
Abstract
Pancreatic cancer (PC) remains one of the most extremely lethal malignancies worldwide due to late diagnosis and early metastasis, with a 1-year overall survival rate of approximately 20%. The hypoxic microenvironment, induced by intratumoral hypoxia, promotes tumor invasion and progression, leading to chemotherapy or radiotherapy resistance and eventual mortality after treatment of PC. However, the role of the hypoxic microenvironment in PC is complicated and requires further investigation. In this article, we review recent advances regarding the regulation of malignant behaviors in PC, which provide insight into the potential of hypoxic microenvironment activation therapy for the therapeutic agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pancreatic cancer (PC) is the fourth leading cause of cancer-related mortality in the United States, with an overall survival rate of only 8% [1]. However, clinical outcomes are quite modest due to acquired and inherent chemoresistance. Although research advances have been made to develop chemotherapy options and certain biomarkers have been used for patient-targeted therapeutic strategies, there has been no significant improvement in preventing PC progression and metastasis. The limited availability of diagnostic channels and the use of surgery as the only existing curative option with an overall survival of only 10% of patients diagnosed contribute to the terrible nature of this disease [2].
Increasing interest has been concentrated on the tumor microenvironment of PC. Cancer cells, stromal cells, and extracellular components are significant constituents of the tumor microenvironment. Compared with most solid tumors, PC is recognized as lowest level of oxygen [3]. This is possibly a common phenomenon given the fibrotic vasculature, which is ineffective. Recent studies have begun to delineate that the hypoxia microenvironment, a fundamental characteristic in PC, plays a critical role in PC development [4]. In this review, we summarize the current information about the role of hypoxia in promoting the progression of PC and provide new insights for developing targeted therapies for PC.
Hypoxia promotes tumorigenesis in PC
Hypoxia, which is defined as reduced oxygen levels, occurs in 50–60% of locally advanced solid tumors, including PC. In PC, there is a reduction in the tissue oxygen partial pressure in tumors, with a median partial pressure of oxygen (PO2) of 0–5.3 mmHg (0–0.7%); this is distinct from the median PO2 in normal pancreas (24.3–92.7 mmHg, 3.2–12.3%) [3, 5]. A signature feature of PC is the formation of a compressed desmoplastic stroma with poor tumor cellularity that surrounds the tumor [6]. Hypoxia gives rise to several changes in gene expression in tumor cells. This adaptive response to hypoxia is activated by transcription factors such as hypoxia-inducible factors (HIFs), which in turn stimulate the expression of related genes involved in angiogenesis and glycolysis, thus prompting PC cells to adapt to the hypoxic conditions.
HIFs are heterodimeric transcription factors composed of an unstable α subunit (HIFα) and a stable β subunit (HIFβ) [7]. In normoxia, HIFα protein subunits have a rapid half-life (< 5 min) because they are continually transcribed and rapidly degraded. This turnover is mediated by the posttranslational hydroxylation of highly conserved proline residues (Pro564 and Pro402 in HIF1α, Pro405 and Pro531 in HIF2α) within their N-terminal oxygen-dependent degradation domains (NODDs) and C-terminal oxygen-dependent degradation domains (CODDs) by proteins containing proline hydroxylase domains (PHDs) [8]. Under hypoxia, HIFα subunits translocate into the nucleus to bind with HIFβ due to the suppression of factor inhibiting HIF (FIH) and PHD activities. The heterodimeric HIFα:HIFβ transcription factor complex binds to hypoxia-responsive elements (HREs) of its target genes and adjusts their transcription [9].
Previous studies have demonstrated that HIF1 could induce a large number of downstream transactivating genes encoding glucose transporters and glycolytic enzymes in response to hypoxia [10]. With the decrease in oxygen levels, the production of adenosine triphosphate (ATP) changes from the oxidative phosphorylation pathway to the oxygen-independent pathway of cytoplasmic glycolysis, a phenomenon known as the Warburg effect [11, 12]. Regarding the generation of ATP, glycolysis is less efficient than oxidative phosphorylation. However, due to the increase in glycolytic enzyme activity, ATP production could be sustained by sufficient glycolysis. To increase the glucose supply, HIF1 promotes the increase in the transcription of glucose transporters 1 and 3 (GLUT1 and GLUT3) and facilitates the production of pyruvate and lactate dehydrogenase [13]. The lactate generated from glycolysis is further utilized as energy for surrounding cells and weakens the production of T-cell cytokines. Pyruvate dehydrogenase kinase 1 (PDK1) can prevent pyruvate dehydrogenase from using pyruvate to fuel the mitochondrial tricarboxylic acid cycle (TCA cycle), causing a drop in mitochondrial oxygen consumption [14]. In a hypoxic environment, low oxygen levels cannot support oxidative phosphorylation in mitochondria, and HIF1 reduces mitochondrial oxygen consumption via activation of pyruvate dehydrogenase kinase 1 (PDK1) [15]. Another characteristic of a hypoxic environment in PC is an increase in tumor cell migration. There are multiple pathways activated by HIF1 that promote epithelial to mesenchymal transition (EMT), which is the transformation of cells from a polarized epithelial phenotype to fibroblast-like mesenchymal phenotype. Hypoxia and HIFs promote EMT by upregulating transcriptional repressors such as SNAIL, TWIST, and ZFHX1B and induce degradation of the extracellular matrix (ECM) via various proteins such as cathepsin and matrix metalloproteinase-1 and -2. In addition, hypoxia-induced expression of vascular endothelial growth factor (VEGF) promotes lymphangiogenesis and angiogenesis, which drive tumor cell intravasation and dissemination [16].
Furthermore, an increasing number of findings supports the essential role of noncoding RNAs because they are aberrantly expressed under hypoxia and participate in tumor cell migration and metastasis. Based on the complicated roles of noncoding RNAs in tumor metastasis, targeting them in signaling pathways related to hypoxia has been emphasized recently [17]. Noncoding RNAs include microRNAs (miRNAs, 18–22 nucleotide) and long noncoding RNAs (lncRNAs, ≥ 200 nucleotides). MiRNA research offers molecular insight into tumor aggression and carcinogenesis. The mature form of these small noncoding RNAs fit into the RNA-induced silencing complex [18]. MiRNAs cause either translational inhibition or mRNA degradation by binding to a conserved sequence of the 3′untranslated region of the target gene. Prior studies have noted evidence of the dysregulation of the expression of specific miRNAs in PC. Given the various roles of miRNAs in multiple aspects of cancer biology, it could conceivably be hypothesized that they also play a major role in the neoplastic hypoxia microenvironment [19]. Recently, a subset of hypoxia-inducible miRNAs have been reported to be abnormally expressed in PC, suggesting that inducing the expression of these miRNAs may influence important target genes involved in tumor survival, invasion, and metastasis [20]. Migration and invasion inhibitory protein (MIIP) has recently been noted as an inhibitor of tumor development. Niu et al. reported that HIF1 could regulate MIIP expression at the posttranscriptional level by upregulating miR-646 transcription. Dysregulated miR-646 and MIIP expression were correlated with tumor stage, metastasis, and lymphatic invasion in PC patients [21]. Mitochondrial fission is an important procedure for the development and progression of PC. Suppressed mitochondrial fission could lead to a reduction in proapoptotic protein content, which hampers mitochondria-related apoptosis pathways. A recent in vitro study described mitochondrial fission as a tumor suppression process that could be regulated by the HIF1/miR-125a/Mfn2 pathway to restrict PANC-1 cell survival and migration [22]. LncRNAs may act as transcriptional modulators, enhancers, and molecular decoys for miRNAs and protein–RNA interactions [23]. LncRNAs can also act as an oncogene or tumor suppressor via modulation of cancer-related signaling pathways in various manners [24]. ZEB1, a key regulator of EMT, could be activated by the binding of lncRNA-BX111887 (BX111) to the promoter region of the transcriptional factor Y-box protein (YB1). The hypoxic microenvironment is a crucial factor in the generation of pathological EMT-induced BX111 transcription via HIF1 overexpression [25]. Furthermore, lncRNAs can work as competing endogenous RNAs (ceRNAs) or “RNA sponges” that interact with miRNAs and reduce their regulatory activity on target mRNAs. NORAD (annotated as LINC00657 in RefSeq), a highly conserved mammalian noncoding RNA, could act as in an oncogenic capacity in the pathogenesis of PC via competition for miR-125a-3p, was upregulated during hypoxia and promoted EMT in PC cells [26]. These results are similar to a study reported by Ou et al. FEZF1-AS1, a recently described oncogenic long noncoding RNA, has been associated with poor prognosis of gastric cancer and shown to accelerate progression in pancreatic ductal adenocarcinoma. Ou et al. reported that FEZF1-AS1 could stimulate PC cell proliferation and invasion via the miR-142/HIF1α axis under hypoxic conditions [27].
Role of hypoxia and HIF in drug resistance
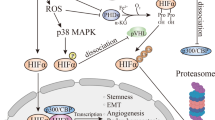
In the past two decades, there was no significant progress in the prognosis of PC. Surgery is the only curative option, but 5-year survival rates after surgical resection alone are still pessimistic (approximately 15–27%; median survival period, 17–23 months) [28]. The combination of fluorouracil, leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) and nab-paclitaxel plus gemcitabine has been administered as a first-line treatment in patients with metastatic PC [29, 30]. However, the current treatments are correlated with a median progression-free survival of approximately 6 months, and fewer than 10% of patients are alive 5 years after initial diagnosis [31]. Hypoxia and/or HIFs can mediate chemotherapy resistance through additional mechanisms, including the following (Fig. 1):
-
a)
Extrinsic resistance. Hypoxia in PC is produced partly through fibrogenic effects of cancer-associated fibroblasts (CAFs) and dense ECM components [32]. Elevated interstitial fluidic pressure and inappropriate blood flow lead to hypoxic niches in the tumors and a decrease in circulating drug perfusion into the tumor. The ECM is a complicated network of macromolecules that provides a substantial scaffold to maintain tissue architecture [33]. All the proteins, glycoproteins, and polysaccharide elements of the ECM are generally produced by epithelial and stromal cells, which account for the fibrotic area in the PC. Posttranslational modifications of fibrillar collagens require the assistance of collagen-modifying enzymes, some of which are regulated by HIFs. Therefore, reducing tissue tension and intratumoral pressure by modifying the expression of HIFs may result in improved perfusion of chemotherapeutic agents and therapeutic response [34].
-
b)
The regulation of drug efflux. A growing body of literature supports the participation of HIF1 in drug resistance by stating that HIF1 is able to stimulate the expression of the multidrug resistance 1 (MDR1) gene in response to hypoxia. MDR1, which is translated into the membrane-bound protein P-glycoprotein (P-gp), is associated with a family of ATP-binding cassette (ABC) transporters. P-gp can decrease the intracellular concentration of chemotherapeutic drugs, such as paclitaxel and anthracyclines, and can be used as a drug efflux pump [35].
-
c)
Metabolic reprogramming. HIF manages a diverse range of metabolic pathways that are overactivated in cancer, such as mitochondrial activity, intracellular pH regulation, and glycolysis. Mitochondria also plays an important role in cell death by regulating intrinsic apoptotic pathways and producing reactive oxygen species (ROS) [36]. Experimental data for enhanced ROS formation in response to chemotherapy under hypoxic conditions were recently provided by Maher Y. Abdalla et al. Heme oxygenase-1 (HO-1) is the rate-limiting enzyme of cellular heme catabolism. This microsomal enzyme acts on heme moieties to generate equimolar amounts of carbon monoxide and iron. These HO-1 degradation products were shown to modulate metabolism and promote tumor growth. Maher Y. Abdalla et al. suggested that hypoxia could upregulate HO-1 expression in PC cells. Under in vitro hypoxia conditions, the application of the HO-1 inhibitors zinc protoporphyrin (ZnPP) and tin protoporphyrin IX (SnPP) could increase ROS production, enhance apoptosis, inhibit PC cell proliferation, and sensitize these cells to gemcitabine [37]. Similarly, enolase 1 (ENO1), a multifunctional glycolytic enzyme, sensitizes PC cells to hypoxia-induced resistance by altering ROS homeostasis. The mechanisms might be related to the influence of increase ROS on apoptosis and cell cycle progression [38].
-
d)
Hypoxia-driven alterations in apoptosis and cell survival. Some studies have shown that HIF1 could regulate proapoptotic factors (BNIP3, NIX, and NOXA) as well as antiapoptotic factors (Bcl-xL, Bcl-2, Bid, Mcl-1, NF-κB, and p53) to modulate defective apoptosis and changes in cell cycle progression [39,40,41]. HIF1 functions as a strong suppressor of apoptosis, and functional intervention with HIF1 in PC cells results in changes in programmed cell death upon treatment with chemotherapeutic agents. For example, Nagaraju et al. suggested that inhibition of HSP90 with ganetespib, a small-molecule inhibitor (SMI) of HSP90, downregulated the expression of HIF1 and induced the activation of signaling pathways, including proliferative, angiogenic, and antiapoptotic pathways. Notably, the promotion of apoptosis was also observed in gemcitabine- and 5-FU-resistant pancreatic cell lines when HSP90 was inhibited [42].
-
e)
Inhibition of DNA damage. DNA damage remains the foundation of many cancer treatments, and inducing DNA damage is the fundamental mode of action for the majority of classical chemotherapeutic agents. Despite their effectiveness in suppressing tumors, DNA repair pathways can also allow tumor cells to escape from genotoxic assault [43]. Complementing these results, a study by FF Blanco et al. showed that the proto-oncogene proviral integration site for Moloney murine leukemia virus 1 (PIM1), a serine–threonine kinase, could regulate hypoxia-induced chemoresistance by phosphorylating targets and promoting apoptosis. Upon PIM1-mediated phosphorylation of CDC25a, PC cells undergo dynamic reprogramming that allows them to acquire a chemoresistant response via DNA repair mechanisms. HuR (Hu antigen R; ELAVL1), an RNA-binding protein fundamental to posttranscriptional gene regulation in PC, is an element of the embryonic lethal and Drosophila-like protein family. HuR can mediate the regulation of PIM1 and consequently prevent DNA damage induced by chemotherapeutics. Importantly, inhibiting HuR could reverse hypoxia-induced PIM1 overexpression and notably enhance PC cell sensitivity to oxaliplatin and 5-fluorouracil in a hypoxic microenvironment [44].
-
f)
Hypoxia induction of stemness. Cancer stem cells (CSCs) are a group of cancer cells with stem cell-like characteristics. In solid tumors, CSCs are functionally defined by their traits of self-renewal, differentiation, and tumor generation [45, 46]. If the CSC subpopulation is not eliminated during chemoradiotherapy of PC, tumor recurrence and subsequent clinical progression may manifest due to the drug resistance of CSCs. Components in the stromal microenvironment, such as pancreatic stellate cells (PSCs), endothelial cells and immune cells, could develop into an ideal niche for pancreatic CSCs [47]. Hypoxic niches further facilitate the remodeling of the ECM, and changes in biochemical secreted factors can promote tumor invasive capacities and tumorigenicity and help preserve progenitor CSCs. PC stem cell markers, such as ESA, CD133, CD24 and CD44, may be negative prognostic factors that are related to poor outcomes and resistance to standard treatment. Moreover, the abnormal activation of multiple signaling pathways in the hypoxic microenvironment, including the Wnt, Notch and PI3K/Akt/mTOR pathways, may play an important role in acquiring chemoresistance to gemcitabine through the induction of stemness [48]. Zhang et al. reported that the Akt/Notch1 signaling pathway intervened to induce gemcitabine resistance in PC via stemness induction, which was exacerbated by the universal hypoxic niche in cancer cells [49]. Additionally, CD133 is a transmembrane protein expressed in lipid rafts, with an extracellular ganglioside-binding domain and cytoplasmic activity akin to tyrosine phosphorylation [50]. By promoting HIF1α expression under hypoxia, CD133 could influence tumor cell migration and invasion through EMT gene expression. Confirmation of the CD133/HIF1α-signaling axis bolsters, the future possibility of a supplemental HIF1α inhibitor combined with conventional therapy to increase the efficacy of treating CD133+ CSCs in PC [51].

Summary of mechanisms and pathways underlying HIF-mediated chemotherapy resistance
Strategies to inhibit the HIF1 pathway
As the key factors that correlate with treatment resistance and PC, hypoxia and HIFs have attracted increasing attention. Because the key regulator during hypoxia is HIF, agents that can suppress its function are still eagerly anticipated and should be examined for clinical activity in PC. Minnelide, a water-soluble analog of triptolide that has potent antiproliferative activity against multiple tumor types, inhibits HIF1 transcriptional activity and decreases stemness in PC cells. A phase I dose escalation and pharmacokinetic study with single-agent minnelide were recently completed in patients with advanced gastrointestinal tumors [52]. In addition, minnelide is currently in phase II clinical trials for the treatment of PC, which has stimulated increased interest in this promising agent [53]. Digoxin, a well-known cardiac glycoside, was previously verified to be a potent inhibitor of hypoxia-induced HIF1α production. In an in vitro study, Zhou et al. reported that digoxin could be used as a potential sensitizer to reverse chemoresistance in PC, but there is no official clinical trial that confirms this activity [54]. Another compound extracted from melphalan, PX-478, which inhibits HIF1α expression in PC, resulted in reduced preclinical tumor growth but showed restricted clinical activity in a phase I study [55]. However, in a recent in vitro study, Lang et al. found that PX-478 plus arsenic trioxide (ATO) could be a promising strategy to promote ROS-induced apoptosis in the treatment of PC [56].
Conclusion
The contribution of the tumor microenvironment to the lethal outcomes of PC is substantial. One of the elements in the PC microenvironment is hypoxia, which shifts the expression patterns in cancer cells to reduce intrinsic and extrinsic damage originating from rapid tumor growth under disadvantageous conditions. It is noteworthy that tumor hypoxia and its main driver, HIFs, promote the capacity for PC invasiveness and metastasis by activating EMT- and CSC-related pathways, which all contribute to the aggressive phenotype of this disease. Furthermore, hypoxia regulates many important biological hallmarks of cancer, ranging from tumor cell differentiation and metabolic reprogramming to chemotherapy resistance. Despite significant advances in recent years, chemotherapy resistance and the subsequent need for novel therapeutic approaches remain the main barrier in clinical oncology. It is now commonly accepted that the specialized hypoxia microenvironment of PC is largely accountable for the insufficient in vivo efficacy of many treatments. To optimize the effects of these compounds, it is of chief importance to identify suitable patient subgroups and tumor cell subpopulations for precision medicine in PC. Moreover, identification of predictive markers for “hypoxia-based” substances and the development of combination therapies comprising multiple compounds that interfere with HIFs and hypoxia-related pathways with traditional chemotherapeutics are urgently needed to permit their investigation in clinical trials.
Abbreviations
- ATP:
-
Adenosine triphosphate
- CAF:
-
Cancer-associated fibroblast
- CODD:
-
C-terminal oxygen-dependent degradation domain
- CSC:
-
Cancer stem cell
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial to mesenchymal transition
- ENO1:
-
Enolase 1
- HIF:
-
Hypoxia-inducible factor
- MDR1:
-
Multidrug resistance 1
- MIIP:
-
Migration and invasion inhibitory protein
- NODD:
-
N-terminal oxygen-dependent degradation domain
- PC:
-
Pancreatic cancer
- PDK1:
-
Pyruvate dehydrogenase kinase 1
- PHD:
-
Proline hydroxylase domain
- ROS:
-
Reactive oxygen species
- VEGF:
-
Vascular endothelial growth factor
References
Siegel RL, Miller KD, Jemal A (2019) Cancer statistics, 2019. CA Cancer J Clin 69(1):7–34. https://doi.org/10.3322/caac.21551
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM (2014) Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74(11):2913–2921. https://doi.org/10.1158/0008-5472.CAN-14-0155
Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, Bastidas AJ, Vierra M (2000) Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys 48(4):919–922. https://doi.org/10.1016/s0360-3016(00)00803-8
Colbert LE, Fisher SB, Balci S, Saka B, Chen Z, Kim S, El-Rayes BF, Adsay NV, Maithel SK, Landry JC, Curran WJ Jr (2015) High nuclear hypoxia-inducible factor 1 alpha expression is a predictor of distant recurrence in patients with resected pancreatic adenocarcinoma. Int J Radiat Oncol Biol Phys 91(3):631–639. https://doi.org/10.1016/j.ijrobp.2014.11.004
Chen J, Luo H, Liu Y, Zhang W, Li H, Luo T, Zhang K, Zhao Y, Liu J (2017) Oxygen-self-produced nanoplatform for relieving hypoxia and breaking resistance to sonodynamic treatment of pancreatic cancer. ACS Nano 11(12):12849–12862. https://doi.org/10.1021/acsnano.7b08225
Ye LY, Zhang Q, Bai XL, Pankaj P, Hu QD, Liang TB (2014) Hypoxia-inducible factor 1alpha expression and its clinical significance in pancreatic cancer: a meta-analysis. Pancreatology 14(5):391–397. https://doi.org/10.1016/j.pan.2014.06.008
Jiang BH, Rue E, Wang GL, Roe R, Semenza GL (1996) Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem 271(30):17771–17778. https://doi.org/10.1074/jbc.271.30.17771
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC (2003) Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol 23(24):9361–9374. https://doi.org/10.1128/mcb.23.24.9361-9374.2003
Tamama K, Kawasaki H, Kerpedjieva SS, Guan J, Ganju RK, Sen CK (2011) Differential roles of hypoxia inducible factor subunits in multipotential stromal cells under hypoxic condition. J Cell Biochem 112(3):804–817. https://doi.org/10.1002/jcb.22961
Israel M, Schwartz L (2011) The metabolic advantage of tumor cells. Mol Cancer 10:70. https://doi.org/10.1186/1476-4598-10-70
Yang J, Ren B, Yang G, Wang H, Chen G, You L, Zhang T, Zhao Y (2019) The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. https://doi.org/10.1007/s00018-019-03278-z
Smith H, Board M, Pellagatti A, Turley H, Boultwood J, Callaghan R (2016) The effects of severe hypoxia on glycolytic flux and enzyme activity in a model of solid tumors. J Cell Biochem 117(8):1890–1901. https://doi.org/10.1002/jcb.25488
Wise DR, Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35(8):427–433. https://doi.org/10.1016/j.tibs.2010.05.003
Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, Miyazawa H, Yamaguchi Y, Miura M, Jenkins DM, Choi H, Kim JW, Asagiri M, Cowburn AS, Abe H, Soma K, Koyama K, Katoh M, Sayama K, Goda N, Johnson RS, Manabe I, Nagai R, Komuro I (2016) HIF-1alpha-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun 7:11635. https://doi.org/10.1038/ncomms11635
Denko NC, Fontana LA, Hudson KM, Sutphin PD, Raychaudhuri S, Altman R, Giaccia AJ (2003) Investigating hypoxic tumor physiology through gene expression patterns. Oncogene 22(37):5907–5914. https://doi.org/10.1038/sj.onc.1206703
Wu X, Qiao B, Liu Q, Zhang W (2015) Upregulation of extracellular matrix metalloproteinase inducer promotes hypoxia-induced epithelial-mesenchymal transition in esophageal cancer. Mol Med Rep 12(5):7419–7424. https://doi.org/10.3892/mmr.2015.4410
Wu Z, Liu X, Liu L, Deng H, Zhang J, Xu Q, Cen B, Ji A (2014) Regulation of lncRNA expression. Cell Mol Biol Lett 19(4):561–575. https://doi.org/10.2478/s11658-014-0212-6
Lai XL, Huang YH, Li YS, Li GN, Wang LP, Sun R, Ma YS, Feng SY, Chang ZY, Wang XH, Fu D, Han X, Cong XL, Li WP (2015) Differential expression profiling of microRNAs in para-carcinoma, carcinoma and relapse human pancreatic cancer. Clin Transl Oncol 17(5):398–408. https://doi.org/10.1007/s12094-014-1249-8
Hwang HW, Mendell JT (2007) MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer 96(Suppl):R40–44
Liu B, Yang H, Taher L, Denz A, Grutzmann R, Pilarsky C, Weber GF (2018) Identification of prognostic biomarkers by combined mRNA and miRNA expression microarray analysis in pancreatic cancer. Transl Oncol 11(3):700–714. https://doi.org/10.1016/j.tranon.2018.03.003
Niu Y, Jin Y, Deng SC, Deng SJ, Zhu S, Liu Y, Li X, He C, Liu ML, Zeng Z, Chen HY, Zhong JX, Ye Z, Wang CY, Zhao G (2018) MiRNA-646-mediated reciprocal repression between HIF-1alpha and MIIP contributes to tumorigenesis of pancreatic cancer. Oncogene 37(13):1743–1758. https://doi.org/10.1038/s41388-017-0082-2
Pan L, Zhou L, Yin W, Bai J, Liu R (2018) miR-125a induces apoptosis, metabolism disorder and migrationimpairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int J Oncol 53(1):124–136. https://doi.org/10.3892/ijo.2018.4380
Duguang L, Jin H, Xiaowei Q, Peng X, Xiaodong W, Zhennan L, Jianjun Q, Jie Y (2017) The involvement of lncRNAs in the development and progression of pancreatic cancer. Cancer Biol Ther 18(12):927–936. https://doi.org/10.1080/15384047.2017.1385682
Chandra Gupta S, Nandan Tripathi Y (2017) Potential of long non-coding RNAs in cancer patients: from biomarkers to therapeutic targets. Int J Cancer 140(9):1955–1967. https://doi.org/10.1002/ijc.30546
Deng SJ, Chen HY, Ye Z, Deng SC, Zhu S, Zeng Z, He C, Liu ML, Huang K, Zhong JX, Xu FY, Li Q, Liu Y, Wang CY, Zhao G (2018) Hypoxia-induced LncRNA-BX111 promotes metastasis and progression of pancreatic cancer through regulating ZEB1 transcription. Oncogene 37(44):5811–5828. https://doi.org/10.1038/s41388-018-0382-1
Li H, Wang X, Wen C, Huo Z, Wang W, Zhan Q, Cheng D, Chen H, Deng X, Peng C, Shen B (2017) Long noncoding RNA NORAD, a novel competing endogenous RNA, enhances the hypoxia-induced epithelial-mesenchymal transition to promote metastasis in pancreatic cancer. Mol Cancer 16(1):169. https://doi.org/10.1186/s12943-017-0738-0
Ou ZL, Zhang M, Ji LD, Luo Z, Han T, Lu YB, Li YX (2019) Long noncoding RNA FEZF1-AS1 predicts poor prognosis and modulates pancreatic cancer cell proliferation and invasion through miR-142/HIF-1alpha and miR-133a/EGFR upon hypoxia/normoxia. J Cell Physiol. https://doi.org/10.1002/jcp.28188
Siegel RL, Miller KD, Jemal A (2017) Cancer statistics, 2017. Cancer J Clin 67(1):7–30. https://doi.org/10.3322/caac.21387
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF (2013) Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369(18):1691–1703. https://doi.org/10.1056/NEJMoa1304369
Vaccaro V, Sperduti I, Milella M (2011) FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 365(8):768–769. https://doi.org/10.1056/NEJMc1107627author reply 769
Chiorean EG, Cheung WY, Giordano G, Kim G, Al-Batran SE (2019) Real-world comparative effectiveness of nab-paclitaxel plus gemcitabine versus FOLFIRINOX in advanced pancreatic cancer: a systematic review. Ther Adv Med Oncol 11:1758835919850367. https://doi.org/10.1177/1758835919850367
Thomas D, Radhakrishnan P (2019) Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol Cancer 18(1):14. https://doi.org/10.1186/s12943-018-0927-5
Gilkes DM, Semenza GL, Wirtz D (2014) Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer 14(6):430–439. https://doi.org/10.1038/nrc3726
Katagiri T, Kobayashi M, Yoshimura M, Morinibu A, Itasaka S, Hiraoka M, Harada H (2018) HIF-1 maintains a functional relationship between pancreatic cancer cells and stromal fibroblasts by upregulating expression and secretion of Sonic hedgehog. Oncotarget 9(12):10525–10535. https://doi.org/10.18632/oncotarget.24156
Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP (2002) Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 62(12):3387–3394
Semenza GL (2010) HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20(1):51–56. https://doi.org/10.1016/j.gde.2009.10.009
Abdalla MY, Ahmad IM, Rachagani S, Banerjee K, Thompson CM, Maurer HC, Olive KP, Bailey KL, Britigan BE, Kumar S (2019) Enhancing responsiveness of pancreatic cancer cells to gemcitabine treatment under hypoxia by heme oxygenase-1 inhibition. Transl Res 207:56–69. https://doi.org/10.1016/j.trsl.2018.12.008
Wang L, Bi R, Yin H, Liu H, Li L (2019) ENO1 silencing impaires hypoxia-induced gemcitabine chemoresistance associated with redox modulation in pancreatic cancer cells. Am J Transl Res 11(7):4470–4480
Chen N, Chen X, Huang R, Zeng H, Gong J, Meng W, Lu Y, Zhao F, Wang L, Zhou Q (2009) BCL-xL is a target gene regulated by hypoxia-inducible factor-1{alpha}. J Biol Chem 284(15):10004–10012. https://doi.org/10.1074/jbc.M805997200
Erler JT, Cawthorne CJ, Williams KJ, Koritzinsky M, Wouters BG, Wilson C, Miller C, Demonacos C, Stratford IJ, Dive C (2004) Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol 24(7):2875–2889. https://doi.org/10.1128/mcb.24.7.2875-2889.2004
Trollmann R, Richter M, Jung S, Walkinshaw G, Brackmann F (2014) Pharmacologic stabilization of hypoxia-inducible transcription factors protects developing mouse brain from hypoxia-induced apoptotic cell death. Neuroscience 278:327–342. https://doi.org/10.1016/j.neuroscience.2014.08.019
Nagaraju GP, Zakka KM, Landry JC, Shaib WL, Lesinski GB, El-Rayes BF (2019) Inhibition of HSP90 overcomes resistance to chemotherapy and radiotherapy in pancreatic cancer. Int J Cancer 145(6):1529–1537. https://doi.org/10.1002/ijc.32227
Bolderson E, Richard DJ, Zhou BB, Khanna KK (2009) Recent advances in cancer therapy targeting proteins involved in DNA double-strand break repair. Clin Cancer Res 15(20):6314–6320. https://doi.org/10.1158/1078-0432.CCR-09-0096
Blanco FF, Jimbo M, Wulfkuhle J, Gallagher I, Deng J, Enyenihi L, Meisner-Kober N, Londin E, Rigoutsos I, Sawicki JA, Risbud MV, Witkiewicz AK, McCue PA, Jiang W, Rui H, Yeo CJ, Petricoin E, Winter JM, Brody JR (2016) The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene 35(19):2529–2541. https://doi.org/10.1038/onc.2015.325
Yoshida GJ, Saya H (2016) Therapeutic strategies targeting cancer stem cells. Cancer Sci 107(1):5–11. https://doi.org/10.1111/cas.12817
Ning X, Shu J, Du Y, Ben Q, Li Z (2013) Therapeutic strategies targeting cancer stem cells. Cancer Biol Ther 14(4):295–303. https://doi.org/10.4161/cbt.23622
Rodriguez-Aznar E, Wiesmuller L, Sainz B Jr, Hermann PC (2019) EMT and stemness-key players in pancreatic cancer stem cells. Cancers. https://doi.org/10.3390/cancers11081136
Gzil A, Zarebska I, Bursiewicz W, Antosik P, Grzanka D, Szylberg L (2019) Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol Biol Rep. https://doi.org/10.1007/s11033-019-05058-1
Chen S, Zhang J, Chen J, Wang Y, Zhou S, Huang L, Bai Y, Peng C, Shen B, Chen H, Tian Y (2019) RER1 enhances carcinogenesis and stemness of pancreatic cancer under hypoxic environment. J Exp Clin Cancer Res 38(1):15. https://doi.org/10.1186/s13046-018-0986-x
Roper K, Corbeil D, Huttner WB (2000) Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat Cell Biol 2(9):582–592. https://doi.org/10.1038/35023524
Maeda K, Ding Q, Yoshimitsu M, Kuwahata T, Miyazaki Y, Tsukasa K, Hayashi T, Shinchi H, Natsugoe S, Takao S (2016) CD133 modulate HIF-1alpha expression under hypoxia in EMT phenotype pancreatic cancer stem-like cells. Int J Mol Sci. https://doi.org/10.3390/ijms17071025
Greeno E, Borazanci E, Gockerman J, Korn R, Saluja A, Von Hoff D (2015) Abstract CT207: Phase I dose escalation and pharmokinetic study of 14-O-phosphonooxymethyltriptolide. Can Res 75(15 Supplement):CT207. https://doi.org/10.1158/1538-7445.am2015-ct207
Noel P, Von Hoff DD, Saluja AK, Velagapudi M, Borazanci E, Han H (2019) Triptolide and its derivatives as cancer therapies. Trends Pharmacol Sci 40(5):327–341. https://doi.org/10.1016/j.tips.2019.03.002
Zhou Y, Zhou Y, Yang M, Wang K, Liu Y, Zhang M, Yang Y, Jin C, Wang R, Hu R (2019) Digoxin sensitizes gemcitabine-resistant pancreatic cancer cells to gemcitabine via inhibiting Nrf2 signaling pathway. Redox Biol 22:101131. https://doi.org/10.1016/j.redox.2019.101131
Zhao T, Ren H, Jia L, Chen J, Xin W, Yan F, Li J, Wang X, Gao S, Qian D, Huang C, Hao J (2015) Inhibition of HIF-1alpha by PX-478 enhances the anti-tumor effect of gemcitabine by inducing immunogenic cell death in pancreatic ductal adenocarcinoma. Oncotarget 6(4):2250–2262. https://doi.org/10.18632/oncotarget.2948
Lang M, Wang X, Wang H, Dong J, Lan C, Hao J, Huang C, Li X, Yu M, Yang Y, Yang S, Ren H (2016) Arsenic trioxide plus PX-478 achieves effective treatment in pancreatic ductal adenocarcinoma. Cancer Lett 378(2):87–96. https://doi.org/10.1016/j.canlet.2016.05.016
Funding
This study was jointly funded by the National Science Foundation for Distinguished Young Scholars of China (No. 81625016), the National Natural Science Foundation of China (No. 81602085 and 81902428) and the Shanghai Sailing Program (No. 17YF1402500 and 19YF1409400).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tan, Z., Xu, J., Zhang, B. et al. Hypoxia: a barricade to conquer the pancreatic cancer. Cell. Mol. Life Sci. 77, 3077–3083 (2020). https://doi.org/10.1007/s00018-019-03444-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03444-3