD (Cdc4 phosphodegron), followed by subsequent polyubiquitylational degradation [50]. In our study, we identified LSD1 serves as a pseudo-substrate of FBXW7, which was neither for targeted degradation nor for altered function of LSD1, rather unexpectedly triggering FBXW7 self-ubiquitylation and subsequent degradation [48] (Fig. 3). Specifically, LSD1 directly binds to the CPD-binding sites of FBXW7 via its C-terminal region (AA415–852), which competitively attenuated the binding ability of FBXW7 on its bona fide substrates, such as c-MYC, cyclin E and NOTCH-1 (51). More importantly, LSD1 with both wild-type form and enzymatic-dead mutant LSD1–K661A were able to reduce the protein level and half-life of FBXW7, which cannot be blocked by LSD1 inhibitors, compound 6b [31], or GSK2879552 [32]. In contrast, two LSD1-binding dead mutants LSD1-M1 or LSD1-M2 had no effect on FBXW7 levels. Therefore, it proves that LSD1–FBXW7 binding destabilizes FBXW7 in a manner dependent of the CPD-binding sites contained in LSD1, but independent of the demethylase activity of LSD1. Mechanistic study indicated that LSD1 particularly disrupts FBXW7 dimerization and accelerates the self-ubiquitylation of FBXW7 with monomeric form, followed by the rapid degradation through both proteasome and p62-mediated lysosomal pathways. Similar with the effect on FBXW7 degradation, LSD1 also abrogates FBXW7 functions in growth suppression, NHEJ (nonhomologous end-joining) repair and radiation protection in a demethylase-independent manner [48].

Non-canonical functions of LSD1 beyond its demethylase activity: (1) LSD1 destabilizes FBXW7 by promoting FBXW7 self-ubiquitylation. (2) LSD1 protects ERRα from proteasome-dependent degradation, and ERRα enhances the H3K9 demethylation activity of LSD1. (3) LSD1 destabilizes p62 and inhibits autophagy in gynecologic cancers. (4) LSD1 collaborates with ZNF217 to activate a lethal prostate cancer gene network, thus promoting the cell survival of cancer cells
Collectively, FBXW7 is a novel binding partner of LSD1 which serves as a pseudo-substrate triggering FBXW7 self-ubiquitylation and degradation. This study established a novel demethylase-independent oncogenic mechanism of LSD1 via targeting FBXW7.
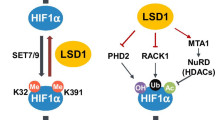
LSD1 protects ERRα from proteasome-dependent degradation in a demethylation-independent manner
Estrogen-related receptor α (ERRα) is an orphan nuclear receptor without natural ligand, acts as a transcriptional regulator [51]. Carnesecchi et al. first reported that ERRα physically bind with LSD1 to induce the H3K9 demethylation activity of LSD1, thus transcriptionally repressing the expression of several target genes to regulate cell migration [52].
A later study from this group further revealed that LSD1 is able to, independently of its demethylase activity, stabilizes ERRα via protecting ERRα from proteasome-dependent degradation [53]. The authors first found that the high protein expression of LSD1 in cancer is correlated to the elevation of its mRNA level in the comparisons of breast cancer vs normal tissues, while the mRNA level of ERRα was not significantly altered in cancer samples. Next, they found that genetic or pharmacologic inactivation of LSD1 results in decreased ERRα protein stabilization. Mechanistic study showed that LSD1-mediated regulation does not occur in the mRNA level, but is a post-transcriptional event, since siRNA-mediated LSD1 depletion promotes the ubiquitylation of ERRα and shortens its protein half-life. More importantly, the enzymatic-dead mutant LSD1-K661A also actively binds with ERRα and exhibits similar stabilizing effect on it, indicating that the enzymatic activity of LSD1 is not involved in ERRα stabilization.
Collectively, this study build up a positive feedback loop between ERRα and LSD1: on the one hand, ERRα enhances the H3K9 demethylation activity of LSD1; On the other hand, LSD1 is able to stabilize ERRα independent of its demethylation activity (Fig. 3). However, how does LSD1 non-catalytical interfere with ERRα degradation remains elusive, further works is needed to elucidate the involved mechanism.
LSD1 destabilizes p62 and inhibits autophagy in gynecologic cancers
p62, also termed as sequestasome 1 (SQSTM1), is a key component of autophagic machinery that promotes autophagy activation [54]. Owing to the presence of several functional domains, p62 has been reported to interact with different proteins [55]. Recently, a study reported that LSD1 is capable of destabilizing p62 and inhibits autophagy in gynecologic cancers [19] (Fig. 3). In this study, LSD1 was observed to highly express in tumors compared to the surrounding normal tissues detected from the ovarian and endometrial cancer tissue arrays. And LSD1 inactivation obviously activates autophagy, while the protein levels of p62 were increased in cancer cells. As it is known that p62 is an autophagy substrate digested in autophagosomes during a completed autophagic process, the observation of accumulated p62 in a LSD1-induced autophagy process suggests the potential regulation axis between LSD1 and p62. Indeed, it was found that LSD1 directly binds to N-terminal PB1 domain of p62 via C-terminal AOL domain. However, this binding is unable to demethylate p62, but resulting in promoting the ubiquitylation of p62 and the following proteasomal degradation. Biologically, LSD1 inhibition suppresses the cell growth by inducing apoptosis in uterine serous carcinoma ARK2 cells, which is magnified by p62 knockdown. And the combination of LSD1 inhibitor with autophagy inhibitor shows a synergistic effect on cancer cell death.
This study reveals that LSD1 destabilizes p62 and inhibits autophagy in gynecologic malignant cells, in a manner without demethylating p62. However, the specific mechanism involved in LSD1-induced p62 degradation remains less clear.
LSD1 collaborates with ZNF217 to activate a lethal prostate cancer gene network
It is well established that LSD1 regulates the transcriptional activities of AR by demethylating H3K9, by doing so, LSD1 plays a pivotal role in AR-associated disease especially in prostate cancer where AR is highly expressed [5, 32, 56, 57]. Most recently, Sehrawat et al. showed that LSD1 promotes the survival of castration-resistant prostate cancer cells independently of its demethylase function and of the AR [58] (Fig. 3). Indeed, the authors found that LSD1 increases the expression of the gene network enriched in lethal prostate cancer, which contributes to LSD1-promoted cell survival in cancer cells. Most importantly, this effect is independent on the demethylase enzymatic activity of LSD1, which is demonstrated by the unchanged H3K4me2 and H3K9me2 marks following LSD1 silencing and by rescue of LSD1-deficient cells with an enzymatic-dead LSD1 mutant. Rather, LSD1 activates this lethal prostate cancer gene network in a manner of non-catalytically interacting with its binding protein ZNF217. Another important finding is that LSD1′s effect even occurs in those prostate cancer cells grown in the absence of androgens, resistant to the AR antagonist enzalutamide, or those without AR expression. Therefore, it is rational that LSD1 may be an effective target in both AR+ and AR− prostate cancer, where cellular reprogramming drives resistance to androgen-deprivation therapy.
As expected, treatment with enzymatic inhibitors of LSD1, such as GSK2879552, GSK-LSD1, and RN1 [59, 60], exert no survival suppression effect on those cell models sensitive to LSD1 silencing. However, treatment with SP-2509, a reversible inhibitor acting as a non-MAO (monoamine oxidase) inactivator which blocks key demethylase-independent functions of LSD1 [61, 62], potently suppresses tumor growth in the models detected. The molecular docking studies indicate that SP-2509 particularly binds to the H3 pocket within LSD1, which may interrupt the binding of LSD1-ZNF217. It suggests that protein–protein interactions beyond demethylation activity may be key mediators of non-canonical functions of LSD1 in this study.
Collectively, this study defines non-canonical and AR-independent functions of LSD1 in prostate cancer, and considers that antagonizing its protein–protein binding ability may be a novel strategy in cancer therapy.
Targeting demethylase-independent roles of LSD1 as potential cancer therapies
Growing studies indicate that LSD1 is actively involved in human tumorigenesis, and its ubiquitously overexpressed expression is proven to correlate with the poor prognosis of various cancers [63,64,65,66,67]. Accordingly, LSD1 is emerging as a promising anticancer target and a line of LSD1 inhibitors are currently in clinical trial studies, such as ORY-1001, GSK-2879552, RG6016, INCB059872, IMG-7289 and CC-90011 [68, 69]. However, most of these inhibitors were based on blocking its demethylase activity.
As characterized above, LSD1 is endowed with non-canonical functions independent of its demethylase activity, it is reasonable to expect that this will innovatively provide novel directions and prospective sites for LSD1-based drug design by targeting LSD1-involved protein interactions, not only confined to the current limitation on its demethylase activity. For example, for those human cancers with LSD1 overexpression, targeting LSD1 protein for degradation by PROTAC technique [70], rather than inhibiting its enzymatic activity, may be a novel strategy to reactivate FBXW7 to prevent tumor development. And blockage of non-canonical functions of LSD1 through allosteric inhibition such as SP-2509 or protein degradation may be a potential approach to control prostate cancer or re-sensitize tumors to androgen deprivation. Additionally, searching for compounds that typically disrupt interaction of LSD1–SNAG domain might yield potent therapeutic efficacy in those malignancies consequent upon the activity of SNAG domain transcription repressors. Interestingly, it was reported that OG86, an inhibitor disrupting LSD1–SNAG domain, potently induces differentiation in AML, which particularly results from physical separation of LSD1 from GFI1, rather than inhibiting the demethylase activity of LSD1. Therefore, pharmacologic displacement of LSD1 from its various cofactors might be another promising mechanism for drug development targeting non-catalytic LSD1.
Conclusions and perspectives
LSD1 has been long known as an eraser enzyme regulating the methylation dynamics of both histones and non-histone proteins, and its biological roles has been attributed to this demethylase activity. The biological functions about its demethylase-independent activity is relatively unexplored. In this present review, we focus on summarizing recent findings about the non-canonical functions of LSD1 beyond its demethylase activity. We first overview LSD1′s structural complexity which is the backbone supporting its diverse functions. With the variety of the non-catalytic domains and the lobed feature of its catalytic core, LSD1 has been shown to possess potent ability for different protein–protein interactions. Then, we briefly describe the canonical functions of LSD1 serving as a demethylase targeting both histones and non-histone proteins and its interacting cofactors. Particularly, we devote a large chunk of efforts to summarize those findings about the non-canonical functions of LSD1 beyond its demethylase activity. First, LSD1 roles as a FBXW7 pseudo-substrate, which triggers FBXW7 self-ubiquitylation and degradation and thus negatively regulates FBXW7 functions in growth suppression, NHEJ repair and radiation protection [18, 71]. Second, LSD1 was shown to protect ERRα from proteasome-dependent degradation in a demethylation-independent manner. Third, LSD1 is able to non-catalytically destabilize p62 and inhibit autophagy in gynecologic cancers. Last but not least, ZNF217 was identified to role as a binding partner of LSD1, the collaboration of LSD1 and ZNF217 promotes survival of castration-resistant prostate cancer cells by activating a lethal prostate cancer gene network, independently of the demethylase activity of LSD1 and of the AR. Overall, these studies largely extend the biological roles and establish novel oncogenic mechanisms of LSD1 via interacting different proteins independent of its demethylase activity. Lastly, we discuss the translational significance of non-catalytic roles of LSD1 in cancer therapies.
However, there are still some questions to be fully addressed for these non-catalytical protein interactions. For example, what is the specific mechanism for that LSD1-FBXW7 binding disrupts FBXW7 dimerization? Further structural analysis is needed to verify that whether there is a conformational change induced by LSD1-FBXW7 binding, or any other mechanisms involved. Also, it remains unknown the reason that why FBXW7 fail to promote the ubiquitylation of LSD1 which contains a typical CPD bind site. For the LSD1–ERRα interaction, why does enzymatic inhibition of LSD1 by pharmaceutical compounds also destabilize ERRα in the case of that LSD1-induced ERRα stabilization does not depend on LSD1 demethylase activity? Though the authors claim that compounds might merely act as a disruptor of ERRα–LSD1 physical contacts, rather than as inhibitors of LSD1 activity [53], further investigation is still needed to explain this contradictory and exclude the possibility that ERRα may be modulated by LSD1-involved methylation dynamic. Another questions is that how does LSD1–ERRα interaction impair the protein stability of ERRα without the demethylation reaction? The author discussed that LSD1 may act as steric hindrance, which shuts down the accessibility of contacting domain within ERRα for ubiquitin ligase [53]. However, the ubiquitin ligase responsible for ERRα degradation is still unknown. Nonetheless, though there are still many questions and challenges existed, we believe that more and more proteins will be identified as LSD1′s binding partners, and the non-catalytic functions of LSD1 is emerging to endow this enzyme with a second life beyond its demethylase activity.
Abbreviations
- AR:
-
Androgen receptor
- AML:
-
Acute myeloid leukemia
- AOL:
-
Amine oxidase-like
- CoREST:
-
REST (RE1-silencing transcription factor) corepressor
- CPD:
-
Cdc4 phosphodegron
- CREB:
-
CAMP-response element-binding protein
- CtBP:
-
C-terminal-binding protein 1
- Dnmt1:
-
DNA methyltransferase 1
- E2F1:
-
Transcription factor
- EMT:
-
Epithelial-to-mesenchymal transition
- ER:
-
Estrogen receptor
- ERRα:
-
Estrogen-related receptor α
- GFI:
-
Growth factor independence
- H3K4me1/2:
-
Mono- and di-methylated histone 3 lysine 4
- H3K9me1/2:
-
Mono- and di-methylated histone 3 lysine 9
- H4K20me2:
-
Di-methylated histone 4 lysine 20
- HIF-1α:
-
Hypoxia-inducible factor-1
- LSD1:
-
Lysine-specific demethylase 1
- MAO:
-
Monoamine oxidase
- MEF2:
-
Myocyte enhancer factor 2
- NHEJ:
-
Nonhomologous end-joining
- NuRD:
-
Nucleosome remodeling and deacetylase
- PTMs:
-
Post-translational modifications
- SCF:
-
SKP1-CUL1-F-box protein
- STAT3:
-
Signal transducer and activator of transcription 3
- SQSTM1:
-
Sequestasome 1
References
Shi Y et al (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119(7):941–953
Shi YJ et al (2005) Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 19(6):857–864
Lee MG, Wynder C, Cooch N, Shiekhattar R (2005) An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437(7057):432–435
Laurent B et al (2015) A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol Cell 57(6):957–970
Metzger E et al (2005) LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437(7057):436–439
Wang J et al (2007) Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446(7138):882–887
Huang J et al (2007) p53 is regulated by the lysine demethylase LSD1. Nature 449(7158):105–108
He Y et al (2018) LSD1 promotes S-phase entry and tumorigenesis via chromatin co-occupation with E2F1 and selective H3K9 demethylation. Oncogene 37(4):534–543
Zhang J et al (2011) Cyclophosphamide perturbs cytosine methylation in Jurkat-T cells through LSD1-mediated stabilization of DNMT1 protein. Chem Res Toxicol 24(11):2040–2043
Lee JY et al (2017) LSD1 demethylates HIF1alpha to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene 36(39):5512–5521
Yang JB et al (2010) Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci USA 107(50):21499–21504
Lan F, Nottke AC, Shi Y (2008) Mechanisms involved in the regulation of histone lysine demethylases. Curr Opin Cell Biol 20(3):316–325
Ambrosio S, Sacca CD, Majello B (2017) Epigenetic regulation of epithelial to mesenchymal transition by the Lysine-specific demethylase LSD1/KDM1A. Biochim Biophys Acta Gene Regul Mech 1860(9):905–910
Lv S et al (2010) LSD1 is required for chromosome segregation during mitosis. Eur J Cell Biol 89(7):557–563
Sakamoto A et al (2015) Lysine demethylase LSD1 coordinates glycolytic and mitochondrial metabolism in hepatocellular carcinoma cells. Cancer Res 75(7):1445–1456
Whyte WA et al (2012) Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482(7384):221–225
Amente S, Lania L, Majello B (2013) The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim Biophys Acta 1829(10):981–986
Lan H et al (2019) LSD1 destabilizes FBXW7 and abrogates FBXW7 functions independent of its demethylase activity. Proc Natl Acad Sci USA 116(25):12311–12320
Chao A et al (2017) Lysine-specific demethylase 1 (LSD1) destabilizes p62 and inhibits autophagy in gynecologic malignancies. Oncotarget 8(43):74434–74450
Si W, Zhao Y, Zhou J, Zhang Q, Zhang Y (2019) The coordination between ZNF217 and LSD1 contributes to hepatocellular carcinoma progress and is negatively regulated by miR-101. Exp Cell Res 379(1):1–10
Aravind L, Iyer LM (2002) The SWIRM domain: a conserved module found in chromosomal proteins points to novel chromatin-modifying activities. Genome Biol 3(8):reserch0039-1
Chen Y et al (2006) Crystal structure of human histone lysine-specific demethylase 1 (LSD1). P Natl Acad Sci USA 103(38):13956–13961
Niwa H, Umehara T (2017) Structural insight into inhibitors of flavin adenine dinucleotide-dependent lysine demethylases. Epigenetics 12(5):340–352
Fang R et al (2013) LSD2/KDM1B and its cofactor NPAC/GLYR1 endow a structural and molecular model for regulation of H3K4 demethylation. Mol Cell 49(3):558–570
Zhang Q et al (2013) Structure-function analysis reveals a novel mechanism for regulation of histone demethylase LSD2/AOF1/KDM1b. Cell Re 23(2):225–241
Peng B et al (2015) Modulation of LSD1 phosphorylation by CK2/WIP1 regulates RNF168-dependent 53BP1 recruitment in response to DNA damage. Nucleic Acids Res 43(12):5936–5947
Metzger E et al (2016) Assembly of methylated KDM1A and CHD1 drives androgen receptor-dependent transcription and translocation. Nat Struct Mol Biol 23(2):132–139
Stavropoulos P, Blobel G, Hoelz A (2006) Crystal structure and mechanism of human lysine-specific demethylase-1. Nat Struct Mol Biol 13(7):626–632
Shi Y et al (2003) Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422(6933):735–738
Wang Y et al (2009) LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138(4):660–672
Perillo B et al (2008) DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319(5860):202–206
Cai CM et al (2014) Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell Rep 9(5):1618–1627
Wang J et al (2015) LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat Neurosci 18(9):1256–1264
Hojfeldt JW, Agger K, Helin K (2013) Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov 12(12):917–930
Saleque S, Kim JW, Rooke HM, Orkin SH (2007) Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell 27(4):562–572
Wu ZQ et al (2012) Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc Natl Acad Sci USA 109(41):16654–16659
Lin YW et al (2010) The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. Embo J 29(11):1803–1816
Ferrari-Amorotti G et al (2013) Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer Res 73(1):235–245
Egolf S et al (2019) LSD1 inhibition promotes epithelial differentiation through derepression of fate-determining transcription factors. Cell Rep 28(8):1981
Maiques-Diaz A et al (2018) Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep 22(13):3641–3659
Majello B, Gorini F, Sacca CD, Amente S (2019) Expanding the role of the histone lysine-specific demethylase LSD1 in cancer. Cancers 11(3):324
Shi Y (2007) Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet 8(11):829–833
Zheng YC et al (2015) A Systematic review of histone lysine-specific demethylase 1 and its inhibitors. Med Res Rev 35(5):1032–1071
Cho HS et al (2011) Demethylation of RB regulator MYPT1 by histone demethylase LSD1 promotes cell cycle progression in cancer cells. Cancer Res 71(3):655–660
Kontaki H, Talianidis I (2010) Lysine methylation regulates E2F1-induced cell death. Mol Cell 39(1):152–160
Baek SH, Kim KI (2016) Regulation of HIF-1 alpha stability by lysine methylation. Bmb Rep 49(5):245–246
Sheng W et al (2018) LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174(3):549–563e519
Lan H et al (2019) LSD1 destabilizes FBXW7 and abrogates FBXW7 functions independent of its demethylase activity. Proc Natl Acad Sci USA 116:12311–12320
Welcker M, Clurman BE (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8(2):83–93
Wang Z, Liu P, Inuzuka H, Wei W (2014) Roles of F-box proteins in cancer. Nat Rev Cancer 14(4):233–247
Horard B, Vanacker JM (2003) Estrogen receptor-related receptors: orphan receptors desperately seeking a ligand. J Mol Endocrinol 31(3):349–357
Carnesecchi J et al (2017) ERRalpha induces H3K9 demethylation by LSD1 to promote cell invasion. Proc Natl Acad Sci USA 114(15):3909–3914
Carnesecchi J, Cerutti C, Vanacker JM, Forcet C (2017) ERR alpha protein is stabilized by LSD1 in a demethylation-independent manner. PLoS One 12(11):e0188871
Moscat J, Diaz-Meco MT (2009) p62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell 137(6):1001–1004
Moscat J, Diaz-Meco MT (2012) p62: a versatile multitasker takes on cancer. Trends Biochem Sci 37(6):230–236
Green SM, Mostaghel EA, Nelson PS (2012) Androgen action and metabolism in prostate cancer. Mol Cell Endocrinol 360(1–2):3–13
Yuan X et al (2014) Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 33(22):2815–2825
Sehrawat A et al (2018) LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Cancer Res 78(13):E4179–E4188
Mohammad HP et al (2015) A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 28(1):57–69
McGrath JP et al (2016) Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res 76(7):1975–1988
Fiskus W et al (2014) Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 28(11):2155–2164
Theisen ER et al (2014) Reversible inhibition of lysine specific demethylase 1 is a novel anti-tumor strategy for poorly differentiated endometrial carcinoma. Bmc Cancer 14:752
Kahl P et al (2006) Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res 66(23):11341–11347
Schulte JH et al (2009) Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res 69(5):2065–2071
Kauffman EC et al (2011) Role of androgen receptor and associated lysine-demethylase coregulators, LSD1 and JMJD2A, in localized and advanced human bladder cancer. Mol Carcinog 50(12):931–944
Harris WJ et al (2012) The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21(4):473–487
Lv T et al (2012) Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE 7(4):e35065
Fu X, Zhang P, Yu B (2017) Advances toward LSD1 inhibitors for cancer therapy. Future Med Chem 9(11):1227–1242
Hosseini A, Minucci S (2017) A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics 9(8):1123–1142
Deshaies RJ (2015) Protein degradation: prime time for PROTACs. Nat Chem Biol 11(9):634–635
Lan H, Sun Y (2019) FBXW7 E3 ubiquitin ligase: degrading, not degrading, or being degraded. Protein Cell 10:861–863
Wang J et al (2009) The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 41(1):125–129
Zhang X et al (2013) Regulation of estrogen receptor alpha by histone methyltransferase SMYD2-mediated protein methylation. Proc Natl Acad Sci USA 110(43):17284–17289
Nair SS, Li DQ, Kumar R (2013) A core chromatin remodeling factor instructs global chromatin signaling through multivalent reading of nucleosome codes. Mol Cell 49(4):704–718
Acknowledgements
This study was supported in part by the Natural Science Foundation of Zhejiang Province (20160171) (YZW), by the Ningbo Natural Science Foundation, China Grant (No. 2016A610148) (ZYY), and by the Medical Scientific Research Foundation of Zhejiang Province, China (Grant No. 2018KY689) (ZYY). Authors’ contributions: HYL had the idea for the article and providing the final approval of the version to be published. YXL was involved in performed the literature search and revision work, FYG was involved in drafting the manuscript. XXW and ZYY were involved in checking the language problems. YZW was involved in revising the manuscript critically for important scientific content. And the authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gu, F., Lin, Y., Wang, Z. et al. Biological roles of LSD1 beyond its demethylase activity. Cell. Mol. Life Sci. 77, 3341–3350 (2020). https://doi.org/10.1007/s00018-020-03489-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03489-9