
Abstract
During embryonic development, one of the two X chromosomes of a mammalian female cell is randomly inactivated by the X chromosome inactivation mechanism, which is mainly dependent on the regulation of the non-coding RNA X-inactive specific transcript at the X chromosome inactivation center. There are three proteins that are essential for X-inactive specific transcript to function properly: scaffold attachment factor-A, lamin B receptor, and SMRT- and HDAC-associated repressor protein. In addition, the absence of X-inactive specific transcript expression promotes tumor development. During the process of chromosome inactivation, some tumor suppressor genes escape inactivation of the X chromosome and thereby continue to play a role in tumor suppression. A well-functioning tumor suppressor gene on the idle X chromosome in women is one of the reasons they have a lower propensity to develop cancer than men, women thereby benefit from this enhanced tumor suppression. This review will explore the mechanism of X chromosome inactivation, discuss the relationship between X chromosome inactivation and tumorigenesis, and consider the consequent sex differences in cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
In 1949, the American scholar Barr et al. found a deeply stained corpuscle in the nucleus of the mitotic interphase of female cats, but not in male cats. He named the corpus the “Barr body” [1]; later studies confirmed that the “Barr body” is an abnormally condensed and inactive X chromosome, the compact shape of which is thought to be the result of its nearly complete inactivation [2]. Further studies determined that while maintaining an active X chromosome in mammalian diploid cells, all remaining X chromosomes are inactivated—a phenomenon called X chromosome inactivation (XCI) [3, 4]. It is achieved by transcriptional silencing of one of the two X chromosomes in the early stages of embryonic development: one of the two X chromosomes in the female is silenced, resulting in the same gene expression as a male with only one X chromosome [3]. The dosage compensation effect based on XCI is a very delicate, systematic process, the core function of which involves the XCI center (XIC) as the main switch seat of XCI. The Xist (X-inactive specific transcript) gene located in the XIC plays a major regulatory role throughout the entire process of XCI.
XCI is a normal developmental regulatory process; however, if the process is abnormal, it will cause interference with X-linked gene silencing at the local and chromosomal levels. Thus, the expression of cancer-related and other genes is altered by XCI—a process that may ultimately cause tumors [5]. At the same time, sex differences in cancer occurrence and progression have long been known [6], with men experiencing a higher incidence of malignancies than women [7]; furthermore, male cancer mortality rates are significantly higher than those in females. These findings led us to consider whether sex differences in cancer may be related to abnormal XCI. This review will describe the mechanism of XCI and the relationship between XCI abnormalities and tumorigenesis, while investigating whether sex differences in cancer patients may also be closely related to abnormal inactivation of the X chromosome.
Mechanism of XCI
Traditional genetic studies have shown that the X chromosome is controlled by a single cis-acting total switching site known as the XIC; it is the main regulatory region for XCI. The X chromosome carrying the XIC can initiate inactivation by homeopath. On the contrary, the X chromosome lacking the XIC is not inactivated. The XIC ensures the appropriate random inactivation of the X chromosome. This locus produces a large non-coding RNA called Xist, which is the major regulatory gene of XCI. It engages in cis-binding and accumulates along the entire chromosome from the transcription site [8].
X chromosome dosage compensation mechanism
In XY sex-determined organisms, female individuals have two X chromosomes, while male individuals have only one X chromosome. Although the Y chromosome was the original homologous chromosome of the X chromosome, it has degraded with time, thus creating an imbalance of X-linked genes between the sexes. Dosage compensation is the mechanism that balances the expression of unequal doses of X-linked genes between males and females and ensures a balance between the expression of a single X chromosome and two autosomal genes. Female mammals contain two X chromosomes, one of which is "closed" to avoid gene overexpression. XCI follows the n − 1 rule: no matter the number of X chromosomes, only one can be randomly retained, thereby balancing the dosage compensation effect between XX and XY chromosomes. The dosage compensation effect is widespread in the biological world. This phenomenon was first discovered in Drosophila melanogaster M. and subsequently confirmed in mammals and Caenorhabditis elegans M.; based on these findings, three possible dose compensation mechanisms were proposed [9]: (1) male single X chromosome expression is doubled in male Drosophila; (2) female mammals inactivate an X chromosome; (3) Caenorhabditis elegans reaches equilibrium by halving the expression of two X chromosomes in females. By virtue of this mechanism, not only is the expression of the X chromosome between the sexes balanced but the expression between the X chromosome and the autosomes is also balanced [10].
The regulatory mechanism of Xist in the process of XCI
Long non-coding RNA (lncRNA) is a class of genes that are longer than 200 nt and do not encode proteins [11]. Among them, lncRNA Xist is the main regulator of mammalian X inactivation [8]; it is necessary for transcriptional silencing of an X chromosome during the development of a female mammal [12, 13]. In normal human tissues, a slightly lower expression level of Xist may lead to a decrease in chromosomal coating efficiency, resulting in the destruction of silencing nuclear compartments in somatic cells [14]; its inappropriate silencing leads to qualitative abnormalities in stem cells [15,16,17,18]. The current view is that Xist is essential for the initiation of XCI in embryonic stem (ES) cell [19] and mouse models [20]; furthermore, Xist deletions lead to long-term inactive X (Xi) maintenance failure in vivo [21, 22].
The RNF12 gene acts as an activator of Xist upstream and its protein product—the ubiquitin ligase that causes the degradation of the Xist repressor protein Rex1—has been shown to play a key role in the XX dose-dependent activation of Xist [23, 24]. Xist is able to aggregate specific proteins and interact with them to cover and silence an X chromosome in each female cell [25], ultimately preventing women from possessing an additional functional X chromosome. Three proteins: SAFA (scaffold attachment factor-A), LBR (lamin B receptor), and SHARP (SMRT- and HDAC-associated repressor protein) are essential for XCI. All three proteins are indispensable; if even one is inactive, Xist will not silence the X chromosome during development. Further analysis has shown that these three proteins play three different but vital roles: (1) the localization of Xist is controlled by the hnRNP U/SAFA family of molecules [26], which bind directly to the transmembrane protein LBR anchored to the inner nuclear membrane [27]; (2) LBR is rich in silencing protein [28, 29] immobilized on the nuclear membrane and interacts with chromatin regulatory proteins and lamin B [30]; (3) LBR recruits inactive X chromosomes to the nuclear layer and changes the three-dimensional structure of DNA. LBR binds to Xist, allowing Xist and its silencing proteins to spread on the X chromosome to silence transcription, thereby remodeling the chromosome and making its genes less likely to be expressed. For a gene to produce a functional protein, the gene must first be transcribed into RNA by RNA polymerase II [13, 14, 25, 26]. The actual "silencing" work is done by the third protein SHARP, which excludes polymerase from DNA, thus preventing transcription and gene expression. The function of RNA-Xist, which does not encode proteins, on the XIC is multifaceted [31]. On the one hand, Xist acts as a modular RNA scaffold in the assembly of inhibitory protein factors. Polycomb repressive complex 1 (PRC1) maintains chromatin stability in a repressed state; it can catalyze the ubiquitination of tyrosine at position 119 of H2A histone9 (H2AK119ub) and participate in the transcriptional silencing of genes. Polycomb repressive complex 2 (PRC2) can catalyze the double or triple methylation of the 27th tyrosine on H3 histones. The methylation of H3K27 (H3K27me3) is a marker of gene silencing caused by PcG protein [32,33,34,35,36,37,38]. The Xist and polycomb complexes are interdependent and propagate during Xi [31]. On the other hand, Xist forms an inhibitory compartment by rejecting transcription and building factors to create a unique Xi chromosome conformation [2, 36, 39,40,41].
The antisense regulatory factor Tsix of Xist is localized in the XIC region, which plays a key role in the expression of Xist [42]. Tsix is upregulated on one of the X chromosomes and is responsible for recruiting the polycomb protein to cis-coat the X chromosome to induce X chromosome silencing. The accumulation of Xist RNA rapidly produces a silent nuclear compartment [43]. The mutual regulatory mechanism of Xist and Tsix has been studied and confirmed by many researchers. Both participate in the physiological process of XCI during early embryo development, including the three stages of initiation, transmission, and maintenance: first, the Xist gene encodes Xist RNA and then the Xist RNA encapsulates the X chromosome, which prepares to initiate XCI [44]. As Xist RNA expands on the X chromosome, DNA methylation and histone modifications (such as H3K9Ac, H3K4me2, and H3K4me3) occur immediately; H4 hypoacetylation occurs shortly thereafter [45], which establishes and maintains XCI. Importantly, inactivated chromosomes continue to synthesize Xist RNA to maintain their inactivation [42].
Xist expression loss and tumorigenesis
A large number of lncRNAs are reportedly associated with cancer [46]; indeed, a direct causal relationship between lncRNA Xist and tumors has been demonstrated [47]. Xist is a key regulator of dosage compensation that randomly inactivates an X chromosome in a mammal during embryonic development. Given that the X chromosome harbors important cell differentiation and proliferation genes—as well as cancer-related genes—genetic changes occurring on vulnerable sex chromosomes may cause immediate damage; moreover, such mutated cells are more likely to develop into tumor cells in the genetic process [3]. Xist-led XCI processes silence hundreds of genes (including oncogenes); therefore, the loss of Xist expression promotes tumor development [48]. Downregulation of Xist expression and loss of XCI are commonly observed in basal-like cancer, breast cancer susceptibility gene 1-null triple-negative breast cancer [49,50,51,52,53,54,55], and ovarian cancer cell lines. In addition, some patients with testicular germ cell tumors were found to have significantly higher levels of Xist demethylation [56,57,58].
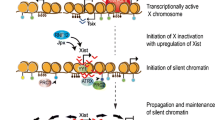
Xist is critical in maintaining XCI, with the deletion of Xist expression causing the inactivated X chromosome to be reactivated, triggering a series of unfavorable genome-wide changes including involvement in DNA replication, chromosome segregation, cell cycle checkpoints, and hematopoietic genetic disorders. As mature hematopoietic cells accumulate, they can potentially cause a wide range of X-linked genes to include tumor-associated gene de-suppression, thereby driving tumorigenesis. Related studies have revealed that the loss of Xist in mouse hematopoietic cells leads to the reactivation of the inactivated X chromosome leading to a genome-wide change in cancer, inducing aggressive and fatal leukemia [47]. Xist has been found to be disordered in a variety of human cancers [59]; many cancer cell lines derived from female breast, cervical, and ovarian tumors show a loss of Xist expression levels [53, 60, 61]. In addition, the use of human induced pluripotent stem cells (iPSCs) to explore the role of Xist RNA shows that the loss of Xist expression in iPSCs is significantly associated with upregulation of X-linked oncogenes, with a higher rate of growth of oncogenes in vivo and poorer differentiation in vitro [17]. Therefore, we conclude that Xist is involved in epigenetic, transcriptional, and post-transcriptional regulation; furthermore, it is abnormally expressed in many diseases, especially in tumors. It is a novel molecule involved in carcinogenesis and tumor suppressor pathways in tumor pathology (Fig. 1).

The regulation of Xist in X chromosome inactivation. a RNF12 acts as an activator of Xist and secretes a protein that degrades the Xist repressor protein Rex1. b The SAFA family controls the positioning of Xist and can prompt Xist to directly bind to the lamin B receptor; LBR binds to Xist, allowing Xist and its silencing protein to spread on the X chromosome to silence transcription, thereby remodeling the chromosome so that it does not express. c SHARP excludes polymerase from DNA, thus preventing transcription and gene expression; the Xist and polycomb complexes are interdependent, spread on Xi, and form inhibitory compartments by repelling transcription and building factors to create a unique Xi chromosome conformation. Xist guarantees that the X chromosome inactivation process is carried out in an orderly manner. If Xist is deleted, the originally inactivated X chromosome may be reactivated, causing the degeneration of tumor suppression-associated genes, thereby inducing cancer
Escape from X-inactivation tumor suppressor genes and tumorigenesis
Tumor suppressor genes escape XCI
Most of the genes located on the X chromosome of female mammals are silenced by the inactivation of the X chromosome [62, 63]. However, not all genes on Xi are silenced, as it is reported that approximately 15% of X-linked genes evade XCI to some extent and continue to be expressed on Xa and Xi [64, 65]. Many chromosomal regions of the X chromosome including Xp11-22, Xq25-26, and Xq27-28 have been proposed as potential loci for tumor suppressor genes and many tumor suppressor genes in this region can escape XCI [66,67,68,69,70,71,72,73,74,75,76,77,78]. These escaped genes are often located outside the Xist RNA-coating region [14, 79]. Given that they escape the XCI mechanism, the X chromosome tumor suppressor genes can be expressed and thus have a tumor suppressor function. We call these genes “escape X chromosome loss” or “escape from X-inactivation tumor-suppressor (EXITS)” genes.
Escaped gene sequences often exist in the pseudoautosomal region (PAR) on the X sex chromosome. PAR is a region of mammalian X and Y chromosomes that is homologous to autosomes, mainly at the two ends of the sex chromosome, and plays a crucial role in the pairing and genetic recombination of sex chromosomes during meiosis. There are two groups of major escapers on the X chromosome: one group contains escapers located in the PAR and has precise homologs on the Y chromosome, showing equal expression levels between the sexes [80]; the second group consists of genes located outside of the PAR, which have Y-chromosome homologs also located outside of the PAR and may represent evolutionary residues from the original sex chromosomes. There is a large difference in the actual degree of escape of genes from these different tissues or species in the XCI process [81,82,83].
Tumor suppressor gene mutation
The traditional Knudson’s two-hit mechanism assumes that the occurrence of tumors requires more than two mutations. As age increases, the probability of cancer increases; that is, the occurrence of cancer requires the accumulation of multiple mutations [84,85,86,87]. However, the study of X-linked tumor suppressor genes challenges the "two-hit inactivation" theory of tumor suppressor genes and introduces a new concept: a single genetic hit can also result in the loss of tumor suppressor function. The loss of function of the tumor suppressor locus is followed by a loss of heterozygosity (LOH), thus losing the wild-type tumor suppressor gene [88,89,90]. LOH is generally associated with tumor suppressor genes (such as p53) that inhibit the development of malignant tumors when both alleles are present. In a female cell, when an X chromosome has been inactivated, the tumor suppressor gene is in an inactive state; when the other tumor suppressor allele on the active X chromosome is mutated or deleted, the malignant state is no longer inhibited, resulting in a decrease in the level of tumor suppressor function during tumor development and progression, and the cells are transformed into cancer cells. This phenomenon is common in retinoblastoma, breast cancer, and other cancers caused by mutations in tumor suppressor genes [91,92,93]. LOH on the active X chromosome may result in the complete loss of tumor suppressor function in these X-linked genes, making the individual susceptible to cancer formation [3, 94]; this is in contrast to the biallelic inactivation of the autosomal tumor suppressor gene, the expression levels of which remain sufficient to function in human cancers.
Based on a comprehensive analysis of the mutation status of human cancer genes, genes such as ATRX (alpha-thalassemia retardation syndrome, X-linked), KDM6A (lysine demethylase 6A), CNKSR2 (connector enhancer of kinase suppressor of ras 2), DDX3X (DEAD-box helicase 3, X-linked), KDM5C (lysine demethylase 5C), and MAGEC3 (melanoma-associated antigen C3)—located on the X chromosome—can escape XCI and thus play a role in inhibiting tumorigenesis. However, these genes have a higher frequency of mutations in cancer and are presumed to be important candidate EXITS genes [95, 96]. Among these six genes, research on the protein KDM6A (also known as UTX) is currently more extensive. The KDM6A gene is mainly located in Xp11.2 and encodes histone H3 lysine 27 (H3K27) demethylase expressed by two X chromosomes [97]; it was found to be an EXITS gene in a mouse lymphoma model [98] that is mainly responsible for preventing the uncontrolled division of cells. In cancer patients, low expression of KDM6A results in a significant survival rate, while the absence of KDM6A not only accelerates the onset of lymphoma, but also strongly promotes tumor progression [99]. Ultimately, the mutation of the gene may weaken this constraint system and cause cancer (Fig. 2).

The study of X-linked tumor suppressor genes challenges the two-hit inactivation theory. a The traditional Knudson’s two-hit hypothesis holds that malignant tumor occurs after two or more mutations, with the first mutation occurring in germ or somatic cells and the second occurring in somatic cells. b There are tumor suppressor genes on both the female and male X chromosomes. For females, if the tumor suppressor gene on the active X chromosome is mutated, the EXITS gene can function normally; males do not have the protection of the corresponding copy and a harmful mutation of the tumor suppressor gene can cause cancer
Sex differences in XCI and cancer
Abnormal inactivation of the X chromosome promotes the development of tumors, at the same time, tumors occur in different environments in males and females. An increasing number of studies report that the occurrence, progression, molecular phenotype, and response to treatment are generally biased based on sex [100]. The incidence of most tumor types in men is on the rise [101]; many malignant tumors including esophageal cancer are diagnosed more often in men and are characterized by worse prognosis leading to higher mortality [102]. The link between the X chromosome and cancer applies to men. For example, XXY males carrying the BRCA1 mutation have a 20- to 50-fold increased risk of breast cancer [103].
The biallelic expression of the EXITS gene may explain the reduction in cancer incidence in women [17]. When an X chromosome is inactivated, the tumor suppressor gene on it is inactivated; meanwhile, another tumor suppressor gene on another active X chromosome is also inactivated and a mutation or deletion is required to allow cancer to occur [104,105,106,107]. Although the EXITS gene is located on the X chromosome, it escapes from the inactivation of the X chromosome and continues to play a role in suppressing cancer, thereby reducing the risk of cancer in women. In males, there is only one X chromosome, and the inactivation of a single copy of the X chromosome tumor suppressor gene can allow cancer to occur; this may be one of the reasons why male cancer incidence and mortality are higher than those in females. Evidence has shown that these six important candidate EXITS genes evade XCI, leading to sex bias [108]. Compared to women, mutation in the EXITS gene is a common phenomenon in men [109] with ATRX, CNKSR2, DDX3X, KDM5C, KDM6A, and MAGEC3 having a higher frequency of mutation loss [95, 110,111,112]. To date, little is known about this mechanism that leads to differences in individuals and cell types [64, 80, 113,114,115]. We conclude that the expression of the biallelic gene of the female EXITS gene reduces female cancer mortality in different tumor types relative to men.
The lack of an idle X chromosome that bears a well-functioning tumor suppressor gene is one of the reasons why men have a greater propensity to develop cancer. The genes that are more frequently mutated in men are found only on the X chromosome, some of which are known to be tumor suppressor genes that evade X inactivation; the protection provided by working copies of these genes in female cells may help explain why many cancers have a lower incidence among women. One of the common causes of cancer in men is that male cells need only one harmful mutation in a tumor suppressor gene to cause cancer. In contrast, female cells need two copies to be mutated to cause cancer. In males, the homolog of KDM6A on the Y chromosome is called KDM6C (also known as UTY); its catalytic activity is very low or non-existent [116]. Furthermore, the inactivation mutation of UTX located on the male X chromosome is not compensated by UTY. An X chromosome in a female cell is closed during embryogenesis and remains inactive throughout life. The inactivated KDM6A gene on the X chromosome “avoids” this dormant state and functions normally. A “good” copy of the gene is sufficient to prevent the cell from becoming cancerous [95]. There is no doubt that the EXITS mutation is not the only explanation for the difference in the incidence of cancer in men and women. However, the environmental and hormonal factors associated with sex-specific differences in cancer may interact with the EXITS locus or its gene products to modulate cancer risk [95].
Conclusions and perspectives
XCI is closely related to the occurrence and development of tumors [5]. There are more than 1000 genes on the X chromosome, accounting for 5% of the entire genome. A large number of tumor suppressor genes are located on the X chromosome [117]. Epigenetic instability of inactivated X chromosomes appears to occur extensively in breast cancer types [43]. The heterochromatin structure of Barr bodies often disappears in invasive tumors such as breast cancer cells [103, 118, 119]; a common cause of its disappearance may be due to the overall disturbance of nuclear tissue in cancer cells and the destruction of its heterochromatic structure [43]. In theory, the use of IF and RNA FISH to detect X-linked gene reactivation and abnormal chromatin status in breast tumors can provide valuable biological indicators to assess epigenetic status and tumor response to drug therapy [120]. Given that the loss of Xist expression may potentially drive tumorigenesis [47, 121, 122], it would be reasonable to recommend Xist reactivation as a treatment strategy for cancer. In tumors, Xist expression can be reactivated by small molecules (such as Xist expression vectors), providing a novel therapeutic approach to target epigenetically functional lncRNA. In ovarian cancer cell lines, Xist expression can serve as a prognostic marker associated with chemotherapy response [123]. If a molecule that induces Xist expression is identified, improved patient prognosis may become feasible [124].
The EXITS gene escapes the inactivation of the X chromosome, allowing for the expression of X chromosome tumor suppressor genes and thereby invoking a tumor suppressing function. Perhaps during tumor treatment, the tumor suppressor gene on the X chromosome can be inactivated by artificial escape. It is believed that this will bring about a qualitative leap and breakthrough in related diseases, especially the diagnosis and treatment of female diseases.
Among the various individual differences, sex differences are one of the most interesting research topics. Sex is an important factor in the occurrence, diagnosis, and prognosis of many diseases [125]. Numerous clinical studies have shown that the incidence of many tumors involves significant sex differences. If we want to better understand different cancers, the key is to determine the reasons underlying sex differences. At present, there is a lack of targeted individualized prevention and treatment programs in clinical settings. To understand the different responses of men and women to cancer treatment due to their genetic differences, future clinical studies on cancer management should involve adequate numbers of patients and tumor tissue samples. To better understand the pathogenesis of certain diseases in humans, the sex ratio of the disease should be further clarified. This will help optimize treatment for both sexes, identify possible sex-biased protections or harmful factors, and may assist the development of new treatment strategies.
References
Barr ML, Bertram EG (1949) A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 163:676
Giorgetti L, Lajoie BR, Carter AC et al (2016) Structural organization of the inactive X chromosome in the mouse. Nature 535:575–579
Spatz A, Borg C, Feunteun J (2004) X-chromosome genetics and human cancer. Nat Rev Cancer 4:617
Graves JA (2003) Mammals that break the rules: genetics of marsupials and monotremes. Annu Rev Genet 30:233–260
Chaligne R, Heard E (2014) X-chromosome inactivation in development and cancer. FEBS Lett 588:2514–2522
Clemmesen J, Busk T (1947) Cancer mortality among males and females in Denmark, England and Switzerland; Danish towns and rural areas. Cancer Res 7:286
Wei F, Wu Y, Tang L et al (2017) Trend analysis of cancer incidence and mortality in China. Sci China Life Sci 60:1271–1275
Brown CJ, Ballabio A, Rupert JL et al (1991) A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349:38–44
Lucchesi JC, Kelly WG, Panning B (2005) Chromatin remodeling in dosage compensation. Annu Rev Genet 39:615–651
Gupta V, Parisi M, Sturgill D et al (2006) Global analysis of X-chromosome dosage compensation. J Biol 5:3
Bo H, Fan L, Li J et al (2018) High Expression of lncRNA AFAP1-AS1 promotes the progression of colon cancer and predicts poor prognosis. J Cancer 9:4677–4683
Lee JT (2009) Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev 23:1831–1842
Wutz A (2011) Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet 12:542–553
Chaumeil J, Le Baccon P, Wutz A, Heard E (2006) A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 20:2223–2237
Shen Y, Matsuno Y, Fouse SD et al (2008) X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc Natl Acad Sci USA 105:4709–4714
Silva SS, Rowntree RK, Mekhoubad S et al (2008) X-chromosome inactivation and epigenetic fluidity in human embryonic stem cells. Proc Natl Acad Sci 105:4820–4825
Anguera MC, Sadreyev R, Zhang Z et al (2012) Molecular signatures of human induced pluripotent stem cells highlight sex differences and cancer genes. Cell Stem Cell 11:75–90
Mekhoubad S, Bock C, de Boer AS et al (2012) Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 10:595–609
Penny GD, Kay GF, Sheardown SA et al (1996) Requirement for Xist in X chromosome inactivation. Nature 379:131–137
Marahrens Y, Panning B, Dausman J et al (1997) Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev 11:156–166
Brown CJ, Willard HF (1994) The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature 368:154–156
Csankovszki G, Panning B, Bates B et al (1999) Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat Genet 22:323–324
Gontan C, Achame EM, Demmers J et al (2012) RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature 485:386–390
Barakat TS, Gunhanlar N, Pardo CG et al (2011) RNF12 activates Xist and is essential for X chromosome inactivation. PLoS Genet 7:e1002001
Engreitz JM, Pandya-Jones A, McDonel P et al (2013) The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341:1237973
Sakaguchi T, Hasegawa Y, Brockdorff N et al (2016) Control of chromosomal localization of Xist by hnRNP U family molecules. Dev Cell 39:11–12
Chen CK, Blanco M, Jackson C et al (2016) Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science 354:468
Kind J, van Steensel B (2010) Genome-nuclear lamina interactions and gene regulation. Curr Opin Cell Biol 22:320–325
Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL (2005) The nuclear lamina comes of age. Nat Rev Mol Cell Biol 6:21–31
Worman HJ, Yuan J, Blobel G et al (1988) A lamin B receptor in the nuclear envelope. Proc Natl Acad Sci USA 85:8531–8534
Colognori D, Sunwoo H, Kriz AJ, Wang CY, Lee JT (2019) Xist deletional analysis reveals an interdependency between Xist RNA and polycomb complexes for spreading along the inactive X. Mol Cell 74:101–117
Schoeftner S, Sengupta AK, Kubicek S et al (2006) Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J 25:3110–3122
Zhao J, Sun BK, Erwin JA et al (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322:750–756
Chu C, Zhang QC, da Rocha ST et al (2015) Systematic discovery of Xist RNA binding proteins. Cell 161:404–416
McHugh CA, Chen CK, Chow A et al (2015) The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521:232–236
Wang YA, Li XL, Mo YZ et al (2018) Effects of tumor metabolic microenvironment on regulatory T cells. Mol Cancer 17:168
Moindrot B, Cerase A, Coker H et al (2015) A pooled shRNA screen identifies Rbm15, Spen, and Wtap as factors required for Xist RNA-mediated silencing. Cell Rep 12:562–572
Ren D, Hua Y, Yu B et al (2020) Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Mol Cancer 19:19
Nora EP, Lajoie BR, Schulz EG et al (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485:381–385
Rao SS, Huntley MH, Durand NC et al (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159:1665–1680
Deng X, Xiong F, Li X et al (2018) Application of atomic force microscopy in cancer research. J Nanobiotech 16:102
Kalantry S (2011) Recent advances in X-chromosome inactivation. J Cell Physiol 226:1714–1718
Chaligne R, Popova T, Mendoza-Parra MA et al (2015) The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Res 25:488–503
Wutz A, Jaenisch R (2000) A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol Cell 5:695–705
Keohane AM, O’Neill LP, Belyaev ND et al. (1996) X-inactivation and histone H4 acetylation in embryonic stem cells. Dev Biol 180:0–630.
Spizzo R, Almeida MI, Colombatti A, Calin GA (2012) Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene 31:4577–4587
Patrat C, Ouimette JF, Rougeulle C (2020) X chromosome inactivation in human development. Development. https://doi.org/10.1242/dev.183095
Yildirim E, Kirby JE, Brown DE et al (2013) Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 152:727–774
Pageau GJ, Hall LL, Lawrence JB (2007) BRCA1 does not paint the inactive X to localize XIST RNA but may contribute to broad changes in cancer that impact XIST and Xi heterochromatin. J Cell Biochem 100:835–850
Silver DP, Dimitrov SD, Feunteun J et al (2007) Further evidence for BRCA1 communication with the inactive X chromosome. Cell 128:991–1002
Vincent-Salomon A, Ganem-Elbaz C, Manie E et al (2007) X inactive-specific transcript RNA coating and genetic instability of the X chromosome in BRCA1 breast tumors. Cancer Res 67:5134–5140
Sirchia SM, Tabano S, Monti L et al (2009) Misbehaviour of XIST RNA in breast cancer cells. PLoS ONE 4:e5559
Sirchia SM (2005) Loss of the inactive X chromosome and replication of the active X in BRCA1-defective and wild-type breast cancer cells. Cancer Res 65:2139–2146
Ganesan S, Silver DP, Greenberg RA et al (2002) BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell 111:393–405
Fan C, Tang Y, Wang J et al (2017) Role of long non-coding RNAs in glucose metabolism in cancer. Mol Cancer 16:130
Looijenga LHJ, Gillis AJM, Gurp RJV et al (1997) X inactivation in human testicular tumors: XIST expression and androgen receptor methylation status. Am J Pathol 151:581–590
Lobo J, Nunes SP, Gillis AJM et al (2019) XIST-promoter demethylation as tissue biomarker for testicular germ cell tumors and spermatogenesis quality. Cancers (Basel). https://doi.org/10.3390/cancers11091385
Kawakami T, Okamoto K, Ogawa O, Okada Y (2004) XIST unmethylated DNA fragments in male-derived plasma as a tumour marker for testicular cancer. Lancet 363:40–42
Weakley SM, Wang H, Yao Q, Chen C (2011) Expression and function of a large non-coding RNA gene XIST in human cancer. World J Surg 35:1751–1756
Kawakami T, Zhang C, Taniguchi T et al (2004) Characterization of loss-of-inactive X in Klinefelter syndrome and female-derived cancer cells. Oncogene 23:6163–6169
Richardson AL, Wang ZC, De Nicolo A et al (2006) X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 9:121–132
Lyon MF (1961) Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190:372–373
Plath K, Mlynarczyk-Evans S, Nusinow DA, Panning B (2002) Xist RNA and the mechanism of X chromosome inactivation. Annu Rev Genet 36:233–278
Carrel L, Willard H (2005) X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 434:400–404
Cotton AM, Ge B, Light N et al (2013) Analysis of expressed SNPs identifies variable extents of expression from the human inactive X chromosome. Genome Biol 14(11):R122
Liu R, Kain M, Wang L (2012) Inactivation of X-linked tumor suppressor genes in human cancer. Fut Oncol 8:463–481
Yang-Feng TL, Li S, Han H et al (1992) Frequent loss of heterozygosity on chromosomes Xp and 13q in human ovarian cancer. Int J Cancer 52:575–580
Choi C, Kim MH, Juhng SW (1998) Loss of heterozygosity on chromosome XP22.2-p22.13 and Xq26.1-q27.1 in human breast carcinomas. J Korean Med Sci 13:311
Mo Y, Wang Y, Zhang L et al (2019) The role of Wnt signaling pathway in tumor metabolic reprogramming. J Cancer 10:3789–3797
Feder M, Liu Z, Apostolou S et al (2000) Loss of chromosomes 1 and X in a renal oncocytoma—cancer genetics and cytogenetics. Cancer Genet Cytogenet 123:71–72
Edelson MI, Lau CC, Colitti CV et al (1998) A one centimorgan deletion unit on chromosome Xq12 is commonly lost in borderline and invasive epithelial ovarian tumors. Oncogene 16:197–202
Thrash-Bingham CA, Salazar H, Greenberg RE et al (1996) Loss of heterozygosity studies indicate that chromosome arm Ip harbors a tumor suppressor gene for renal oncocytomas. Genes Chromosomes Cancer 16:64–67
Xu J, Meyers D, Freije D et al (1998) Evidence for a prostate cancer susceptibility locus on the X chromosome. Nat Genet 20:175–179
Wu Y, Wei F, Tang L et al (2019) Herpesvirus acts with the cytoskeleton and promotes cancer progression. J Cancer 10:2185–2193
Tang Y, Wang J, Lian Y et al (2017) Linking long non-coding RNAs and SWI/SNF complexes to chromatin remodeling in cancer. Mol Cancer 16:42
Fujino T, Risinger JI, Collins NK et al (1994) Allelotype of endometrial carcinoma. Cancer Res 54:4294–4298
Buekers TE, Lallas TA, Buller RE (2000) Xp22.2-3 Loss of heterozygosity is associated with germline BRCA1 mutation in ovarian cancer. Gynecol Oncol 76:418-422
Loupart ML, Adams S, Armour JA et al (1995) Loss of heterozygosity on the X chromosome in human breast cancer. Genes Chromosomes Cancer 13:229–238
Simon MD, Pinter SF, Fang R et al (2013) High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 504:465–469
Johnston CM, Lovell FL, Leongamornlert DA et al (2008) Large-scale population study of human cell lines indicates that dosage compensation is virtually complete. PLoS Genet 4:e9
Yang F, Babak T, Shendure J, Disteche CM (2010) Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res 20:614–622
Wu H, Luo J, Yu H et al (2014) Cellular resolution maps of X chromosome inactivation: implications for neural development, function, and disease. Neuron 81:103–119
Calabrese JM, Sun W, Song L et al (2012) Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell 151:951–963
Peng M, Mo Y, Wang Y et al (2019) Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer 18:128
Fan C, Tu C, Qi P et al (2019) GPC6 Promotes cell proliferation, migration, and invasion in nasopharyngeal carcinoma. J Cancer 10:3926–3932
Xiao L, Wei F, Liang F et al (2019) TSC22D2 identified as a candidate susceptibility gene of multi-cancer pedigree using genome-wide linkage analysis and whole-exome sequencing. Carcinogenesis 40:819–827
Ge J, Wang J, Wang H et al (2020) The BRAF V600E mutation is a predictor of the effect of radioiodine therapy in papillary thyroid cancer. J Cancer 11:932–939
Wang W, Zhou R, Wu Y et al (2019) PVT1 promotes cancer progression via MicroRNAs. Front Oncol 9:609
Jin K, Wang S, Zhang Y et al (2019) Long non-coding RNA PVT1 interacts with MYC and its downstream molecules to synergistically promote tumorigenesis. Cell Mol Life Sci 76:4275–4289
Tu C, Zeng Z, Qi P et al (2018) Identification of genomic alterations in nasopharyngeal carcinoma and nasopharyngeal carcinoma-derived Epstein–Barr virus by whole-genome sequencing. Carcinogenesis 39:1517–1528
Knudson AG (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci 68:820–823
Wang S, Gao J, Lei Q et al (2003) Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4:209–221
Wu P, Mo Y, Peng M et al (2020) Emerging role of tumor-related functional peptides encoded by lncRNA and circRNA. Mol Cancer 19:22
Liao DJ, Du QQ, Yu BW et al (2003) Novel perspective: focusing on the X chromosome in reproductive. Cancers 21:641–658
Dunford A, Weinstock DM, Savova V et al (2017) Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat Genet 49:10–16
Clocchiatti A, Cora E, Zhang Y, Dotto GP (2016) Sexual dimorphism in cancer. Nat Rev Cancer 16:330–339
Bellott DW, Hughes JF, Skaletsky H et al (2014) Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 508:494–499
Li X, Zhang Y, Zheng L et al (2018) UTX is an escape from X-inactivation tumor-suppressor in B cell lymphoma. Nat Commun 9:2720
Snijders Blok L, Madsen E, Juusola J et al (2015) Mutations in DDX3X Are a common cause of unexplained intellectual disability with gender-specific effects on Wnt signaling. Am J Hum Genet 97:343–352
Duijf PHG, Schultz N, Benezra R (2013) Cancer cells preferentially lose small chromosomes. Int J Cancer 132:2316–2326
Edgren G, Liang L, Adami HO, Chang ET (2012) Enigmatic sex disparities in cancer incidence. Eur J Epidemiol 27:187–196
Cook MB, McGlynn KA, Devesa SS et al (2011) Sex disparities in cancer mortality and survival. Cancer Epidemiol Biomarkers Prev 20:1629–1637
Fentiman IS, Fourquet A, Hortobagyi GN (2006) Male breast cancer. The Lancet 367:595–604
Greenfield A, Carrel L, Pennisi D et al (1998) The UTX gene escapes X inactivation in mice and humans. Hum Mol Genet 7:737–742
Wu C, Li M, Meng H et al (2019) Analysis of status and countermeasures of cancer incidence and mortality in China. Sci Chin Life Sci 62:640–647
Xiong F, Deng S, Huang HB et al (2019) Effects and mechanisms of innate immune molecules on inhibiting nasopharyngeal carcinoma. Chin Med J (Engl) 132:749–752
Fan CM, Wang JP, Tang YY et al (2019) circMAN1A2 could serve as a novel serum biomarker for malignant tumors. Cancer Sci 110:2180–2188
Tukiainen T, Pirinen M, Sarin AP et al (2014) Chromosome X-Wide association study identifies loci for fasting insulin and height and evidence for incomplete dosage compensation. PLoS Genet 10:e1004127
Miyake N, Koshimizu E, Okamoto N et al (2013) MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am J Med Genet A 161A:2234–2243
Van der Meulen J, Sanghvi V, Mavrakis K et al (2015) The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood 125:13–21
Yoshida K, Sanada M, Shiraishi Y et al (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478:64–69
Van Vlierberghe P, Palomero T, Khiabanian H et al (2010) PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet 42:338–342
Berletch JB, Ma W, Yang F et al (2015) Escape from X inactivation varies in mouse tissues. PLoS Genet 11:e1005079
Berletch JB, Yang F, Xu J, Carrel L et al (2011) Genes that escape from X inactivation. Hum Genet 130:237–245
Talebizadeh Z, Simon SD, Butler MG (2006) X chromosome gene expression in human tissues: male and female comparisons. Genomics 88(6):675–681
Walport LJ, Hopkinson RJ, Vollmar M et al (2014) Human UTY(KDM6C) is a male-specific N-methyl lysyl demethylase. J Biol Chem 289:18302–18313
Ross MT, Grafham DV, Coffey AJ et al (2005) The DNA sequence of the human X chromosome. Nature 434:325–337
Perry M (1972) Evaluation of breast tumour sex chromatin (Barr body) as an index of survival and response to pituitary ablation. Br J Surg 59:731–734
Serdy KM, Leone JP, Dabbs DJ, Bhargava R (2017) Male breast cancer. Am J Clin Pathol 147:110–119
Bennett CL, Christie J, Ramsdell F et al (2001) The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27:20–21
Zhang Y, Xia M, Jin K et al (2018) Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer 17:45.
Rack KA, Chelly J, Gibbons RJ et al (1994) Absence of the XIST gene from late-replicating isodicentric X chromosomes in leukaemia. Hum Mol Genet 3:1053–1059
Huang KC, Rao PH, Lau CC et al (2002) Relationship of XIST expression and responses of ovarian cancer to chemotherapy. Mol Cancer Ther 1:769–776
Mo Y, Wang Y, Xiong F et al (2019) Proteomic analysis of the molecular mechanism of lovastatin inhibiting the growth of nasopharyngeal carcinoma cells. J Cancer 10:2342–2349
Yang L, Tang Y, He Y et al (2017) High Expression of LINC01420 indicates an unfavorable prognosis and modulates cell migration and invasion in nasopharyngeal carcinoma. J Cancer 8:97–103
Acknowledgements
This study was supported by grants from The National Natural Science Foundation of China (81672683, 81672993, 81702907, 81772928, 81803025, 81872278, and 81972776), the Natural Science Foundation of Hunan Province (2018SK21210, 2018SK21211, 2018JJ3704, 2018JJ3815, and 2017SK2105) and the Fundamental Research Funds for the Central Universities of Central South University (2019zzts712 and 2019zzts089).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, D., Tang, L., Wu, Y. et al. Abnormal X chromosome inactivation and tumor development. Cell. Mol. Life Sci. 77, 2949–2958 (2020). https://doi.org/10.1007/s00018-020-03469-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03469-z