
Abstract
The hypoxia-inducible factor (HIF) is a heterodimeric transcription factor governing a transcriptional program in response to reduced O2 availability in metazoans. It contributes to physiology and pathogenesis of many human diseases through its downstream target genes. Emerging studies have shown that the transcriptional activity of HIF is highly regulated at multiple levels and the epigenetic regulators are essential for HIF-mediated transactivation. In this review, we will discuss the comprehensive regulation of HIF transcriptional activity by different types of epigenetic regulators.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The hypoxia-inducible factor (HIF) is a master transcriptional factor consisting of an inducible α subunit and a constitutively expressed β subunit in response to low oxygen in metazoans [1]. Three α subunits (HIF-1α, HIF-2α, and HIF-3α) and two β subunits (HIF-1β, also known as ARNT, and ARNT2) have been cloned thus far [2,3,4,5]. Three HIF-1α mRNA transcripts are encoded by human HIF1A gene, whereas only one HIF-2α mRNA transcript is transcribed from human EPAS1 gene (Fig. 1). HIF-1α and HIF-2α share about 48% overall amino acid sequence identity and contain the same functional domains: basic helix–loop–helix (bHLH) domain, Per-Arnt-Sim (PAS) domain, oxygen-dependent degradation (ODD) domain, N-terminal transactivation domain (N-TAD), inhibitory domain (ID), and C-terminal transactivation domain (C-TAD) (Fig. 1). A total of 19 distinct HIF-3α transcripts exist in the human genome database due to alternative mRNA splicing but only 8 variants may encode HIF-3α proteins. Human HIF-3α isoforms 1 and 9 contain a N-TAD and a unique leucine zipper (LZIP) domain and are shown to activate gene transcription in human cells (Fig. 1) [6]. However, HIF-3α isoform 4 lacks the transactivation domain and LZIP domain, and functions as a transcriptional inhibitor for HIF-1 and HIF-2 [7]. HIF-1β is ubiquitously expressed, but ARNT2 expression is restricted to the brain and kidney in rat and mouse [8, 9]. Hundreds of genes have been discovered to be regulated by HIF in various cell types and their protein products regulate erythropoiesis, angiogenesis, metabolism, pH homeostasis, stem cell maintenance, autophagy, immune evasion, and cell migration/invasion [10]. Therefore, HIF-1 and HIF-2 play an important role in development, physiology, and diseases, such as cancer, heart disease, sleep apnea, and trauma [10]. Recent studies showed that HIF-3α overexpression causes aberrant late branching morphogenesis, alveolar formation, and epithelial differentiation during lung development in transgenic mice [11]. HIF-3α knockout impairs pulmonary endothelial cell proliferation and angiogenic potential [12], suggesting a critical role of HIF-3α in physiology and pathology.

The structure scheme of active human HIF-α isoforms and human HIF-β. The functional domains of each isoform are indicated. bHLH basic helix-loop-helix, PAS Per-Arnt-Sim, ODD oxygen-dependent degradation, N-TAD N-terminal transactivation domain, ID inhibitory domain, C-TAD C-terminal transactivation domain, LZIP leucine zipper
The transcriptional activity of HIF, particularly HIF-1 and HIF-2, is regulated at both mRNA and protein levels. Under normoxic conditions, HIF-α is hydroxylated on proline (Pro) residues (Pro 402 and Pro 564 for human HIF-1α; Pro 405 and Pro 531 for human HIF-2α; Pro 406 and Pro 492 for human HIF-3α isoform 9) by a family of prolyl hydroxylases (PHDs) in the presence of the cofactors (iron, α-ketoglutarate, and ascorbate) and the substrate O2 [13]. Prolyl hydroxylated HIF-α is recognized by von Hippel-Lindau (VHL), which recruits the Cullin-2/Elongin-B/C ubiquitin E3 ligase complex to induce HIF-α protein degradation in the 26S proteasome (Fig. 2) [14]. Under hypoxic conditions, prolyl hydroxylation of HIF-α is impaired, leading to stabilization of HIF-α protein (Fig. 2). HIF-α is then translocated into the nucleus and dimerized with HIF-1β. The heterodimer binds to the hypoxia response element (5′-A/GCGTG-3′) across the genome to enhance gene transcription [15]. Emerging studies have shown that epigenetic regulators, including epigenetic writers, erasers, and readers, and ATP-dependent chromatin remodelers, are essential for HIF-mediated transactivation (Table 1). In this review, we will discuss the comprehensive regulation of HIF transcriptional activity by different types of epigenetic regulators.

O2-dependent regulation of HIF-1 activity. Under normoxia, human HIF-1α is hydroxylated on proline (P) residues 402 and 564 by a family of prolyl hydroxylases (PHDs). Prolyl hydroxylated HIF-1α is recognized by von Hippel-Lindau (VHL), which recruits the Cullin-2/Elongin-B/C ubiquitin E3 ligase complex to induce HIF-1α protein degradation in the 26S proteasome. On the other hand, human HIF-1α is also hydroxylated on asparagine (N) 803 residue by another dioxygenase factor inhibiting HIF-1 (FIH-1), leading to blockade of p300 recruitment to the C-terminal transactivation domain of HIF-1α, thereby preventing HIF-1-dependent gene transcription. Under hypoxia, PHDs and FIH-1 lose their enzymatic activity. As such, HIF-1α protein is stabilized and the epigenetic regulators (e.g., p300/CBP, JMJD2C) are recruited to promote HIF-1-mediated transactivation
Regulation of HIF transcriptional activity by epigenetic writers
Acetyltransferases, methyltransferases, protein kinases, and ubiquitin E3 ligases function as epigenetic writers by adding epigenetic marks onto histones, DNA, or RNA. p300/CBP possess the intrinsic histone acetyltransferase activity that induces histone acetylation to relax the chromatin [16]. They bind to the transactivation domain of HIF-α to coactivate HIF-mediated transactivation, and are responsible for expression of about 30–50% of global HIF-1 downstream target genes [17]. Post-translational modifications of HIF-α are known to modulate p300 coactivator functions. Asparagine 803 of HIF-1α is hydroxylated by factor inhibiting HIF-1 (FIH-1) under nonhypoxic conditions, thereby blocking p300 binding to HIF-1α to inhibit HIF-1 transcriptional activity (Fig. 2) [18, 19]. In contrast, S-nitrosylation of cysteine 800 enhances p300 recruitment to HIF-1α to increase HIF-1-mediated transactivation [20]. Our previous study showed that the recruitment of p300 to the hypoxia response element is enhanced by pyruvate kinase M2, a HIF coactivator [21]. Several other HIF-1α-interacting proteins, including CITED2, EAF2, protein kinase C zeta, FOXO3a, ORF3, p65, histone deacetylase 4 (HDAC4), HDAC5, and FHL1, are also shown to regulate HIF-1α-p300 interaction, leading to altered HIF-1 transcriptional activity [22,23,24,25,26,27,28,29].
Similarly, the histone acetyltransferase TIP60 was recently reported to enhance HIF-1 transcriptional activity and activate about 25% of HIF-1 downstream target genes in colorectal cancer HCT116 cells [30]. TIP60 is recruited by HIF-1α to the hypoxia response element of the HIF-1 target gene ANKRD37. Knockdown of TIP60 decreases RNA polymerase II loading and activation at the promoter of ANKRD37. Acetylation of histone H3 at lysine (K) 9 and histone H4 at the promoter of ANKRD37 is also reduced in TIP60 knockdown HCT116 cells. TIP60 is known to acetylate the N-terminal lysine residues of histone H4 [31], and thus TIP60 may regulate chromatin reprogramming to enhance HIF-1 transcriptional activity.
In addition, histone acetyltransferases directly acetylate HIF-α to modulate HIF transcriptional activity. p300 acetylates HIF-1α at K709 and reduces HIF-1α ubiquitination, leading to increased HIF-1α protein stability [32]. p300/CBP-associated factor (PCAF) also acetylates HIF-1α at K674 and increases transcription of the HIF target genes BID, CA9 and VEGFA in human osteosarcoma cells [33, 34]. Jeong et al. showed that the acetyltransferase arrest-defective-1 (ARD1) induces acetylation of K532 of HIF-1α to increase HIF-1α ubiquitination and subsequent protein degradation by the PHD/VHL pathway, which diminishes HIF-1 transcriptional activity [35]. However, ARD1 fails to acetylate HIF-1α in vitro [36, 37]. It was recently suggested that prolyl hydroxylation and K391 methylation are prerequisite for ARD1-mediated K532 acetylation of HIF-1α [38].
Histone methyltransferases also play a critical role in HIF transcriptional activity. An inhibitor of the lysine methyltransferase G9a, BIX01294, increases prolyl hydroxylation of HIF-1α under hypoxic conditions, leading to increased HIF-1α ubiquitination and decreased HIF-1α protein stability in human hepatocellular carcinoma HepG2 cells [39]. Treatment of BIX01294 blocks expression of the HIF target gene VEGFA. Pharmacological or genetic inhibition of G9a prevents tumorigenesis in mice [40, 41]. G9a is induced by hypoxia at both transcriptional and post-translational levels in embryonic stem cells and cancer cells and may mediate hypoxia-induced gene repression through increasing dimethyl K9 of histone H3 in cancer cells [41,42,43]. However, the direct effect of G9a on HIF transcriptional activity remains unknown.
Regulation of HIF activity by the lysine methyltransferase SET7/9 has been the focus of recent studies, but its role is controversial. Liu et al. found that SET7/9 interacts with HIF-1α and blocks the ubiquitin E3 ligase CHIP-mediated degradation of HIF-1α protein in human cancer cells [44]. SET7/9 is also enriched at the hypoxia response elements of a subset of HIF-1 regulated genes LDHA, HK2, and PDK1, and increases transcription of these glycolytic genes in hypoxic cells [44]. In contrast, Xiao and his colleagues recently showed that SET7/9 directly induces monomethylation of HIF-1α at K32 and of HIF-2α at K29, leading to suppression of HIF transcriptional activity without affecting their protein levels [45]. Knockdown of SET7/9 increases HIF-1α binding to the hypoxia response element to promote HIF-1 transcriptional activity and expression of the glycolytic genes in human clear cell renal carcinoma RCC4 cells, thereby increasing glucose uptake and ATP production. SET7/9-mediated monomethylation of HIF-1α at K32 was later confirmed by Kim et al. in vitro and in human cervical carcinoma HeLa cells [46]. SET7/9 also catalyzes dimethylation of HIF-1α at K391 in HEK293T cells [38]. However, these two recent studies showed that SET7/9-mediated HIF-1α methylation decreases HIF-1α protein levels through increasing VHL-HIF-1α interaction and HIF-1α ubiquitination, thereby inhibiting HIF-1 transcriptional activity in transfected cells [38, 46]. Upregulated HIF-1α protein and the HIF-1 target genes Epo, Vegfa, and Slc2a1 are observed in knockin mice bearing mutant Hif1a K32A/K32A [46]. These mice display elevated erythrocytosis and angiogenesis in the retina as well as increased tumorigenesis [46], suggesting the important physiological and pathological functions of monomethyl K32 of HIF-1α. The discrepancies in regulation of HIF by SET7/9 in these studies remain unknown.
Recent studies showed that the protein arginine methyltransferase (PRMT) regulates HIF-α mRNA stability to alter HIF transcriptional activity. PRMT1 inhibits HIF-1α mRNA transcription through its methyltransferase activity in HeLa cells [47]. Knockdown of PRMT1 increases the transcriptional activity of Sp1 and Sp3, which are known to bind to the HIF1A promoter to control its mRNA transcription [48], leading to upregulation of HIF-1α mRNA transcription, thereby enhancing HIF-1 binding to the hypoxia response element and expression of the HIF target genes under hypoxia. Another member of the PRMT family PRMT9 (also named as FBXO11) impairs stability of HIF-1α mRNA to decrease de novo synthesis of HIF-1α protein and its transcriptional activity in human glioblastoma U87MG cells [49]. Interestingly, the ubiquitin E3 ligase activity of PRMT9 is responsible for HIF-1α mRNA destabilization. In contrast, PRMT5 promotes cap-dependent translation of HIF-1α mRNA, leading to increased de novo synthesis of HIF-1α protein in A549 cells [50].
Apart from PRMTs, DNA methyltransferase is also involved in the regulation of HIF-α mRNA transcription. The CpG within the hypoxia response element at the HIF1A promoter is highly methylated in human colon tissues, but is hypomethylated in human embryonic tissues and colorectal cancer tissues [51, 52]. Treatment of the DNA methylation inhibitor 5-Aza-2′-deoxycytidine blocks CpG methylation at the HIF1A promoter, leading to increased autoregulation by HIF-1 and subsequent its downstream target gene expression [52]. The transcription factor Kaiso binds methylated HIF1A promoter and suppresses HIF-1α mRNA expression under hypoxia to impair HIF-1 transcriptional activity [53]. Recently, it was shown that DNMT3a methylates the CpG at the promoter of the EPAS1 gene and decreases expression of HIF-2α in normal human epithelial cells. DNMT3a is frequently downregulated in primary tumors, which elevates HIF-2α levels to promote tumorigenesis [54]. Methylation of the HIF3A gene was found in adipose tissues and correlated with obesity [55], although its DNA methyltransferase has not yet been identified.
Regulation of HIF transcriptional activity by epigenetic erasers
The deacetylases and demethylases, acting as epigenetic erasers, remove the acetyl and methyl groups from the chromatin, respectively, and are involved in chromatin remodeling and gene regulation. The role of epigenetic erasers in HIF transcriptional activity has been extensively studied. Several HDACs, including HDAC1, HDAC2, HDAC3, HDAC4, and HDAC6, have been shown to increase HIF-1α protein stability to promote HIF-1 transactivation [56,57,58,59]. Consistently, HDAC inhibitors (HDACi), including trichostatin A, apicidin, valproic acid, FK228, sodium butyrate, and LAQ824, dose-dependently decrease HIF-1α protein levels in various cell lines [56, 59]. Ubiquitintation of HIF-1α is elevated upon treatment of HDACi or knockdown of HDAC, leading to HIF-1α protein degradation in the 26S proteasome and inhibition of HIF-1 transcriptional activity. The PHD/VHL pathway is dispensable for HDACi-mediated HIF-1α protein degradation [56]. Interestingly, deacetylation of HIF-1α by HDAC2 or HDAC4 was suggested to increase HIF-1α protein stability and transcriptional activity [57, 58].
Additional mechanisms underlying HDAC-mediated HIF-1 transcriptional activity have been also proposed. HDAC4 and HDAC5 bind to the inhibitory domain of HIF-1α to compete with FIH-1 for binding to HIF-1α, leading to increased recruitment of p300 to the transactivation domain of HIF-1α [29]. HDAC4 or HDAC5 promotes expression of the HIF-1 target gene VEGFA without affecting HIF-1α protein stability. HDAC7 was also shown to physically interact with HIF-1α, p300, and CBP, and enhances HIF-1 transcriptional activity [60].
Sirtuin (SIRT, also known as Sir2 in yeast) was initially characterized as the class III NAD+-dependent HDAC in yeast [61]. This protein is highly conserved from archaea to humans. Mammalian SIRT consists of seven family members and their activity is highly regulated [62]. Several environmental stresses including hypoxia reduces intracellular NAD+ levels in mammalian cells, which results in decreased deacetylase activity of SIRT [34]. SIRT1 is the most studied SIRT family member in gene regulation, but its effect on HIF-1 transcriptional activity is still under debate [34, 63,64,65,66]. Dioum et al. reported that SIRT1 selectively interacts with HIF-2α and deacetylates HIF-2α to increase its transcriptional activity in human hepatoma Hep3B cells [65]. SIRT1 knockout reduces HIF-2-dependent Epo gene expression in vitro and in mice. Distinct to its effect on HIF-2, SIRT1 does not associate with HIF-1α nor affect HIF-1 transcriptional activity [65]. However, the Park group found that SIRT1 is able to bind to HIF-1α and reverses PCAF-induced acetylation of K674 of HIF-1α, leading to blockade of p300 recruitment to the transactivation domain of HIF-1α and subsequent inhibition of HIF-1 transcriptional activity in multiple cancer cell lines [34, 66]. HIF-2 activation by SIRT1 was shown to be cell type-dependent [66]. Forced expression of SIRT1 decreases hypoxia-induced expression of PDK1, VEGFA, and CA9, and impairs tumor growth in xenograft mice [34]. Subsequent studies indicate that SIRT1 indirectly acts on HIF-1 transcriptional activity through HIF-1α protein stability [63, 64]. Genetic or pharmacological inhibition of SIRT1 increases the acetylation of HIF-1α to decrease stabilization of HIF-1α protein and HIF-1-dependent gene expression in human cancer cell lines. In contrast, SIRT1 inhibits HIF-1α expression in primary vascular smooth muscle cells under hypoxia as well as in the femoral artery of SIRT1 transgenic mice after wire injury [67], suggesting differential regulation of HIF-1 by SIRT1 in the disease context. Finally, SIRT1 was recently shown to deacetylate K14 of histone H3 at the promoter of HIF1A gene to attenuate HIF-1α mRNA expression in SH-SY5Y cells treated with methyl-4-phenylpyridinium, a toxic chemical causing symptoms of Parkinson’s disease [68].
SIRT2 interacts with and deacetylates HIF-1α at K709 to facilitate PHD2-HIF-1α interaction in HeLa cells, which increases HIF-1α ubiquitination and protein degradation, thereby inhibiting HIF-1 transcriptional activity [69]. Similarly, SIRT7 also destabilizes HIF-1α and HIF-2α proteins and blocks their transcriptional activity [70]. This effect is independent of its deacetylase activity. SIRT6 negatively regulates the de novo protein synthesis and stability of HIF-1α in embryonic stem cells and blocks HIF-1-dependent glucose uptake and glycolysis [71]. Finally, SIRT3 knockdown increases HIF-1α protein levels in human cancer cells and tumorigenesis in mice [72]. SIRT3 is mainly localized in mitochondria and loss of SIRT3 increases reactive oxygen species (ROS) in mouse embryonic fibroblasts. ROS is known to increase HIF-1α protein stability [73]. Elimination of ROS by N-acetyl-cysteine abolishes increased HIF-1 transcriptional activity in SIRT3 knockdown cells [72], suggesting that SIRT3 is involved in indirect regulation of HIF-1 activity.
The Jumonji domain (JMJD) containing protein family consists of 32 members in humans and possesses lysine demethylase activity with histone H3 and non-histone proteins as substrates [74,75,76,77]. Iron, α-ketoglutarate and O2 are required for their enzymatic activity like other α-ketoglutarate-dependent dioxygenases [74]. Our previous work showed that JMJD2C selectively coactivates HIF-1 in breast cancer cells [78]. JMJD2C demethylates trimethyl K9 of histone H3 at the hypoxia response element to enhance HIF-1 binding to its target genes and subsequent transcription of LDHA, PDK1, SLC2A1, LOXL2, and L1CAM. Knockdown of JMJD2C reduces breast tumor progression and metastasis in mice. JMJD2C expression is controlled by HIF-1 and HIF-2 [79], and thus JMJD2C represents a positive feedback mechanism amplifying HIF-1 activation in breast cancer. Similarly, JMJD1A (also known as KDM3A) interacts with HIF-1α and demethylates dimethyl K9 of histone H3 at the hypoxia response element of a subset of HIF-1 target genes to enhance their gene transcription, thereby increasing colorectal tumor growth in mice [80, 81]. JMJD1A also activates HIF-1-dependent SLC2A3 gene transcription to enhance glucose uptake in HUVEC cells [80]. Therefore, the histone demethylases JMJD proteins control HIF transcriptional activity through altering histone methylation at the HIF target genes.
LSD1 is a flavin adenine dinucleotide (FAD)-dependent lysine demethylase. Unlike JMJD proteins, LSD1 indirectly promotes HIF-1 transcriptional activity through altering HIF-1α protein degradation machinery [38, 46, 82]. LSD1 counteracts SET7/9-induced methylation of HIF-1α at K32 and K391 and also inhibit PHD2-mediated prolyl hydroxylation of HIF-1α to decrease HIF-1α ubiquitination and protein degradation [38, 46]. In addition, LSD1 interacts with and demethylates RACK1, which attenuates RACK1 binding to HIF-1α [82]. RACK1 competes with Hsp90 to mediate O2-independent but proteasome-dependent HIF-1α protein degradation [83]. Consequently, inhibition of LSD1 by its inhibitors or small interfering RNAs enhances RACK1-mediated HIF-1α protein degradation [82]. LSD1 potentiates transcription of HIF-1-dependent VEGFA and glycolytic genes in human cancer cells, leading to increased tumor angiogenesis [38, 82]. Interestingly, the FAD levels are reduced during prolonged hypoxia, which diminishes the demethylase activity of LSD1 [82]. Thus, LSD1 represents a molecular mechanism of reduced HIF-1 transcriptional activity during prolonged hypoxia.
The DNA demethylase TET is another subgroup of α-ketoglutarate-dependent dioxygenases and consists of three family members (TET1-3) [84]. TET hydroxylates 5-methylcytosine to generate 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxycytosine in the consecutive biochemical reactions [85]. It has been reported that TET1 functions as a HIF coactivator and controls HIF-1-dependent epithelial-mesenchymal transition in breast cancer cells [86]. The protein interaction of TET1 with HIF-1α and HIF-2α, but not its DNA demethylase activity, is critical for HIF activation. Like JMJD protein, TET1 is induced by HIF-1 and HIF-2 in breast cancer cells and provides a positive feedback regulation of HIF in breast cancer [86].
On the other hand, hypoxia causes reprogramming of the chromatin landscape by altering expression and activity of epigenetic erasers. Hypoxia induces expression of HDAC3 and decreases the global levels of acetyl K4 of histone H3 in breast cancer cells [87]. HDAC3 further recruits the methyltransferase WDR5, which increases dimethyl and trimethyl K4 of histone H3 in breast cancer cells under hypoxia [87]. Changes of these histone marks are associated with altered expression of E-cadherin, plakoglobin, N-cadherin, and Vimentin that are involved in epithelial-mesenchymal transition [87]. In addition, the demethylase activity of JMJD proteins such as JMJD3 and JARID1A/B, is impaired under hypoxic conditions, due to limited availability of their substrate O2 [88], which contributes to increased trimethyl K27 and K4 of histone H3, respectively [88, 89]. The global increases of dimethyl and trimethyl K9 of histone H3, trimethyl K36 of histone H3, dimethyl K79 of histone H3, acetyl K14 of histone H3, and dimethyl arginine 3 of histone H4 are also found under hypoxic conditions [87, 90]. In contrast, hypoxia decreases the global levels of acetyl K5 of histone H4, acetyl K12 of histone H4, acetyl K5 of histone H2A in breast cancer cells [87]. Interestingly, knockdown of JMJD2A increases levels of trimethyl K9 of histone H3 on the HIF-1A gene in RKO cells under hypoxia, leading to inhibition of HIF-1α mRNA transcription and subsequent HIF-1 activity [91].
Regulation of HIF transcriptional activity by epigenetic readers
The role of the epigenetic reader in HIF transcriptional activity is less studied. Emerging studies have elucidated an essential role of bromodomain and extra-terminal (BET) proteins, particularly BRD4, in gene regulation [92, 93]. The epigenetic reader BRD4 recognizes acetylated lysine of histones and recruits the transcription factors and epigenetic regulators to control gene transcription [94]. A recent study found that a BET inhibitor JQ1 downregulates hypoxia-induced global transcriptome by blocking HIF binding to the hypoxia response element in breast cancer cells [95]. Decreased expression of the HIF target genes CA9 and VEGFA is also observed in JQ1-treated breast tumors in mice, leading to inhibition of angiogenesis and tumor growth [95]. Although hypoxia increases BRD4 recruitment to the hypoxia response element of CA9 and VEGFA [95], the direct effect of BRD4 on HIF transcriptional activity is still unclear.
Regulation of HIF transcriptional activity by ATP-dependent chromatin remodelers
The ATP-dependent chromatin remodelers modulate chromatin structure and assembly and have an important role in gene regulation. Four chromatin remodeler complex families, including SWI/SNF complex, ISWI complex, INO80 complex, and NuRD complex, have been well characterized [96]. Transient ischemia causes chromatin condensation in accompany with decreased cellular ATP levels, which is reversible upon reoxygenation [97]. Although little is known about the role of chromatin remodelers in ischemia-induced chromatin condensation, several ATP-dependent chromatin remodelers have been shown to regulate HIF transcriptional activity. The key components of the SWI/SNF complex, BRG1 and BRM, promote expression of a subset of HIF target genes in human cancer cell lines in an ATPase-dependent manner [98]. Upon hypoxia, BRG1 is recruited by HIF to the hypoxia response elements of CA9 and EPO and induces nucleosome remodeling at these gene promoters [98, 99]. BRG1 and BRM also enhance the recruitment of RNA polymerase II to the EPO gene in hypoxic Hep3B cells [100]. On the other hand, knockdown of BRG1 decreases transcription of HIF1A and EPAS1 genes in hypoxic Hep3B cells [98, 101]. The other component of the SWI/SNF complex, BAF57, is also required for HIF-1α mRNA expression [101]. Thus, the SWI/SNF complex regulates HIF transcriptional activity through controlling HIF-α mRNA levels and HIF transactivation machinery.
Unlike SWI/SNF complex, ISWI negatively modulates HIF-1 transcriptional activity through the FIH-1-dependent pathway [102]. Knockdown of ISWI decreases RNA polymerase II binding to the promoter of the HIF1AN gene (encoding FIH-1) in hypoxic U2OS cells, which may lead to decreased FIH-1 expression. ISWI selectively inhibits expression of HIF-1 target genes BNIP3, CA9, and EGLN1, but not BHLHE40, EGLN3, and SLC2A1 [102]. It was shown that FIH-1 preferentially downregulates expression of the HIF-1 target genes CA9, EGLN3, SLC2A1, but not BNIP3 [103], suggesting that FIH-1 may be not the only mechanism underlying ISWI-mediated inhibition of HIF-1.
Reptin and Pontin belong to AAA+ ATPases with a similar conserved structure and are present in multiple protein complexes, including the INO80 complex [104]. The data from the Baek group indicate that Reptin and Pontin both are monomethylated by G9a in a hypoxia-dependent manner, and methylated Reptin and Pontin exhibit the opposite functions of HIF transcriptional activity in human breast cancer cells [43, 105]. Reptin is enriched at the hypoxia response elements of a subset of HIF-1 target genes and recruits HDAC1 to decrease RNA polymerase II occupancy at the promoter of these genes, thereby blocking HIF-1-dependent gene transcription and breast tumorigenesis [43]. In contrast, Pontin enhances the recruitment of p300 to the HIF-1 target genes to promote HIF-1 transcriptional activity and breast cancer cell proliferation and mobility in vitro [105]. Pontin’s coactivator function may be independent of the INO80 complex as its ATPase dead mutant still enhances HIF-1-mediated transactivation [105]. This scenario has been also suggested in a recent study, showing that Reptin and Pontin are present in the TIP60 complex to coactivate HIF-1 and knockdown of INO80 has no significant effect on HIF-1 transcriptional activity in Drosophila S2 cells [30].
Metastasis-associated protein 1 (MTA1) is a component of the NuRD complex and mediates loading of the NuRD complex on the genome through interacting with transcription factors. MTA1 was shown to bind to the C-terminus of HIF-1α and recruit HDAC1 to decrease K532 acetylation of HIF-1α [106]. MTA1 counteracts ARD1′s effect to increase HIF-1α protein stability [106]. Overexpression of MTA1 potentiates HIF-1 transcriptional activity and VEGFA expression in human cancer cells, leading to increased angiogenesis [106, 107]. MTA1 is upregulated by HIF-1 in human breast cancer cell lines under hypoxia [106]. The LSD1-c-Myc axis also activates MTA1 mRNA transcription [38]. LSD1 additionally increases MTA1 protein stability by blocking ubiquitin-dependent proteasomal degradation of MTA1 [38].
Concluding remarks
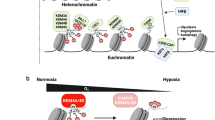
HIF governs the transcriptional program of hypoxic responses and mediates physiology and pathogenesis of many human diseases, particularly cancers. Accumulating studies have shown that epigenetic regulators play a key role in HIF-mediated transactivation (Table 1). Multiple epigenetic regulators cooperate to modulate HIF transcriptional activity through controlling chromatin reprogramming or HIF-α mRNA or protein levels (Fig. 3). Importantly, epigenetic regulators determine the specificity of the transcriptional activity of HIF-1 and HIF-2 and their downstream target genes. Therefore, understanding of epigenetic regulation of HIF activity may uncover the fundamental mechanisms of human physiology and disease progression and provide new targets and approaches for treatment of human diseases.

Mechanisms of HIF transcriptional activity by epigenetic regulators. Epigenetic regulators modulate HIF transcriptional activity through controlling HIF-α mRNA or protein stability or chromatin reprogramming. A list of epigenetic regulators within three subgroups is shown
References
Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92(12):5510–5514
Semenza GL, Wang GL (1992) A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12(12):5447–5454
Tian H, McKnight SL, Russell DW (1997) Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 11(1):72–82
Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA (1998) Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr 7(3):205–213
Keith B, Adelman DM, Simon MC (2001) Targeted mutation of the murine aryl hydrocarbon receptor nuclear translocator 2 (Arnt2) gene reveals partial redundancy with Arnt. Proc Natl Acad Sci USA 98(12):6692–6697. doi:10.1073/pnas.121494298
Zhang P, Yao Q, Lu L, Li Y, Chen PJ, Duan C (2014) Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep 6(6):1110–1121. doi:10.1016/j.celrep.2014.02.011
Maynard MA, Evans AJ, Shi W, Kim WY, Liu FF, Ohh M (2007) Dominant-negative HIF-3 alpha 4 suppresses VHL-null renal cell carcinoma progression. Cell Cycle 6(22):2810–2816. doi:10.4161/cc.6.22.4947
Hirose K, Morita M, Ema M, Mimura J, Hamada H, Fujii H, Saijo Y, Gotoh O, Sogawa K, Fujii-Kuriyama Y (1996) cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt). Mol Cell Biol 16(4):1706–1713
Drutel G, Kathmann M, Heron A, Schwartz JC, Arrang JM (1996) Cloning and selective expression in brain and kidney of ARNT2 homologous to the Ah receptor nuclear translocator (ARNT). Biochem Biophys Res Commun 225(2):333–339. doi:10.1006/bbrc.1996.1176
Semenza GL (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148(3):399–408. doi:10.1016/j.cell.2012.01.021
Huang Y, Kapere Ochieng J, Kempen MB, Munck AB, Swagemakers S, van Ijcken W, Grosveld F, Tibboel D, Rottier RJ (2013) Hypoxia inducible factor 3alpha plays a critical role in alveolarization and distal epithelial cell differentiation during mouse lung development. PLoS One 8(2):e57695. doi:10.1371/journal.pone.0057695
Kobayashi S, Yamashita T, Ohneda K, Nagano M, Kimura K, Nakai H, Poellinger L, Ohneda O (2015) Hypoxia-inducible factor-3alpha promotes angiogenic activity of pulmonary endothelial cells by repressing the expression of the VE-cadherin gene. Genes Cells 20(3):224–241. doi:10.1111/gtc.12215
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr (2001) HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292(5516):464–468. doi:10.1126/science.1059817
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399(6733):271–275. doi:10.1038/20459
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A (1996) Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271(51):32529–32537
Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM (1996) An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA 93(23):12969–12973
Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino TA, Cleveland JL, Brindle PK (2005) Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J 24(22):3846–3858. doi:10.1038/sj.emboj.7600846
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16(12):1466–1471. doi:10.1101/gad.991402
Mahon PC, Hirota K, Semenza GL (2001) FIH-1: a novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 15(20):2675–2686. doi:10.1101/gad.924501
Yasinska IM, Sumbayev VV (2003) S-nitrosation of Cys-800 of HIF-1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett 549(1–3):105–109. doi:10.1016/S0014-5793(03)00807-X
Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL (2011) Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145(5):732–744. doi:10.1016/j.cell.2011.03.054
Chen Z, Liu X, Mei Z, Wang Z, Xiao W (2014) EAF2 suppresses hypoxia-induced factor 1alpha transcriptional activity by disrupting its interaction with coactivator CBP/p300. Mol Cell Biol 34(6):1085–1099. doi:10.1128/MCB.00718-13
Hubbi ME, Gilkes DM, Baek JH, Semenza GL (2012) Four-and-a-half LIM domain proteins inhibit transactivation by hypoxia-inducible factor 1. J Biol Chem 287(9):6139–6149. doi:10.1074/jbc.M111.278630
Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM (1999) Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev 13(1):64–75
Datta K, Li J, Bhattacharya R, Gasparian L, Wang E, Mukhopadhyay D (2004) Protein kinase C zeta transactivates hypoxia-inducible factor alpha by promoting its association with p300 in renal cancer. Cancer Res 64(2):456–462. doi:10.1158/0008-5472.CAN-03-2706
Emerling BM, Weinberg F, Liu JL, Mak TW, Chandel NS (2008) PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). Proc Natl Acad Sci USA 105(7):2622–2627. doi:10.1073/pnas.0706790105
Moin SM, Chandra V, Arya R, Jameel S (2009) The hepatitis E virus ORF3 protein stabilizes HIF-1alpha and enhances HIF-1-mediated transcriptional activity through p300/CBP. Cell Microbiol 11(9):1409–1421. doi:10.1111/j.1462-5822.2009.01340.x
Mendonca DB, Mendonca G, Aragao FJ, Cooper LF (2011) NF-kappaB suppresses HIF-1alpha response by competing for P300 binding. Biochem Biophys Res Commun 404(4):997–1003. doi:10.1016/j.bbrc.2010.12.098
Seo HW, Kim EJ, Na H, Lee MO (2009) Transcriptional activation of hypoxia-inducible factor-1alpha by HDAC4 and HDAC5 involves differential recruitment of p300 and FIH-1. FEBS Lett 583(1):55–60. doi:10.1016/j.febslet.2008.11.044
Perez-Perri JI, Dengler VL, Audetat KA, Pandey A, Bonner EA, Urh M, Mendez J, Daniels DL, Wappner P, Galbraith MD, Espinosa JM (2016) The TIP60 complex is a conserved coactivator of HIF1A. Cell Rep 16(1):37–47. doi:10.1016/j.celrep.2016.05.082
Kimura A, Horikoshi M (1998) Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells 3(12):789–800. doi:10.1016/j.celrep.2016.05.082
Geng H, Liu Q, Xue C, David LL, Beer TM, Thomas GV, Dai MS, Qian DZ (2012) HIF1alpha protein stability is increased by acetylation at lysine 709. J Biol Chem 287(42):35496–35505. doi:10.1074/jbc.M112.400697
Xenaki G, Ontikatze T, Rajendran R, Stratford IJ, Dive C, Krstic-Demonacos M, Demonacos C (2008) PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional activity in hypoxia. Oncogene 27(44):5785–5796. doi:10.1038/onc.2008.192
Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW (2010) Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 38(6):864–878. doi:10.1016/j.molcel.2010.05.023
Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW (2002) Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 111(5):709–720. doi:10.1016/S0092-8674(02)01085-1
Murray-Rust TA, Oldham NJ, Hewitson KS, Schofield CJ (2006) Purified recombinant hARD1 does not catalyse acetylation of Lys532 of HIF-1alpha fragments in vitro. FEBS Lett 580(8):1911–1918. doi:10.1016/j.febslet.2006.02.012
Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, Sang N, Caro J, Lillehaug JR (2005) Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS Lett 579(28):6428–6432. doi:10.1016/j.febslet.2005.10.036
Lee JY, Park JH, Choi HJ, Won HY, Joo HS, Shin DH, Park MK, Han B, Kim KP, Lee TJ, Croce CM, Kong G (2017) LSD1 demethylates HIF1alpha to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene. doi:10.1038/onc.2017.158
Oh SY, Seok JY, Choi YS, Lee SH, Bae JS, Lee YM (2015) The histone methyltransferase inhibitor BIX01294 inhibits HIF-1alpha stability and angiogenesis. Mol Cells 38(6):528–534. doi:10.14348/molcells.2015.0026
Ueda J, Ho JC, Lee KL, Kitajima S, Yang H, Sun W, Fukuhara N, Zaiden N, Chan SL, Tachibana M, Shinkai Y, Kato H, Poellinger L (2014) The hypoxia-inducible epigenetic regulators Jmjd1a and G9a provide a mechanistic link between angiogenesis and tumor growth. Mol Cell Biol 34(19):3702–3720. doi:10.1128/MCB.00099-14
Casciello F, Al-Ejeh F, Kelly G, Brennan DJ, Ngiow SF, Young A, Stoll T, Windloch K, Hill MM, Smyth MJ, Gannon F, Lee JS (2017) G9a drives hypoxia-mediated gene repression for breast cancer cell survival and tumorigenesis. Proc Natl Acad Sci USA 114(27):7077–7082. doi:10.1073/pnas.1618706114
Chen H, Yan Y, Davidson TL, Shinkai Y, Costa M (2006) Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res 66(18):9009–9016. doi:10.1158/0008-5472.CAN-06-0101
Lee JS, Kim Y, Kim IS, Kim B, Choi HJ, Lee JM, Shin HJ, Kim JH, Kim JY, Seo SB, Lee H, Binda O, Gozani O, Semenza GL, Kim M, Kim KI, Hwang D, Baek SH (2010) Negative regulation of hypoxic responses via induced Reptin methylation. Mol Cell 39(1):71–85. doi:10.1016/j.molcel.2010.06.008
Liu Q, Geng H, Xue C, Beer TM (1853) Qian DZ (2015) Functional regulation of hypoxia inducible factor-1alpha by SET9 lysine methyltransferase. Biochim Biophys Acta 5:881–891. doi:10.1016/j.bbamcr.2015.01.011
Liu X, Chen Z, Xu C, Leng X, Cao H, Ouyang G, Xiao W (2015) Repression of hypoxia-inducible factor alpha signaling by Set7-mediated methylation. Nucleic Acids Res 43(10):5081–5098. doi:10.1093/nar/gkv379
Kim Y, Nam HJ, Lee J, Park DY, Kim C, Yu YS, Kim D, Park SW, Bhin J, Hwang D, Lee H, Koh GY, Baek SH (2016) Methylation-dependent regulation of HIF-1alpha stability restricts retinal and tumour angiogenesis. Nat Commun 7:10347. doi:10.1038/ncomms10347
Lafleur VN, Richard S, Richard DE (2014) Transcriptional repression of hypoxia-inducible factor-1 (HIF-1) by the protein arginine methyltransferase PRMT1. Mol Biol Cell 25(6):925–935. doi:10.1091/mbc.E13-07-0423
Minet E, Ernest I, Michel G, Roland I, Remacle J, Raes M, Michiels C (1999) HIF1A gene transcription is dependent on a core promoter sequence encompassing activating and inhibiting sequences located upstream from the transcription initiation site and cis elements located within the 5′UTR. Biochem Biophys Res Commun 261(2):534–540. doi:10.1006/bbrc.1999.0995
Ju UI, Park JW, Park HS, Kim SJ, Chun YS (2015) FBXO11 represses cellular response to hypoxia by destabilizing hypoxia-inducible factor-1alpha mRNA. Biochem Biophys Res Commun 464(4):1008–1015. doi:10.1016/j.bbrc.2015.07.037
Lim JH, Choi YJ, Cho CH, Park JW (2012) Protein arginine methyltransferase 5 is an essential component of the hypoxia-inducible factor 1 signaling pathway. Biochem Biophys Res Commun 418(2):254–259. doi:10.1016/j.bbrc.2012.01.006
Mistry IN, Smith PJ, Wilson DI, Tavassoli A (2015) Probing the epigenetic regulation of HIF-1alpha transcription in developing tissue. Mol BioSyst 11(10):2780–2785. doi:10.1039/c5mb00281h
Koslowski M, Luxemburger U, Tureci O, Sahin U (2011) Tumor-associated CpG demethylation augments hypoxia-induced effects by positive autoregulation of HIF-1alpha. Oncogene 30(7):876–882. doi:10.1038/onc.2010.481
Pierre CC, Longo J, Bassey-Archibong BI, Hallett RM, Milosavljevic S, Beatty L, Hassell JA, Daniel JM (2015) Methylation-dependent regulation of hypoxia inducible factor-1 alpha gene expression by the transcription factor Kaiso. Biochim Biophys Acta 1849 12:1432–1441. doi:10.1016/j.bbagrm.2015.10.018
Lachance G, Uniacke J, Audas TE, Holterman CE, Franovic A, Payette J, Lee S (2014) DNMT3a epigenetic program regulates the HIF-2alpha oxygen-sensing pathway and the cellular response to hypoxia. Proc Natl Acad Sci USA 111(21):7783–7788. doi:10.1073/pnas.1322909111
Pfeiffer S, Kruger J, Maierhofer A, Bottcher Y, Kloting N, El Hajj N, Schleinitz D, Schon MR, Dietrich A, Fasshauer M, Lohmann T, Dressler M, Stumvoll M, Haaf T, Bluher M, Kovacs P (2016) Hypoxia-inducible factor 3A gene expression and methylation in adipose tissue is related to adipose tissue dysfunction. Sci Rep 6:27969. doi:10.1038/srep27969
Qian DZ, Kachhap SK, Collis SJ, Verheul HM, Carducci MA, Atadja P, Pili R (2006) Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res 66(17):8814–8821. doi:10.1158/0008-5472.CAN-05-4598
Chang CC, Lin BR, Chen ST, Hsieh TH, Li YJ, Kuo MY (2011) HDAC2 promotes cell migration/invasion abilities through HIF-1alpha stabilization in human oral squamous cell carcinoma. J Oral Pathol Med 40(7):567–575. doi:10.1111/j.1600-0714.2011.01009.x
Geng H, Harvey CT, Pittsenbarger J, Liu Q, Beer TM, Xue C, Qian DZ (2011) HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem 286(44):38095–38102. doi:10.1074/jbc.M111.257055
Kim SH, Jeong JW, Park JA, Lee JW, Seo JH, Jung BK, Bae MK, Kim KW (2007) Regulation of the HIF-1alpha stability by histone deacetylases. Oncol Rep 17(3):647–651. doi:10.3892/or.17.3.647
Kato H, Tamamizu-Kato S, Shibasaki F (2004) Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem 279(40):41966–41974. doi:10.1074/jbc.M406320200
Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403(6771):795–800. doi:10.1038/35001622
Blander G, Guarente L (2004) The Sir2 family of protein deacetylases. Annu Rev Biochem 73:417–435. doi:10.1146/annurev.biochem.73.011303.073651
Joo HY, Yun M, Jeong J, Park ER, Shin HJ, Woo SR, Jung JK, Kim YM, Park JJ, Kim J, Lee KH (2015) SIRT1 deacetylates and stabilizes hypoxia-inducible factor-1alpha (HIF-1alpha) via direct interactions during hypoxia. Biochem Biophys Res Commun 462(4):294–300. doi:10.1016/j.bbrc.2015.04.119
Laemmle A, Lechleiter A, Roh V, Schwarz C, Portmann S, Furer C, Keogh A, Tschan MP, Candinas D, Vorburger SA, Stroka D (2012) Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1alpha protein under hypoxic conditions. PLoS One 7(3):e33433. doi:10.1371/journal.pone.0033433
Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, Garcia JA (2009) Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 324(5932):1289–1293. doi:10.1126/science.1169956
Yoon H, Shin SH, Shin DH, Chun YS, Park JW (2014) Differential roles of Sirt1 in HIF-1alpha and HIF-2alpha mediated hypoxic responses. Biochem Biophys Res Commun 444(1):36–43. doi:10.1016/j.bbrc.2014.01.001
Bae JU, Lee SJ, Seo KW, Kim YH, Park SY, Bae SS, Kim CD (2013) SIRT1 attenuates neointima formation by inhibiting HIF-1alpha expression in neointimal lesion of a murine wire-injured femoral artery. Int J Cardiol 168(4):4393–4396. doi:10.1016/j.ijcard.2013.05.044
Dong SY, Guo YJ, Feng Y, Cui XX, Kuo SH, Liu T, Wu YC (2016) The epigenetic regulation of HIF-1alpha by SIRT1 in MPP(+) treated SH-SY5Y cells. Biochem Biophys Res Commun 470(2):453–459. doi:10.1016/j.bbrc.2016.01.013
Seo KS, Park JH, Heo JY, Jing K, Han J, Min KN, Kim C, Koh GY, Lim K, Kang GY, Uee Lee J, Yim YH, Shong M, Kwak TH, Kweon GR (2015) SIRT2 regulates tumour hypoxia response by promoting HIF-1alpha hydroxylation. Oncogene 34(11):1354–1362. doi:10.1038/onc.2014.76
Hubbi ME, Hu H, Kshitiz Gilkes DM, Semenza GL (2013) Sirtuin-7 inhibits the activity of hypoxia-inducible factors. J Biol Chem 288(29):20768–20775. doi:10.1074/jbc.M113.476903
Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, Kurland I, Dor Y, Weissleder R, Shirihai OS, Ellisen LW, Espinosa JM, Mostoslavsky R (2010) The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140(2):280–293. doi:10.1016/j.cell.2009.12.041
Bell EL, Emerling BM, Ricoult SJ, Guarente L (2011) SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30(26):2986–2996. doi:10.1038/onc.2011.37
Kohl R, Zhou J, Brune B (2006) Reactive oxygen species attenuate nitric-oxide-mediated hypoxia-inducible factor-1alpha stabilization. Free Radic Biol Med 40(8):1430–1442. doi:10.1016/j.freeradbiomed.2005.12.012
Johansson C, Tumber A, Che K, Cain P, Nowak R, Gileadi C, Oppermann U (2014) The roles of Jumonji-type oxygenases in human disease. Epigenomics 6(1):89–120. doi:10.2217/epi.13.79
Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y (2006) Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125(3):467–481
Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K (2006) The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442(7100):307–311
Ponnaluri VK, Vavilala DT, Putty S, Gutheil WG, Mukherji M (2009) Identification of non-histone substrates for JMJD2A-C histone demethylases. Biochem Biophys Res Commun 390(2):280–284. doi:10.1016/j.bbrc.2009.09.107
Luo W, Chang R, Zhong J, Pandey A, Semenza GL (2012) Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci USA 109(49):E3367–E3376. doi:10.1073/pnas.1217394109
Pollard PJ, Loenarz C, Mole DR, McDonough MA, Gleadle JM, Schofield CJ, Ratcliffe PJ (2008) Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1α. Biochem J 416(3):387–394. doi:10.1042/BJ20081238
Mimura I, Nangaku M, Kanki Y, Tsutsumi S, Inoue T, Kohro T, Yamamoto S, Fujita T, Shimamura T, Suehiro J, Taguchi A, Kobayashi M, Tanimura K, Inagaki T, Tanaka T, Hamakubo T, Sakai J, Aburatani H, Kodama T, Wada Y (2012) Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol Cell Biol 32(15):3018–3032. doi:10.1128/MCB.06643-11
Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ (2010) Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol 30(1):344–353. doi:10.1128/MCB.00444-09
Yang SJ, Park YS, Cho JH, Moon B, An HJ, Lee JY, Xie Z, Wang Y, Pocalyko D, Lee DC, Sohn HA, Kang M, Kim JY, Kim E, Park KC, Kim JA, Yeom YI (2017) Regulation of hypoxia responses by flavin adenine dinucleotide-dependent modulation of HIF-1alpha protein stability. EMBO J 36(8):1011–1028. doi:10.15252/embj.201694408
Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL (2007) RACK1 competes with HSP90 for binding to HIF-1α and is required for O2-independent and HSP90 inhibitor-induced degradation of HIF-1α. Mol Cell 25(2):207–217. doi:10.1016/j.molcel.2007.01.001
Shen L, Song CX, He C, Zhang Y (2014) Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem 83:585–614. doi:10.1146/annurev-biochem-060713-035513
Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333(6047):1300–1303. doi:10.1126/science.1210597
Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ, Song C, Teng SC, He C, Wu KJ (2014) TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol 15(12):513. doi:10.1186/s13059-014-0513-0
Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, Teng SC, Wu KJ (2011) Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell 43(5):811–822. doi:10.1016/j.molcel.2011.07.012
Chang S, Park B, Choi K, Moon Y, Lee HY, Park H (2016) Hypoxic reprograming of H3K27me3 and H3K4me3 at the INK4A locus. FEBS Lett 590(19):3407–3415. doi:10.1002/1873-3468.12375
Zhou X, Sun H, Chen H, Zavadil J, Kluz T, Arita A, Costa M (2010) Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer Res 70(10):4214–4221. doi:10.1158/0008-5472.CAN-09-2942
Johnson AB, Denko N, Barton MC (2008) Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat Res 640(1–2):174–179. doi:10.1016/j.mrfmmm.2008.01.001
Dobrynin G, McAllister TE, Leszczynska KB, Ramachandran S, Krieg AJ, Kawamura A, Hammond EM (2017) KDM4A regulates HIF-1 levels through H3K9me3. Sci Rep 7(1):11094. doi:10.1038/s41598-017-11658-3
Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K (2005) The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19(4):523–534. doi:10.1016/j.molcel.2005.06.027
Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM (2013) Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell 49(5):843–857. doi:10.1016/j.molcel.2012.12.006
Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, Rusinova E, Zhang G, Wang C, Zhu H, Yao J, Zeng YX, Evers BM, Zhou MM, Zhou BP (2014) Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 25(2):210–225. doi:10.1016/j.ccr.2014.01.028
da Motta LL, Ledaki I, Purshouse K, Haider S, De Bastiani MA, Baban D, Morotti M, Steers G, Wigfield S, Bridges E, Li JL, Knapp S, Ebner D, Klamt F, Harris AL, McIntyre A (2017) The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 36(1):122–132. doi:10.1038/onc.2016.184
Nair SS, Kumar R (2012) Chromatin remodeling in cancer: a gateway to regulate gene transcription. Mol Oncol 6(6):611–619. doi:10.1016/j.molonc.2012.09.005
Kirmes I, Szczurek A, Prakash K, Charapitsa I, Heiser C, Musheev M, Schock F, Fornalczyk K, Ma D, Birk U, Cremer C, Reid G (2015) A transient ischemic environment induces reversible compaction of chromatin. Genome Biol 16:246. doi:10.1186/s13059-015-0802-2
Sena JA, Wang L, Hu CJ (2013) BRG1 and BRM chromatin-remodeling complexes regulate the hypoxia response by acting as coactivators for a subset of hypoxia-inducible transcription factor target genes. Mol Cell Biol 33(19):3849–3863. doi:10.1128/MCB.00731-13
Wang F, Zhang R, Beischlag TV, Muchardt C, Yaniv M, Hankinson O (2004) Roles of Brahma and Brahma/SWI2-related gene 1 in hypoxic induction of the erythropoietin gene. J Biol Chem 279(45):46733–46741. doi:10.1074/jbc.M409002200
Wang F, Zhang R, Wu X, Hankinson O (2010) Roles of coactivators in hypoxic induction of the erythropoietin gene. PLoS One 5(4):e10002. doi:10.1371/journal.pone.0010002
Kenneth NS, Mudie S, van Uden P, Rocha S (2009) SWI/SNF regulates the cellular response to hypoxia. J Biol Chem 284(7):4123–4131. doi:10.1074/jbc.M808491200
Melvin A, Mudie S, Rocha S (2011) The chromatin remodeler ISWI regulates the cellular response to hypoxia: role of FIH. Mol Biol Cell 22(21):4171–4181. doi:10.1091/mbc.E11-02-0163
Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM (2006) The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res 66(7):3688–3698. doi:10.1158/0008-5472.CAN-05-4564
Huber O, Menard L, Haurie V, Nicou A, Taras D, Rosenbaum J (2008) Pontin and reptin, two related ATPases with multiple roles in cancer. Cancer Res 68(17):6873–6876. doi:10.1158/0008-5472.CAN-08-0547
Lee JS, Kim Y, Bhin J, Shin HJ, Nam HJ, Lee SH, Yoon JB, Binda O, Gozani O, Hwang D, Baek SH (2011) Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc Natl Acad Sci USA 108(33):13510–13515. doi:10.1073/pnas.1106106108
Yoo YG, Kong G, Lee MO (2006) Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J 25(6):1231–1241. doi:10.1038/sj.emboj.7601025
Moon HE, Cheon H, Chun KH, Lee SK, Kim YS, Jung BK, Park JA, Kim SH, Jeong JW, Lee MS (2006) Metastasis-associated protein 1 enhances angiogenesis by stabilization of HIF-1alpha. Oncol Rep 16(4):929–935. doi:10.3892/or.16.4.929
Acknowledgements
We thank Carole Baas for language proofreading. Work in authors’ laboratories was supported by Grants from NIH (R00CA168746), CPRIT (RR140036), Susan G. Komen® (CCR16376227), Welch Foundation (I-1903-20160319), and American Cancer Society and UTSW Simmons Cancer Center (ACS-IRG-02-196) to W.L.; and NIH (R00NS078049, R35GM124693), Welch Foundation (I-1939-20170325), CPRIT-HIHR RP170671, Darrell K Royal Research Fund, TIBIR pilot Grant, and UTSW startup funds to Y. W.. W. L. is a CPRIT Scholar in Cancer Research.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Luo, W., Wang, Y. Epigenetic regulators: multifunctional proteins modulating hypoxia-inducible factor-α protein stability and activity. Cell. Mol. Life Sci. 75, 1043–1056 (2018). https://doi.org/10.1007/s00018-017-2684-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2684-9