
Abstract
Glucose avidity, high glycolysis and l-lactate production, regardless of oxygen availability, are the main traits of cancer metabolic reprogramming. The idea that mitochondria are dysfunctional in cancer, thus causing a glycolysis increase for ATP production and l-lactate accumulation as a dead-end product of glucose catabolism, has oriented cancer research for many years. However, it was shown that mitochondrial metabolism is essential for cancer cell proliferation and tumorigenesis and that l-lactate is a fundamental energy substrate with tumor growth-promoting and signaling capabilities. Nevertheless, the known ability of mitochondria to take up and oxidize l-lactate has remained ignored by cancer research. Beginning with a brief overview of the metabolic changes occurring in cancer, we review the present knowledge of l-lactate formation, transport, and intracellular oxidation and underline the possible role of l-lactate metabolism as energetic, signaling and anabolic support for cancer cell proliferation. These unexplored aspects of cancer biochemistry might be exploited for therapeutic benefit.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Otto Warburg first observed that high glucose avidity and high glycolysis with final l-lactate (l-LAC) production regardless of oxygen availability (Warburg effect) are the main traits of tumor cells. It is now well-established that an overall reprogramming of cell metabolism is the most common feature of cancer cells. According to the Warburg hypothesis, aerobic glycolysis, i.e. l-LAC production in the presence of oxygen, in cancer, was associated with impaired mitochondrial function. However, this paradigm has been challenged because of the following: (1) Only little evidence of damaged mitochondria was found in cancer cells. (2) Tumors can rely on a different magnitude of respiration and often require the inhibition of both oxidative phosphorylation (OXPHOS) and glycolysis to be killed via energy deprivation. (3) Mitochondrial function plays an important role in the development of chemoresistance [see 1, 2]. Currently, the Warburg effect is mainly explained by an altered regulation of glucose uptake and metabolism and mitochondrial activity due to aberrant signaling pathways, rather than by mitochondrial defects, although these might occur in certain tumors [3, 4]. Whatever the compromised cell signaling pathway and cancer-driving gene expression, they finally lead to the establishment of the peculiar metabolic strategies that tumor cells must adopt to proliferate. That is why the peculiar metabolism of cancer cells is emerging as an effective target for elaborating novel anticancer therapies [5]. However, some basic biochemical aspects of cancer cell metabolism remain to be fully elucidated. This certainly applies to the mitochondrial oxidation of l-LAC shown to occur in several mammalian tissues and in human normal and cancer cells. In particular, the existence of the mitochondrial l-lactate dehydrogenase (ml-LDH), a putative mitochondrial lactate oxidase (LOX), and mitochondrial l-LAC carriers must be taken into account to fully understand how mitochondrial function correlates with important cytosolic pathways such as glycolysis and the biosynthetic pathways. This particularly applies to cancer, in which the above pathways are enhanced. Of growing interest in cancer are the intracellular and intercellular l-LAC shuttles [6,7,8] that describe the possible role of l-LAC in cellular reduction–oxidation (redox) balance and cell–cell interaction, respectively. Remarkably, certain cancer cells can escape the immune response by causing the acidification of their environment via l-LAC export [9]. Moreover, l-LAC appears to be involved in oxidative stress regulation and cell signaling, as shown by both l-LAC-dependent production of reactive oxygen species (ROS) [8, 10] and expression of certain ROS-responding genes [6, 8].
Here, we will review the present knowledge of l-LAC uptake, production and intracellular utilization, highlighting the possible implications in tumor cell metabolism, signaling and proliferation.
The reprogramming of cancer cell metabolism: an overview
The advantages coming from increased glycolysis in cancer remain at the core of the scientific debate. In addition, several cancers, such as breast, uterine, lung, liver, and skin cancer, show a highly oxidative phenotype, thus deriving a significant amount of energy from OXPHOS [4, 8, 11]. The dramatic cancer metabolic flexibility that underpins drug resistance [see 4] is based on a strict dynamic interconnection between glycolysis and mitochondrial function, and explains why key steps in both pathways must be inhibited simultaneously to effectively kill cancer cells via energy deprivation [3, 12, 13]. This can be achieved using metabolic inhibitors with multimodal mechanisms of action, such as 3-bromopyruvate [14, 15] or combinations of glycolytic and mitochondrial inhibitors [16].
It must be stressed that there is a high metabolic variability between cancer types and within the same tumor. This can depend on several factors, among which are tissue-specific bioenergetics, the tumor microenvironment, and the distance to blood, from which cancer cells obtain nutrients and oxygen [5, 13, 17]. The extremely different incidences of cancer among organs in the body—rare in liver and small intestine, more common in pancreas and kidney, and very common in breast, prostate, and lung—clearly show that tissue-specific metabolism plays a key role in carcinogenesis and supports the emerging concept of oncobioenergetics [18].
Glucose uptake, glycolysis and biosynthesis of macromolecules
Cancer cells take up much more glucose than normal cells from the extracellular phase due to tissue-specific overexpression of glucose transporters (GLUTs) and sodium-dependent glucose transporters (SGLT) that transport sugars along and against the concentration gradient, respectively [19]. In particular, GLUT1, the prevalent type of GLUT, is commonly overexpressed in cancers and often associates with aggressive, metastatic phenotypes [8, 20, 21], thus representing a possible target to impair cancer growth [22,23,24,25,26,27].
Cancer cells also possess potentiated metabolic pathways that allow an optimized utilization of glucose for cell growth and proliferation purposes. Since the overall flux through a metabolic pathway in vivo is influenced by the rate-limiting steps of the pathway [see 12], we will focus here on the three allosteric enzymes that catalyze the main regulatory steps of glycolysis, namely, hexokinase (HK), 6-phosphofructo-1-kinase (PFK1) and pyruvate kinase (PK), the control of which typically changes in several tumors (Scheme 1).

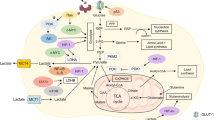
Primary metabolic changes occurring in cancer cells. Glycolysis, the oxidative phase of the pentose phosphate pathway, OXPHOS, the Krebs cycle and glutamine and l-lactate metabolism are schematically represented to emphasize their contributions to biomass synthesis, ROS scavenging and histone/protein acetylation. The black-filled arrows indicate the increase/decrease in the activity/expression of key enzymes and metabolites occurring in most cancers and playing a critical role in cancer cell metabolism (for details and references see the text). Glucose and glutamine are shown as the major carbon sources in proliferating cells. l-LAC is shown to enter mitochondria in a carrier-mediated manner and become oxidized by the ml-LDH. These processes lead to energy production via OXPHOS and PYR formation in the matrix (a more detailed representation is given in Scheme 2). The preferential access of HK-2 to mitochondrial ATP and its linkage to VDAC are also schematically represented. Abbreviations not included in the text: e− electrons; carriers: ASCT2 alanine, serine, cysteine-preferring transporter 2, TRIC tricarboxylate carrier, ? unknown mitochondrial glutamine carrier. Enzymes: CL citrate lyase, GAPDH glyceraldehyde-3-phosphate dehydrogenase, GDH glutamate dehydrogenase, GLS glutamine synthase, GR glutathione reductase, MDH malate dehydrogenase, ME malic enzyme, PC pyruvate carboxylase. Metabolites: AC-CoA acetyl coenzyme A, ASP aspartate, CITR citrate, CoASH coenzyme A, FUM fumarate, FRU-6-P fructose-6-phosphate, FRU-1,6-BP fructose-1,6-bisphosphate, GLU glutamate, IC isocitrate, α-OG α-oxoglutarate, MAL malate, PEP phosphoenolpyruvate, 3-PG 3-phosphoglycerate, 1,3-BPG 1,3-bisphosphoglycerate, RIB-5-P ribose-5-phosphate, SUCC succinate, SUCC-CoA succinyl-CoA. Respiratory chain complexes: I NADH dehydrogenase, II succinate dehydrogenase, Q coenzyme Q, III complex III, c cytochrome c, IV cytochrome c oxidase, V ATP synthase
HK catalyzes glucose phosphorylation to glucose-6-phosphate (G6P), consuming one molecule of ATP, and is normally feedback-inhibited by G6P. Among the four mammalian HK isoenzymes, tumor cells mainly overexpress the HK-2 isoform, which is resistant to G6P inhibition. Moreover, the HK-2 isoform binds reversibly to the voltage-dependent anion channel (VDAC), a porin-like protein of the mitochondrial outer membrane (MOM) involved in the trafficking of OXPHOS substrates [28, 29] and in the release of cytochrome c during the intrinsic pathway of apoptosis. Since the association between HK-2 and VDAC causes the closure of the latter, determining cancer resistance to apoptosis [30], HK-2 overexpression in tumor cells has been proposed as a possible explanation of how aerobic glycolysis correlates with apoptosis resistance in cancer. Undoubtedly, HK-2 overexpression is a central step in cell transformation, since increased glucose phosphorylation efficiency enhances both glycolysis and the pentose phosphate pathway (PP pathway). The latter is fundamental for both reductive biosynthesis and the maintenance of an adequate level of reduced glutathione (GSH) needed for ROS detoxification (Scheme 1).
PFK1 catalyzes the conversion of fructose-6-phosphate into fructose-1,6-bisphosphate, consuming one ATP molecule. Citrate and ATP that reflect high mitochondrial activity normally inhibit PFK1. On the other hand, the enzyme is stimulated by a high ADP/AMP ratio and by fructose-2,6-bisphosphate (F2,6BP), the latter produced and degraded by the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB). In cancers, PFK1 activity is potentiated in several ways, including the following: (1) gene overexpression [31], (2) post-translational modifications leading to PFK1 insensitivity to citrate and ATP and increased sensitivity to activators such as F2,6BP [see 12], and (3) overexpression of a PFKFB isoform, namely, PFKFB3, showing the highest ability to maintain elevated intracellular levels of F2,6BP [see 8]. This could nullify in cancer the normal inhibitory effect of increased O2 concentration and mitochondrial activity on glycolysis, thereby explaining the ability of cancer cells to maintain high rates of glycolysis even under normoxic conditions and when citrate is available in the cytosol for the biosynthetic pathways.
PK converts phosphoenolpyruvate into pyruvate (PYR), producing ATP. Interestingly, in several cancers, a less active PK isoform is expressed, namely, PKM2, which is highly regulated by nutrients and growth factor signals and is typical of both rapidly proliferating tissues and tissues with anabolic functions [32, 33]. It has been proposed that the expression of PKM2, by slowing down the last step of glycolysis, could facilitate the production of glycolytic intermediates for anabolic pathways, thus favoring the conversion of glucose into biomass [34]. In this hypothesis, glutamine (GLN) and alanine are considered additional sources of PYR via transamination [35] (Scheme 1).
A possible role of increased glycolysis in cancer is the provision of higher amounts of building blocks in the biosynthetic pathways that branch from it, which are necessary for the synthesis of proteins, nucleotides, and lipids that are required for cell proliferation (Scheme 1). Furthermore, a high cytosolic NADPH/NADP+ ratio occurs in cancer cells [12, 36] that is likely due to the enhanced activity of HK-2, forming G6P, and of enzymes of the oxidative phase of the PP pathway [37,38,39], thus favoring macromolecule biosynthesis and maintenance of GSH levels for the control of oxidative stress.
The essential role of mitochondria in cancer metabolism and signaling
Mitochondria are involved not only in cell energy production via OXPHOS—shown to be the largest quantitative contributor to ATP production in cancer even in a hypoxic environment—but also in the provision of substrates for biosynthetic pathways and in signaling and survival pathways, including the induction of cell death via apoptosis [12, 40, 41].
In addition to the five complexes constituting the mitochondrial respiratory chain (mRC) (see Scheme 1) and all of the enzymes of the Krebs cycle, many other factors take part in the processes that lead to ATP generation. These include mitochondrial carriers that enable metabolite traffic across mitochondrial membranes and mitochondrial regulatory enzymes. The mitochondrial carriers are necessary to fulfil the metabolic functions of mitochondria, but at present, few functional studies have been carried out on the mitochondrial transport of key metabolites in cancer, including citrate, GLN or PYR [42, 43]. As far as mitochondrial regulatory enzymes are concerned, the occurrence of a potentiated expression of pyruvate dehydrogenase kinase 1 (PDK1), which is an inhibitor of the pyruvate dehydrogenase complex (PDH), has been implicated in the reduction of the mitochondrial utilization of glucose in cancer and is garnering considerable interest as an oncology drug target [see 44]. Reduced PDH complex activity and impaired PYR uptake by mitochondria are considered the main causes for the production of higher l-LAC amounts by glycolysis in cancer cells.
Mitochondria also play a fundamental role in the synthesis of macromolecules, since biosynthetic precursors (mainly citrate) are derived from the Krebs cycle, which is fed by anaplerotic substrates such as GLN, for which many cancers are avid [35]. It must be stressed that the anaplerotic role of GLN requires a functional mRC to re-oxidize the reducing equivalents produced by the PDH complex, glutamate dehydrogenase, and Krebs cycle enzymes (see Scheme 1). Moreover, the intramitochondrial oxidation of both PYR and GLN is necessary for obtaining the biosynthetic precursors that are to be used in the macromolecule synthesis that is required for cancer cell proliferation [45].
Finally, mitochondria influence cell life by signaling functions (see Scheme 1) that include the following: (1) the production and export of Krebs cycle intermediates to the cytosol; an example is given by citrate, which can generate acetyl-CoA that, in addition to being a key precursor of lipid synthesis, also serves for protein and histone acetylation, a typical post-translational regulatory modification [46], (2) the release of cytochrome c and other mitochondrial proteins to initiate apoptosis [47], and (3) the production of reactive oxygen species (ROS), mainly linked to mRC activity.
Noteworthy is the role of mitochondrial ROS (mROS) which can act as signaling messengers by the reversible oxidation of protein thiol groups and by promoting DNA mutations and pro-oncogenic signaling pathways [see 48, 49]. Importantly, hypoxia stimulates the production of mROS, which, in turn, activate the hypoxia-inducible factor-1 (HIF-1) [see 48]. HIF-1 is a transcription factor that enhances the survival and progression of tumors by up regulating genes that are involved in cell metabolism and angiogenesis, among which include PDK1, l-lactate dehydrogenase A (l-LDHA), and glucose and l-LAC transporters [50, 51]. It has been proposed that the increased flux through the PP pathway allows cancer cells to maintain an adequate ROS level for cell signaling purposes by providing high amounts of NADPH [50].
Although most cancer cells rely on both a high glycolytic rate and functional mitochondria to generate ATP, mutations in genes encoding Krebs cycle enzymes or mRC complex subunits (for example, succinate dehydrogenase and fumarase) have been shown to occur in certain tumors [52]. In these cancers, ATP is mainly produced by glycolysis due to mitochondrial impairment, but mitochondria still maintain a crucial role in cell macromolecule biosynthesis and ROS generation [for refs, see 53].
The l-lactate affair
The l-LAC affair is a complex and exciting one. l-LAC is formed exclusively from PYR, which derives mainly from glucose via glycolysis and from alanine via the reaction catalyzed by alanine aminotransferase. The reduction of PYR to l-LAC is catalyzed by the cytosolic NAD-dependent l-lactate dehydrogenase (cl-LDH) that oxidizes NADH re-forming NAD+ needed for the glyceraldehyde 3-phosphate dehydrogenase reaction and the continuation of glycolysis (Schemes 1, 2). It was commonly incorrectly believed that l-LAC is formed from PYR only under anaerobic conditions (anaerobic glycolysis) and that PYR entered mitochondria to feed the Krebs cycle and OXPHOS. In contrast, l-LAC is formed regardless of oxygen availability [for refs, see 54] and is simultaneously produced and oxidized by both resting and exercising skeletal muscles and by other tissues such as the brain and heart [see 6, 7, 55, 56]. Therefore, l-LAC—not PYR—is to be considered the actual final product of glycolysis even in the presence of oxygen. This is supported by thermodynamic considerations: the reduction of PYR to l-LAC in the cl-LDH reaction yields free energy (∆G°′ = − 6 kcal/mol) and has an equilibrium constant—the same for all l-LDH isoenzymes—that strongly favors l-LAC formation [57, 58]. In addition, the Km value for PYR, i.e., the concentration of PYR at which the reaction rate is half of the maximal rate (Vmax), is much lower than that for l-LAC, and Vmax of PYR conversion into l-LAC is very high. This ensures that PYR is readily reduced by cl-LDH instead of being transported into mitochondria. This was confirmed recently using a genetically encoded biosensor to monitor the mitochondrial PYR carrier (MPC) activity in real time in a variety of cancer cells: when incubated with glucose, neither PYR formation nor MPC activation was found unless cells were concomitantly exposed to inhibitors of the monocarboxylate transporters MCT1/MCT2, i.e., when l-LAC transport was impaired. This might be explained by l-LAC accumulation in the cell causing cl-LDH feedback inhibition and a consequent increase in the PYR concentration. Then, in the absence of MCT inhibitors, PYR derived from glucose catabolism is likely significantly below its Km value for MPC or, alternatively, a glucose-induced negative control occurs in the MPC-mediated transport [59]. In any case, it is clear that the assumption that the mitochondrial transport of PYR is somehow thermodynamically preferable over its conversion to l-LAC in the cytosol under aerobic conditions is unsubstantiated.

Possible roles of l-LAC formation and mitochondrial oxidation in cell metabolism. A schematic representation of the possible roles (underlined text) of intracellular l-LAC metabolism is shown. The enzymes/translocators/processes generally potentiated in cancer are highlighted in yellow. ∆µH, proton electrochemical gradient. For a detailed description and references, see the text
Furthermore, the reduction of PYR to l-LAC plays a role in the cellular mitigation of the metabolic acidosis that arises from high rates of glycolysis [60], since (1) PYR reduction to l-LAC consumes protons and (2) protons are removed from the cytosol by the plasma membrane and mitochondrial MCTs that facilitate the symport of l-LAC molecules with protons down their concentration gradients [56].
Finally, most cancers overexpress the l-LDHA isoform [for refs, see 6] (typical of glycolytic type II muscle fibers), which favors PYR conversion to l-LAC, has a role in buffering PYR concentration excursions and ultimately increases l-LAC production.
Therefore, l-LAC formation has at least three important functions (Scheme 2):
-
1.
maintenance of adequate cytosolic NAD+ levels for glycolytic flux maintenance;
-
2.
maintenance of normal intracellular pH values;
-
3.
buffering of PYR concentration variations.
In the light of the above considerations and since l-LAC is always present in blood at concentrations ranging from 1 to 5 mM (during exercise) [61] and is taken up from the bloodstream and is oxidized by most tissues [62], it is feasible that l-LAC can be oxidized per se without being re-converted into PYR. Interestingly, several studies using both radioactive and non-radioactive lactate tracers have shown that (1) infused l-LAC is always produced and turned over rapidly even at rest, (2) l-LAC released from working muscle fuels the heart during exercise, and (3) the major effect of training is not the reduction of l-LAC production but rather the improvement of blood l-LAC clearance by muscles, resulting in the lowering of the arterial l-LAC concentration [see 63 and refs therein].
The mitochondrial l-LDH and l-lactate oxidation
Based on the above observations, it is evident that there are missing pieces in the current knowledge of cell bioenergetics. One of these is certainly represented by the mitochondrial transport and metabolism of l-LAC, which explains the aerobic formation of l-LAC and even of the observed ability of high l-LAC levels to induce a marked increase of mitochondrial mass in both normal and cancer cells [for refs, see 64]. The existence of mitochondrial l-LAC carriers and MCTs, the occurrence of the mitochondrial NAD-dependent l-LDH (ml-LDH), which is included in the MitoCarta list (http://www.broadinstitute.org/pubs/MitoCarta/index.html) and the ability of mitochondria to oxidize l-LAC have long been known [see 7]. Early indications were given in 1959 by Sacktor et al., who showed that l-LAC was oxidized at an appreciable rate by rat brain mitochondria, likely due to the existence of ml-LDH [65]. Since then, several studies have revealed the occurrence of mitochondrial l-LAC oxidation and the ml-LDH in mammalian tissues that are active l-LAC consumers, such as the liver, heart, skeletal muscle, and brain [6, 7, 66]. Furthermore, an isoform of l-LDH, LDH-X, is known to be located in the mitochondrial matrix of human and equine sperm, allowing for the conversion of l-LAC to PYR within mitochondria to support motility [67,68,69]. The heart uses l-LAC as a preferred source of energy, especially during exercise when arterial l-LAC increases and is immediately taken up and oxidized to CO2 in the myocardium [7]. Laughlin et al. found that the infusion of [13C]l-LAC in the working dog heart, unlike [13C]PYR, resulted in the labeling of the Krebs cycle intermediate 2-oxoglutarate, but not of cytosolic PYR or alanine [70], thus supporting the occurrence of intramitochondrial l-LAC oxidation. We consistently found that mitochondria isolated from rat heart, liver, and neuronal cells can take up and oxidize l-LAC due to an ml-LDH located in the matrix [see 7]. In particular, l-LAC induced mitochondrial NAD(P)+ reduction, a mitochondrial membrane potential increase and oxygen (O2) consumption coupled with ATP synthesis via OXPHOS when added to rat liver mitochondria (RLM) in the absence of externally added NAD+ [71]. These events were clearly consequent to l-LAC entry and oxidation inside mitochondria (Scheme 2). Pizzuto et al. came to the same conclusion following the application of immunological analyses, confocal microscopy and enzyme activity assays to HepG2 (human hepatocellular carcinoma) cells [72]. Interestingly, Lemire et al. found that even mitochondria from astrocytes, typically l-LAC-producing and exporting cells, can readily oxidize l-LAC as a respiratory substrate due to an ml-LDH localized in the matrix/inner membrane components [73]. With the existence of an intramitochondrial ml-LDH and hence, of a compartmentation of lactate metabolism, Chatham et al. consistently showed that, in the perfused rat heart, exogenously labeled l-LAC was taken up and oxidized by mitochondria, while simultaneously, unlabeled glycolytically derived l-LAC was exported, suggesting the occurrence of two separate metabolic fates for l-LAC depending on its origin [74]. This hypothesis is intriguing in cancer, where extracellular l-LAC might be taken up and used as a mitochondrial respiratory substrate to supply energy, whereas, as suggested by the occurrence of PKM2 (see above), glucose might mainly fuel the branching pathways of glycolysis, such as the PP pathway, supporting cell growth and the antioxidant apparatus.
The exact location of the ml-LDH is still a matter of debate. Moreover, ml-LDH was not detected in skeletal muscle mitochondria or it was found in the outer mitochondrial compartments. This was probably due to technical concerns or to the different ml-LDH distribution patterns depending on the tissue source of the mitochondria [for refs, see 7, 66, 75, 76]. It can also be hypothesized that two ml-LDH isozymes with peculiar functions that are involved with l-LAC and PYR homeostasis may be located on either side of the MIM. Furthermore, one of these two enzymes might be a LOX [10] and not a dehydrogenase.
l-Lactate transport across the mitochondrial inner membrane (MIM)
The intramitochondrial metabolism of l-LAC implies that l-LAC can cross the MIM. The transport of l-LAC across cell membranes is facilitated by MCTs exhibiting different affinities for l-LAC, tissue specificities and intracellular localization [see 7]. Interestingly, certain MCTs located in the MIM have been found in muscle and in normal and breast cancer cells [6,7,8], although the function of l-LAC or PYR transport into the mitochondrial matrix has been poorly investigated. Moreover, in the 1970s, Halestrap reported data that favored the existence of an additional mitochondrial l-LAC carrier [77]. He detected an l-LAC-dependent flux of protons into mitochondria occurring at a rate even higher than that caused by PYR and insensitive to low concentrations of the MCT inhibitor α-cyano-4-hydroxycinnamate that is able to block PYR but not l-LAC accumulation in RLM. We consistently observed that a proton-compensated mitochondrial uptake of l-LAC occurs in RLM, which is mediated by a putative l-LAC/H+ symporter that is different from MCT [71] (Scheme 2).
It is also important to stress that the rate of l-LAC transport across the MIM proved to limit the rate of l-LAC oxidation by mitochondria [71]. This indicates that the intracellular metabolism of l-LAC might be regulated by or depend on the expression level or activity of l-LAC mitochondrial translocators. This provides an additional explanation as to why targeting l-LAC translocators is so effective in cancer and prompts further study and better characterization of l-LAC transport across cell membranes.
Another interesting issue is the “intracellular lactate shuttle” first proposed by Brooks et al., which was based on the ability of the cell to both produce and oxidize l-LAC [see 6]. In particular, Brooks’ group showed that mLDH is anchored to the MIM of the rat brain and rodent and human skeletal muscle mitochondria, in association with the mitochondrial MCT1, the chaperone protein CD147 and cytochrome c oxidase (COX), forming the “mitochondrial lactate oxidation complex” [see 6] (Scheme 3a). They hypothesized that cytosolic NAD+ and l-LAC are consumed in the outer mitochondrial compartments to form NADH plus PYR, which is an endergonic reaction that is coupled to the exergonic redox change occurring at COX during mitochondrial electron transport. Then, PYR could be transported inside mitochondria by the mitochondrial MCT1. Similarly, the “lactate–malate–aspartate shuttle” hypothesis posits that l-LAC is oxidized in the intermembrane space of mitochondria and cooperates with the malate–aspartate shuttle for the transfer of NADH into mitochondria [76]. However, we found that l-LAC addition to mitochondria isolated from rat liver, heart and cerebellar granule cells in the absence of NAD+ caused a carrier-mediated PYR efflux from the matrix due to a putative l-LAC/PYR antiporter separate from the l-LAC/H+ symporter and MCTs. The subtraction of PYR from the matrix by the putative l-LAC/PYR antiporter, or its utilization by the PDH complex or PYR carboxylase, could favor the intramitochondrial oxidation of l-LAC into PYR. Importantly, the existence of an ml-LDH located in the matrix (see above) and cLDH and mitochondrial l-LAC translocators account for the occurrence of the “l-LAC/PYR shuttle” (Scheme 3b), which transfers the reducing equivalents derived from glycolysis from the cytosol to the mitochondrial matrix. This additional mitochondrial NADH shuttle has also been reported in rat and rabbit sperm mitochondria [78], in rat heart mitochondria [see 7] and in HepG2 cells [72]. It can be hypothesized that the l-LAC/PYR shuttle could compensate for the limited ability of the glycerol-phosphate and malate–aspartate shuttles to recycle NADH into NAD+ under a condition of high glycolytic flux, such as that occurring in cancer [79].

Proposed models for l-LAC oxidation by mitochondria: the “mitochondrial lactate oxidation complex” (a) and the l-LAC/PYR shuttle (b). In a, l-LAC is proposed to be oxidized to PYR by the mitochondrial lactate oxidation complex formed by mLDH, the mitochondrial MCT1, cytochrome c oxidase (COX) and the chaperone protein CD147, which are located in the MIM. PYR is then transported into the matrix by MCT1 in symport with a proton. In b, l-LAC enters mitochondria by crossing the MIM via the putative l-LAC/H+ symporter and is oxidized by mLDH in the matrix. As a result, the cytosolic NADH is shuttled into mitochondria for its oxidation by complex I of the mRC. Newly formed PYR can leave mitochondria in exchange for cytosolic l-LAC, a transport process mediated by the putative l-LAC/PYR antiporter. In both a and b, l-LAC conversion into PYR is favored by PYR removal by transport proteins and/or via the Krebs cycle. The transport of PYR (a) or l-LAC (b) across the MIM is favored by the mitochondrial proton gradient generated by mRC activity
l-Lactate-dependent ROS production and signaling
l-LAC is emerging as a powerful signaling molecule acting both inside cells and among cells [8]. In particular, when added at elevated concentrations (10–20 mM) to cultured L6 myoblast cells, l-LAC caused an increase in ROS production and the up-regulation of hundreds of genes, many known to be ROS-responsive and involved in l-LAC mitochondrial transport and oxidation [see 8]. As described above, cells can oxidize l-LAC, which can be derived from PYR (endogenous l-LAC) or imported from the bloodstream. However, there might be a substantial difference between these two cases, represented by the modification of the cell redox state and, in particular, the NADH/NAD+ ratio. Indeed, since PYR can be generated from several sources besides glucose, such as alanine, glutamine, or malate (see Schemes 1, 2), endogenous l-LAC formation can be accompanied by net NADH oxidation/NAD+ formation in the cell cytosol. In contrast, imported l-LAC is oxidized inside cells, most likely at mitochondria, thus causing no NAD+ production but rather its reduction to NADH. Thus, depending on the source of l-LAC, it might differentially influence the cytosolic NADH/NAD+ ratio, which, in turn, plays an important role in the integration of metabolic and signaling events. In particular, NAD-dependent signaling pathways exist that regulate transcription, DNA repair, cell cycle progression, apoptosis and metabolism, many of which are fundamental for cancer development [80, 81]. Importantly, we found that a putative LOX is present in the intermembrane space of RLM, which is an FAD/FMN-dependent enzyme that oxidizes l-LAC into PYR producing hydrogen peroxide (H2O2) [10] (Scheme 2). This latter is a ROS with a long half-life and a high membrane permeability. Hence, this is the most suitable ROS for acting as a signaling molecule. Since LOX activity is inhibited by NAD+ [10], it can be speculated that LOX-dependent H2O2 production and signaling might become significant in the case of pronounced l-LAC import from the extracellular phase, which is a condition that would not cause NAD+ levels to increase. Clearly, ROS can also be derived from l-LAC-induced mRC activity as byproducts of mitochondrial respiration (Scheme 2). However, recently, intermittent treatment with l-LAC was shown to cause a LOX-dependent increase of mitochondrial H2O2 production and a mild inhibition of mRC activity in skin fibroblasts. This type of treatment with l-LAC also led to the activation of key components of cell quality control signaling, AMPK and PGC1, with a final increase in autophagy and anti-age effects. In contrast, the constant l-LAC supply was detrimental to cells due to excessive ROS production and oxidative stress [82]. Thus, l-LAC might have different effects on the life of the cell depending on the duration and extent of the increase in l-LAC levels in the extracellular and intracellular environments.
On the other hand, exogenous l-LAC can act as a signaling molecule by binding and activating a specific plasma membrane G-protein coupled receptor, GPR81, found in adipose tissue, muscle and brain. The binding causes a decrease in intracellular cAMP levels with consequent glucose-saving/antilipolytic effects [83, 84].
Finally, l-LAC plays an important role in neuronal plasticity and memory by shifting the neuronal intracellular redox state toward the reduced state. This causes the modulation of NMDA receptor activity and hence modulation of the downstream Erk1/2 signaling cascade, finally leading to increased expression of synaptic plasticity genes [85].
l-Lactate and gluconeogenesis (GNG)
By forming PYR in the mitochondrial matrix, l-LAC might directly contribute to GNG in specific tissues. Interestingly, it was reported that at low l-LAC concentrations, the rate of GNG was determined by PYR transport across the MIM, whereas this did not occur at high l-LAC concentrations at which a significant alternative pathway was involved [86]. We first revealed that l-LAC addition to intact isolated RLM at a high concentration (10 mM) caused the export from the matrix of the gluconeogenic precursor oxaloacetate (OAA) formed as a result of the intramitochondrial oxidation of l-LAC. OAA efflux was shown to occur by a putative l-LAC/OAA antiporter, different from the l-LAC/H+ symporter and l-LAC/PYR antiporter [71]. Then, l-LAC could trigger GNG per se, i.e., without passing through PYR formation in the cytosol, at least when its cytosolic level increases (Scheme 2). It was consistently shown that in the perfused rat liver stimulated with glucagon, the rate of GNG from l-LAC could reach values that would require a PYR mitochondrial transport rate greater than the maximum MPC rate. Furthermore, considering the low cytosolic PYR concentration under these conditions, the PYR carrier could operate at no more than 75% of its maximum velocity [77]. Since PYR carboxylation to form OAA—but not its mitochondrial transport—increased after glucagon treatment, PYR was proposed to enter mitochondria as alanine [77, 86]. However, this might also depend on increased mitochondrial transport and metabolism of l-LAC, a possibility never investigated.
Starting from the above considerations, the study of whether and how the mitochondrial metabolism of l-LAC changes in cancer cells and contributes to malignancy development, a still unexplored field, is attractive.
The mitochondrial transport and metabolism of l-lactate in cancer
As reported above and depicted in Scheme 1, in most tumors, PYR metabolism appears to be altered in several ways. As a result, l-LAC formation from PYR is generally favored in cancer cells and probably exceeds the mitochondrial capacity of l-LAC clearance. Since l-LAC is both an energy substrate and a signaling molecule, the increase in the cytosolic l-LAC level causes important changes in cell metabolism and in the surrounding cells following export. Thus, the exact knowledge of the metabolic fate of l-LAC is of great importance in cancer research.
l-LAC is a major mobile energy fuel in cancer [87] and can migrate from hypoxic to oxygenated tumor cell subpopulations to support their oxidative metabolism [88]. Accordingly, both MCT1 and MCT4, which preferentially import and export l-LAC, respectively, are overexpressed in several cancers due to the action of transcription factors, such as HIF-1 and l-LAC itself [see 8]. As an energy source, l-LAC is currently assumed to act via PYR formation in the cancer cell cytosol, with PYR then oxidized by mitochondria. However, l-LAC oxidation to PYR via an l-LDH reaction is unlikely to occur in the cytosol (see above) and PYR was found to be unable to enter HepG2 mitochondria [72]. Most likely, transported or intracellularly formed l-LAC is directly oxidized by the mitochondria of cancer cells and/or serves as a signaling molecule by increasing mROS production (see Scheme 2). Using cultured human prostate cancer cells, we observed that l-LAC is taken up from the extracellular environment and causes O2 consumption and mitochondrial pyridine nucleotide reduction at a higher rate in cancer compared to normal cells, whereas PYR failed to do this. Furthermore, we showed that l-LAC enters prostate cancer mitochondria in a proton-compensated manner and is oxidized inside mitochondria by an overexpressed and more active ml-LDH [89]. The occurrence of both ml-LDH and mitochondrial MCT2 and MCT4 isoforms, which might mediate l-LAC or PYR uptake by mitochondria, was also demonstrated in cancerous breast cell lines [see 8]. Similarly, Chen et al. proved that l-LAC fuels mitochondrial respiration in HeLa (epithelioid cervix carcinoma) and H460 (lung cancer) cells by virtue of an ml-LDH associated with the MIM [90].
l-LAC and Krebs cycle anaplerosis in cancer
Cell growth and division require the synthesis of proteins, lipids and nucleic acids. The precursors of these molecules are generated in the Krebs cycle and are then exported from the mitochondria to participate in cytosolic biosynthetic reactions. Therefore, the replenishment of Krebs cycle intermediates (anaplerosis) and the transport of precursors across the MIM are crucial events for cancer cells. GLN-derived α-oxoglutarate is not the only anaplerotic substrate for cancer cells, which also significantly derive Krebs cycle intermediates from glucose (Scheme 1). In particular, a pivotal role in cancer anaplerosis is played by PYR that is converted inside mitochondria into OAA by pyruvate carboxylase [see 91] or acetyl-CoA via the PDH complex [45]. Both enzymes show enhanced flow in lung tumors [92, 93]. However, by labeling either glucose or l-LAC, it was shown that the majority of carbon flow through the TCA cycle in tumors does not arise directly from glucose but from circulating l-LAC [94, 95]. Therefore, the mitochondrial oxidation of l-LAC into PYR can play an important role in Krebs cycle anaplerosis, particularly in rapidly proliferating cells (Scheme 2). In our in vitro study on prostate cancer cells [89], due to a low l-LAC-dependent O2 consumption rate, we proposed that the main function of mitochondrial metabolism of l-LAC in these cells could be Krebs cycle anaplerosis rather than ATP production via OXPHOS. As a result, citrate or isocitrate could be formed and exported from mitochondria to be used for fatty acid, NADPH and amino acid synthesis in the cytosol [96] (Scheme 1). Chen et al. consistently showed that the mitochondrial oxidation of l-LAC provides an important carbon contribution to the total pool of lipids in HeLa and H460 cells by production of Krebs cycle intermediates [90]. This had been previously observed by Laughlin et al. (see above) [70] and by Chatham, who further showed that acetyl-CoA is derived preferentially from exogenous—instead of glycolytically derived—l-LAC in the perfused rat heart [74]. Recently, Faubert et al. demonstrated that circulating labeled l-LAC—to a greater extent than glucose—extensively labels Krebs cycle intermediates in human non-small-cell lung cancers [94]. Similarly, Hui et al. further showed that circulating l-LAC, derived mainly from glucose but also from other sources, is a primary source of carbon for the Krebs cycle in most tissues and tumors [95]. Interestingly, Pizzuto et al. found that l-LAC addition to mitochondria isolated from HepG2 cells induced OAA, malate and citrate appearance in the extramitochondrial phase [72]. Thus, l-LAC might have the additional function of facilitating the efflux from mitochondria of newly synthesized molecules that can importantly contribute to lipid and nucleic acid synthesis and to the cell antioxidant capacity in cancer (see Scheme 2).
It is plausible that the l-LAC contribution to Krebs cycle anaplerosis occurs in concert with GLN. Indeed, glucose and GLN metabolism are known to operate in an orchestrated manner to support fatty acid synthesis through the production of acetyl-CoA, ATP, and NADPH [4, 54]. Furthermore, it can be hypothesized that GLN and l-LAC also act in concert in the provision of aspartate that can derive from glutamate and OAA via transamination (Schemes 1, 2). This interdependence of GLN and l-LAC metabolism might explain the observed l-LAC-driven activation of GLN uptake and metabolism in oxidative cancer cells [97].
l-Lactate-mediated signaling in cancer
As mentioned above, l-LAC has both extracellular and intracellular signaling functions and appears to promote tumor growth, progression and metastasis [8, 98].
It was found that by releasing H2O2, epithelial cancer cells can induce the autophagic destruction of mitochondria in adjacent cancer-associated fibroblasts. This causes an increase in glycolysis and l-LAC overproduction and release by these cells. Released l-LAC is then taken up by cancer cells to feed mitochondrial energy production, a phenomenon called the “Reverse Warburg Effect” [99]. Moreover, it was shown that l-LAC released by co-cultured fibroblasts induced a significant increase in the mitochondrial mass of human breast adenocarcinoma (MCF7) cells [100]. Thus, cancer cells can obtain metabolic advantages from imported l-LAC through l-LAC-dependent energy production and signaling.
On the other hand, cancer cells can release l-LAC to induce the modification of gene expression in “candidate” cancer cells, i.e., normal cells surrounding tumors [see 6]. In addition, l-LAC acts as an important tool for immunosuppression by tumors. High levels of tumor-infiltrating T cells predict a favorable prognosis in patients with different types of cancer [101]. The acidification of the cancer extracellular environment due to l-LAC export in symport with protons and glucose deprivation due to the high rate of glucose consumption by cancer cells inhibit the activation of T cells, which suppresses their anti-tumor function. This increases the capacity of cancer cells to invade host tissues [8, 102, 103]. Moreover, high cancer-derived extracellular l-LAC concentrations can cause T and NK cell apoptosis [see 8], thus explaining the smaller proportion of these cells in high l-LAC-producing tumors.
Intracellularly, a direct correlation might exist between l-LAC overproduction/uptake and ROS generation and ROS-mediated activation of cell signaling pathways that feed tumor invasiveness [50, 51]. Interestingly, as with ROS, l-LAC can activate HIF-1 in oxidative cancers [97], thereby mimicking a condition of hypoxia even when oxygen is available and stimulating tumor growth and angiogenesis (see chapter 2.2). In particular, l-LAC can stabilize hypoxia-inducible factor-2α (HIF-2α) that in turn transactivates the transcription factor c-Myc, which increases the expression of the high affinity glutamine translocator ASCT2 and glutaminase 1 genes, which potentiate oxidative glutaminolysis in cancer cells [97]. Interestingly, c-Myc has also been reported to play a pivotal role in the stimulation of oxygen consumption and mitochondrial biogenesis [104]. Moreover, l-LAC can activate CREB and AMPK, both of which are responsible for increased mitochondrial biogenesis [for refs, see 6]. l-LAC consistently induced a marked increase of mitochondrial mass in MCF7 cells [for refs, see 64] and respiration in human neuroblastoma cells while reducing the glycolytic flux [105]. Thus, the three most widespread peculiarities of aggressive cancers—high glucose uptake, high l-LAC levels, and glutamine utilization—are mutually dependent events, and the chronic accumulation of l-LAC can induce unusual metabolic changes [8].
Conclusions
The aberrant energy metabolism of cancer can differ widely among human tumors [see 106]. Therefore, the establishment of a precise metabolic profile remains the primary way to obtain efficacious and cancer-specific therapeutic strategies targeting the predominant bioenergetic pathway(s) that are altered. Thus, the first step in cancer research should be the determination of the biochemical pathways that are primarily involved in energy production in cancer cells. Therefore, it is surprising that the occurrence of the mitochondrial metabolism of the high-energy metabolite l-LAC remains almost completely ignored. As described in this review article, l-LAC represents a fundamental connection between cytosolic and mitochondrial events, with l-LAC being both the final product of glycolysis and a mitochondrial respiratory substrate in addition to functioning as an anaplerotic and signaling molecule. The predominant role that l-LAC plays probably depends on the genetic heterogeneity that influences cell metabolism, on the extent and duration of increases in l-LAC levels and even on the origin of l-LAC (exogenous or endogenous).
We posit that the very high rate of glucose uptake is likely to be the primary cause of cell transformation, leading to the up-regulation of glycolysis and consequent increase in l-LAC production without an oxidative phosphorylation to glycolysis switch. This also occurs in rapidly proliferating normal cells due to the temporary higher biomass and energy demands [107]. l-LAC induces cell adaptation to the novel metabolic condition probably in an attempt to re-establish the equilibrium between the glycolytic flux and mitochondrial oxidation. For example, l-LAC could power the mitochondrial network [100] and thus increase the contribution of mitochondria to macromolecule generation for cell division, antioxidant cell power, mROS-mediated signaling/genomic instability and energy production. However, the higher catalytic efficiency, sensitivity to activators and/or increased resistance to inhibitors of certain key transporters and glycolytic enzymes of cancer cells [108] might result in an aberrant cross-talk/mutual regulation between cytosolic and mitochondrial pathways [12]. As a consequence, mitochondria of cancer cells do not completely fulfil their role of l-LAC clearance via oxidation, thus causing increases in l-LAC levels and export, which exacerbates the pathology [see 8].
This review aims to encourage further investigations of the mitochondrial transport and metabolism of l-LAC in different cancer types, the knowledge of which will ultimately lead to effective metabolic anticancer therapies.
Abbreviations
- F2,6BP:
-
Fructose-2,6-bisphosphate
- GLN:
-
Glutamine
- GLU:
-
Glucose
- GLUT:
-
Glucose transporter
- G6P:
-
Glucose-6-phosphate
- HK:
-
Hexokinase
- l-LAC:
-
l-Lactate
- l-LDHA:
-
l-Lactate dehydrogenase A
- cl-LDH:
-
Cytosolic l-lactate dehydrogenase
- ml-LDH:
-
Mitochondrial l-lactate dehydrogenase
- MCT:
-
Monocarboxylate carrier
- MIM:
-
Mitochondrial inner membrane
- MOM:
-
Mitochondrial outer membrane
- MPC:
-
Mitochondrial pyruvate carrier
- mRC:
-
Mitochondrial respiratory chain
- OXPHOS:
-
Oxidative phosphorylation
- PDH:
-
Pyruvate dehydrogenase
- PDK:
-
Pyruvate dehydrogenase kinase
- PFK:
-
6-Phosphofructo-1-kinase
- PFKFB:
-
6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase
- PK:
-
Pyruvate kinase
- PKM2:
-
Pyruvate kinase isoform M2
- PP pathway:
-
Pentose phosphate pathway
- PYR:
-
Pyruvate
- RLM:
-
Rat liver mitochondria
- ROS:
-
Reactive oxygen species
- VDAC:
-
Voltage-dependent anion channel
References
Griguer CE, Oliva CR (2011) Bioenergetics pathways and therapeutic resistance in gliomas: emerging role of mitochondria. Curr Pharm Des 17:2421–2427
Ippolito L, Marini A, Cavallini L et al (2016) Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 7:61890–61904
Gogvadze V, Zhivotovsky B, Orrenius S (2010) The Warburg effect and mitochondrial stability in cancer cells. Mol Asp Med 31:60–74
Obre E, Rossignol R (2015) Emerging concepts in bioenergetics and cancer research: metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodelling, signaling and bioenergetic therapy. Int J Biochem Cell Biol 59:167–181
Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP (2017) Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 14:11–31
Brooks GA (2016) Energy flux, lactate shuttling, mitochondrial dynamics, and hypoxia. Adv Exp Med Biol 903:439–455
Passarella S, de Bari L, Valenti D et al (2008) Mitochondria and l-lactate metabolism. FEBS Lett 582:3569–3576
San-Millán I, Brooks GA (2017) Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg effect. Carcinogenesis 38:119–133
Brand A, Singer K, Koehl GE et al (2016) LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab 24:657–671
de Bari L, Valenti D, Atlante A, Passarella S (2010) l-Lactate generates hydrogen peroxide in purified rat liver mitochondria due to the putative l-lactate oxidase localized in the intermembrane space. FEBS Lett 584:2285–2290
Guppy M, Leedman P, Zu X, Russell V (2002) Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J 364:309–315
Moreno-Sánchez R, Marín-Hernández A, Saavedra E et al (2014) Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int J Biochem Cell Biol 50:10–23
Viale A, Corti D, Draetta GF (2015) Tumors and mitochondrial respiration: a neglected connection. Cancer Res 75:3687–3691
Galina A (2014) Mitochondria: 3-bromopyruvate vs. mitochondria? A small molecule that attacks tumors by targeting their bioenergetic diversity. Int J Biochem Cell Biol 54:266–271
Valenti D, Vacca RA, de Bari L (2015) 3-Bromopyruvate induces rapid human prostate cancer cell death by affecting cell energy metabolism, GSH pool and the glyoxalase system. J Bioenerg Biomembr 47:493–506
Kee HJ, Cheong JH (2014) Tumor bioenergetics: an emerging avenue for cancer metabolism targeted therapy. BMB Rep 47:158–166
Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12:685–698
Jose C, Melser S, Benard G, Rossignol R (2013) Mitoplasticity: adaptation biology of the mitochondrion to the cellular redox state in physiology and carcinogenesis. Antioxid Redox Signal 18:808–849
Wood IS, Trayhurn P (2003) Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr 89:3–9
Airley R, Loncaster J, Davidson S et al (2001) Glucose transporter Glut-1 expression correlates with tumor hypoxia and predicts metastasis—free survival in advanced carcinoma of the cervix. Cancer Res 7:928–934
Szablewski L (2013) Expression of glucose transporters in cancers. Biochim Biophys Acta 1835:164–169
Zhao F, Ming J, Zhou Y, Fan L (2016) Inhibition of Glut1 by WZB117 sensitizes radioresistant breast cancer cells to irradiation. Cancer Chemother Pharmacol 77:963–972
Liu Y, Cao Y, Zhang W et al (2012) A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther 11:1672–1682
Liu TQ, Fan J, Zhou L, Zheng S-S (2011) Effects of suppressing glucose transporter-1 by the antisense oligodeoxynucleotide on the growth of human hepatocellular carcinoma cells. Hepatobiliary Pancreat Dis Int 10:72–77
Rastogi S, Banerjee S, Chellappan S, Simon GR (2007) Glut-1 antibodies induce growth arrest and apoptosis in human cancer cell lines. Cancer Lett 257:244–251
Martel F, Guedes M, Keating E (2016) Effect of polyphenols on glucose and lactate transport by breast cancer cells. Breast Cancer Res Treat 157:1–11
Gwak H, Haegeman G, Tsang BK, Song YS (2015) Cancer-specific interruption of glucose metabolism by resveratrol is mediated through inhibition of Akt/GLUT1 axis in ovarian cancer cells. Mol Carcinog 54:1529–1540
Mathupala SP, Ko YH, Pedersen PL (2006) Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25:4777–4786
Mathupala SP, Ko YH, Pedersen PL (2009) Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin Cancer Biol 19:17–24
Pedersen PL (2008) Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J Bioenerg Biomembr 40:123–126
Marín-Hernández A, Rodríguez-Enríquez S, Vital-González PA et al (2006) Determining and understanding the control of glycolysis in fast-growth tumor cells: flux control by an over-expressed but strongly product-inhibited hexokinase. FEBS J 273:1975–1988
Liu VM, Vander Heiden MG (2015) The role of pyruvate kinase M2 in cancer metabolism. Brain Pathol 25:781–783
Dong G, Mao Q, Xia W et al (2016) PKM2 and cancer: the function of PKM2 beyond glycolysis. Oncol Lett 11:1980–1986
Vander Heiden MG, Lunt SY, Dayton TL et al (2011) Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol 76:325–334
DeBerardinis RJ, Cheng T (2010) Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29:313–324
Locasale JW (2012) The consequences of enhanced cell-autonomous glucose metabolism. Trends Endocrinol Metab 23:545–551
Board M, Humm S, Newsholme EA (1990) Maximum activities of key enzymes of glycolysis, glutaminolysis, pentose phosphate pathway and tricarboxylic acid cycle in normal, neoplastic and suppressed cells. Biochem J 265:503–509
Mc Dermott EW, Barron ET, Smyth PP, O’Higgins NJ (1990) Premorphological metabolic changes in human breast carcinogenesis. Br J Surg 77:1179–1182
Zaccarin M, Bosello-Travain V, Di Paolo ML et al (2017) Redox status in a model of cancer stem cells. Arch Biochem Biophys 617:120–128
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Chandel NS (2014) Mitochondria as signaling organelles. BMC Biol 12:34
Arco AD, Satrùstegui J (2005) New mitochondrial carriers: an overview. Cell Mol Life Sci 62:2204–2227
Ozkaya AB, Ak H, Atay S, Aydin HH (2015) Targeting mitochondrial citrate transport in breast cancer cell lines. Anticancer Agents Med Chem 15:374–381
Senyilmaz D, Teleman AA (2015) Chicken or the egg: Warburg effect and mitochondrial dysfunction. F1000Prime Rep 7:41
DeBerardinis RJ, Mancuso A, Daikhin E et al (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104:19345–19350
Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G (2015) Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab 21:805–821
Galluzzi L, Kepp O, Kroemer G (2012) Mitochondria: master regulators of danger signalling. Nat Rev 13:780–788
Gorrini C, Harris IS, Mak TW (2013) Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12:931–947
Schumacker PT (2015) Reactive oxygen species in cancer: a dance with the devil. Cancer Cell 27:156–157
Sabharwal SS, Schumacker PT (2014) Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14:709–721
Okon IS, Zou MH (2015) Mitochondrial ROS and cancer drug resistance: implications for therapy. Pharmacol Res 100:170–174
Zu XL, Guppy M (2004) Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313:459–465
Weinberg SE, Chandel NS (2015) Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 11:9–15
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Adeva-Andany M, López-Ojén M, Funcasta-Calderón R et al (2014) Comprehensive review on lactate metabolism in human health. Mitochondrion 17:76–100
Proia P, Di Liegro CM, Schiera G, Fricano A, Di Liegro I (2016) Lactate as a metabolite and a regulator in the central nervous system. Int J Mol Sci 17:76–100
Schurr A (2006) Lactate: the ultimate cerebral oxidative energy substrate? J Cereb Blood Flow Metab 26:142–152
Quistorff B, Grunnet N (2011) The isoenzyme pattern of LDH does not play a physiological role; except perhaps during fast transitions in energy metabolism. Aging (Albany NY) 3:457–460
Compan V, Pierredon S, Vanderperre B et al (2015) Monitoring mitochondrial pyruvate carrier activity in real time using a BRET-based biosensor: investigation of the Warburg effect. Mol Cell 59:491–501
Hall MM, Rajasekaran S, Thomsen TW, Peterson AR (2016) Lactate: friend or foe. PM R 8:S8–S15
Kemppainen J, Fujimoto T, Kalliokoski KK et al (2002) Myocardial and skeletal muscle glucose uptake during exercise in humans. J Physiol 542:403–412
Chatham JC (2002) Lactate: the forgotten fuel! J Physiol 542:333
Hsu PP, Sabatini DM (2008) Cancer cell metabolism: Warburg and beyond. Cell 134:703–707
Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N et al (2011) Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 10:4047–4064
Sacktor B, Packerl L, Estabrookp R (1959) Respiratory activity in brain mitochondria. Arch Biochem Biophys 80:68–71
Cruz RS, de Aguiar RA, Turnes T et al (2012) Intracellular shuttle: the lactate aerobic metabolism. Sci World J 2012:420984. https://doi.org/10.1100/2012/420984
Blanco A, Zinkham WH (1963) Lactate dehydrogenases in human testes. Science 139:601–602
Goldberg E (1963) Lactic and malic dehydrogenases in human spermatozoa. Science 139:602–603
Swegen A, Curry BJ, Gibb Z et al (2015) Investigation of the stallion sperm proteome by mass spectrometry. Reproduction (Cambridge) 149:235–244
Laughlin MR, Taylor J, Chesnick AS, DeGroot M, Balaban RS (1993) Pyruvate and lactate metabolism in the in vivo dog heart. Am J Physiol 264:H2068–H2079
de Bari L, Atlante A, Valenti D, Passarella S (2004) Partial reconstruction of in vitro gluconeogenesis arising from mitochondrial l-lactate uptake/metabolism and oxaloacetate export via novel l-lactate translocators. Biochem J 380:231–242
Pizzuto R, Paventi G, Porcile C et al (2012) l-Lactate metabolism in HEP G2 cell mitochondria due to the l-lactate dehydrogenase determines the occurrence of the lactate/pyruvate shuttle and the appearance of oxaloacetate, malate and citrate outside mitochondria. Biochim Biophys Acta 1817:1679–1690
Lemire J, Mailloux RJ, Appanna VD (2008) Mitochondrial lactate dehydrogenase is involved in oxidative-energy metabolism in human astrocytoma cells (CCF-STTG1). PLoS One 3:e1550. https://doi.org/10.1371/journal.pone.0001550
Chatham JC, Des Rosiers C, Forder JR (2001) Evidence of separate pathways for lactate uptake and release by the perfused rat heart. Am J Physiol Endocrinol Metab 281:E794–E802
Passarella S, Paventi G, Pizzuto R (2014) The mitochondrial l-lactate dehydrogenase affair. Front Neurosci 8:407
Kane DA (2014) Lactate oxidation at the mitochondria: a lactate–malate–aspartate shuttle at work. Front Neurosci 8:366
Halestrap AP (1975) The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem J 148:85–96
Gallina FG, Gerez de Burgos NM, Burgos C, Coronel CE, Blanco A (1994) The lactate/pyruvate shuttle in spermatozoa: operation in vitro. Arch Biochem Biophys 308:515–519
Chiaretti B, Casciaro A, Minotti G, Eboli ML, Galeotti T (1979) Quantitative evaluation of the activity of the malate-aspartate shuttle in Ehrlich ascites tumor cells. Cancer Res 39:2195–2199
Chiarugi A, Dölle C, Felici R, Ziegler M (2012) The NAD metabolome—a key determinant of cancer cell biology. Nat Rev Cancer 12:741–752
Nikiforov A, Kulikova V, Ziegler M (2015) The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol 50:284–297
Zelenka J, Dvořák A, Alán L (2015) l-Lactate protects skin fibroblasts against aging-associated mitochondrial dysfunction via mitohormesis. Oxid Med Cell Longev 2015:351698
Rooney K, Trayhurn P (2011) Lactate and the GPR81 receptor in metabolic regulation: implications for adipose tissue function and fatty acid utilisation by muscle during exercise. Br J Nutr 106:1310–1316
Morland C, Lauritzen KH, Puchades M et al (2015) The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: expression and action in brain. J Neurosci Res 93:1045–1055
Yang J, Ruchti E, Petit JM et al (2014) Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc Natl Acad Sci 111:12228–12233
Thomas AP, Halestrap AP (1981) The role of mitochondrial pyruvate transport in the stimulation by glucagon and phenylephrine of gluconeogenesis from l-lactate in isolated rat hepatocytes. Biochem J 198:551–560
Bonuccelli G, Tsirigos A, Whitaker-Menezes D et al (2010) Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 9:3506–3514
Sonveaux P, Végran F, Schroeder T et al (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Investig 118:3930–3942
de Bari L, Chieppa G, Marra E, Passarella S (2010) l-Lactate metabolism can occur in normal and cancer prostate cells via the novel mitochondrial l-lactate dehydrogenase. Int J Oncol 37:1607–1620
Chen YJ, Mahieu NG, Huang X et al (2016) Lactate metabolism is associated with mammalian mitochondria. Nat Chem Biol 12:937–943
Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23:27–47
Sellers K, Fox MP, Bousamra M et al (2015) Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Investig 125:687–698
Hensley CT, Faubert B, Yuan Q et al (2016) Metabolic heterogeneity in human lung tumors. Cell 164:681–694
Faubert B, Li KY, Cai L et al (2017) Lactate metabolism in human lung tumors. Cell 171:358–371
Hui S, Ghergurovich JM, Morscher RJ et al (2017) Glucose feeds the TCA cycle via circulating lactate. Nature 551:115–118
Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27:441–464
Pérez-Escuredo J, Dadhich RK, Dhup S et al (2016) Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 15:72–83
Girgis H, Masui O, White NM et al (2014) Lactate dehydrogenase A is a potential prognostic marker in clear cell renal cell carcinoma. Mol Cancer 13:101
Lee M, Yoon JH (2015) Metabolic interplay between glycolysis and mitochondrial oxidation: the reverse Warburg effect and its therapeutic implication. World J Biol Chem 6:148–161
Martinez-Outschoorn UE, Pavlides S, Howell A et al (2011) Stromal–epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol 43:1045–1051
Galon J, Costes A, Sanchez-Cabo F et al (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964
Ho PC, Bihuniak JD, Macintyre AN et al (2015) Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162:1217–1228
Chang CH, Qiu J, O’Sullivan D et al (2015) Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162:1229–1241
Li F, Wang Y, Zeller KI, Potter JJ et al (2005) Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol 25:6225–6234
Lezi E, Swerdlow RH (2016) Lactate’s effect on human neuroblastoma cell bioenergetic fluxes. Biochem Pharmacol 99:88–100
Jose C, Bellance N, Rossignol R (2011) Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim Biophys Acta 1807:552–561
Chen X, Qian Y, Wu S (2015) The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med 79:253–263
Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R (2009) HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem 9:1084–1101
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
de Bari, L., Atlante, A. Including the mitochondrial metabolism of l-lactate in cancer metabolic reprogramming. Cell. Mol. Life Sci. 75, 2763–2776 (2018). https://doi.org/10.1007/s00018-018-2831-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2831-y