
Abstract
CDK5 is a serine/threonine kinase that is involved in the normal function of the adult brain and plays a role in neurotransmission and synaptic plasticity. However, its over-regulation has been associated with Tau hyperphosphorylation and cognitive deficits. Our previous studies have demonstrated that CDK5 targeting using shRNA-miR provides neuroprotection and prevents cognitive deficits. Dendritic spine morphogenesis and forms of long-term synaptic plasticity—such as long-term potentiation (LTP)—have been proposed as essential processes of neuroplasticity. However, whether CDK5 participates in these processes remains controversial and depends on the experimental model. Using wild-type mice that received injections of CDK5 shRNA-miR in CA1 showed an increased LTP and recovered the PPF in deficient LTP of APPswe/PS1Δ9 transgenic mice. On mature hippocampal neurons CDK5, shRNA-miR for 12 days induced increased dendritic protrusion morphogenesis, which was dependent on Rac activity. In addition, silencing of CDK5 increased BDNF expression, temporarily increased phosphorylation of CaMKII, ERK, and CREB; and facilitated calcium signaling in neurites. Together, our data suggest that CDK5 downregulation induces synaptic plasticity in mature neurons involving Ca2+ signaling and BDNF/CREB activation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
CDK5 is a serine/threonine kinase that participates in neuronal development and function and is involved in cognitive functions, such as memory and learning. In addition, deregulation of CDK5 activity is associated with neuronal death in neurodegenerative diseases [1, 2]. CDK5 activity is controlled mainly by p35 and p39 [3, 4]. Cleavage of p35 by calpain results in the formation of a CDK5/p25 complex, which leads to sustained, abnormal CDK5 kinase activity [5]. Evidence suggests that CDK5 and its activator p35 are involved in synaptic plasticity in mature neurons [5–8]. Most excitatory synapses in the brain are formed by dendritic spines, which are small, actin-rich membrane protrusions that are found on dendrites. During development, and in response to environmental stimuli, dendritic spines undergo changes in form and number, which are determining processes in learning and memory [9, 10]. CDK5 is a suspected synapse regulator, because a large number of synaptic proteins isolated from synaptosomes in the adult brain are putative substrates of this kinase [11]. Rho GTPases and their activators are potential targets of CDK5 and may be regulated by this kinase [12–14]. These small GTPases are key regulators of the actin cytoskeleton and play essential roles in organizing and remodeling dendritic spines [15]. RhoA and Rac regulate the morphogenesis of dendritic spines [16–19]. RhoA activation reduces synaptogenesis, whereas Rac1 activation facilitates an increase in the density and number of dendritic spines [16, 20, 21]. However, a recent study suggested that morphological changes of dendritic spines required the initial activation of both Rac1 and RhoA [22].
Two points of view have developed regarding the possible role of CDK5 in synaptic plasticity. On the one hand, CDK5 conditional knockout mice showed increases in hippocampal-dependent spatial memory in the Morris water maze test and contextual fear conditioning [23]. CDK5 conditional knockout mice also presented a decreased LTP induction threshold, which facilitates LTP. The increased spatial memory in CDK5 conditional knockout mice has been attributed to an increase in NR2B availability at the plasma membrane and a decrease in degradation of total NMDA levels [23, 24]. On the other hand, Li and colleagues reported that CDK5 phosphorylation of the NMDA receptor NR2A subunit facilitated NMDA receptor activity, LTP, and calcium channel induction in the hippocampal CA1 region, which was blocked by a pharmacological inhibitor of CDK5 [25]. Likewise, Lai and colleagues demonstrated that TrkB phosphorylation by CDK5 is required for activity-dependent structural plasticity and spatial memory. Specifically, the absence of TrkB phosphorylation by CDK5 decreased BDNF-induced dendritic spine morphogenesis [26]. BDNF was shown to increase the acquisition process for both short-term memory (STM) and long-term memory (LTM). In addition, the phosphorylation and resulting activation of CREB transcription and BDNF production are crucial for transferring information from STM to LTM [27]. An increase in CREB activity in the CA1-hippocampus prevented damage to dendritic spines and rescued spatial memory deficits in murine models of AD [28]. However, we found that CDK5´s silencing in neurons and astrocytes through BDNF regulation prevented neurodegeneration in excitotoxic brain injury [29, 30]. Therefore, the extent to which silencing CDK5 affects synaptic plasticity and the BDNF pathway remains unclear. This could be an interesting target for functional recovery of the adult brain. To determine the possible therapeutic effect of silencing CDK5 and BDNF/CREB regulation, we examined whether CDK5 RNAi affected dendritic spine morphogenesis, calcium modulation, LTP, and the activation of BDNF/CREB.
Materials and methods
Animal procedures
The animals were housed in a specific pathogen-free vivarium at SIU University of Antioquia (Medellín, Colombia) under a 12-h dark-light cycle. The rats and mice were handled according to Law 84 of 1989 and Resolution 8430 of 1993 of Colombian regulations, as well as Public Law 99-158 (November 20, 1985), ‘Animals in Research’ from the National Institutes of Health. Young adult (2–3 months old) C57BL/6 mice and adult (6 months old) APPswe/PS1ΔE9 (Tg) mice and their wild-type (WT) littermates (129/C57BL/6 mixed background) were used for electrophysiological recordings. For C57BL/6 mice: three males and two females were used as control (n = 5), three males and two females were treated with Scr shRNA-miR (n = 5), and two males and three females were treated with CDK5 shRNA-miR (n = 5). For WT mice: two males and three females were treated with Scr shRNA-miR (n = 5), and two males and two females were treated with CDK5 shRNA-miR (n = 4). For Tg mice: two males and two females were Scr shRNA-miR (n = 4), and three males and one female were CDK5 shRNA-miR (n = 4). Genotypes were distinguished by PCR of isolated genomic DNA obtained from each pup. APPswe/PS1ΔE9 Tg mice demonstrate accelerated amyloid pathologies and synaptic dysfunction by 6 months of age [31, 32]. The Institutional Animal Care and Use Committees of Johns Hopkins University approved all procedures involving animals.
AAV viral vector administration in vivo
C57BL/6 mice (WT) (2–3 months old), APPswe/PS1ΔE9 (Tg) mice, and their wild-type (WT) littermates (6 months old) were used for electrophysiological recordings. Mice were anesthetized and injected in the left and right hippocampi with AAV2 viral vector (1 µL) carrying Scr or CDK5 shRNA-miR (the Bregma coordinates were −1.7 antero-posterior, −0.7 lateral, and −1.75 depth). Injections were performed with a Hamilton syringe (10 μL) at a rate of 0.2 µL/min; a 5-min wait was implemented before withdrawal of the syringe.
Preparation of acute hippocampal slices
Acute hippocampal slices were prepared 3 weeks after AAV viral vector injection using young (2 months old) C57BL/6 and adult (6 months old) APPswe/PS1ΔE9 (Tg) mice and their wild-type (WT) littermates. Acute hippocampal slices were prepared from 2-month-old C57BL/6 mice and 6-month-old WT and APPswe/PS1ΔE9 Tg mice, as described previously [33]. Briefly, each mouse was killed by decapitation following an overdose of isoflurane. The hippocampi were rapidly removed and sectioned into 400-μm slices on a vibratome (Vibratome 3000 series; Ted Pella). Hippocampi were dissected using oxygenated ice-cold dissection buffer (composed of (mM) 212.7 sucrose, 5 KCl, 1.23 NaH2PO4, 26 NaHCO3, 10 dextrose, 10 MgCl2, and 0.5 CaCl2) and recovered at room temperature in artificial CSF (ACSF; composed of (mM) 124 NaCl, 5 KCl, 1.23 NaH2PO4, 26 NaHCO3, 10 dextrose, 1.5 MgCl2, and 2.5 CaCl2).
Field-potential recordings from Schaffer collateral inputs to CA1
All recordings from acute hippocampal slices were performed in a submersion-recording chamber perfused with ACSF (29–30 °C, 2 mL/min) bubbled with 95 % O2/5 % CO2. Synaptic stimuli were delivered through a bipolar glass-stimulating electrode that was placed at a location where they activated Schaffer collaterals with a 0.2-ms duration pulse (baseline stimulation at 0.0333 Hz) and were recorded from the dendritic field of CA1. Synaptic responses were digitized and stored on-line using the Igor Pro software (WaveMetrics). Input–output curves were generated by measuring the fiber volley (FV) amplitude and field excitatory postsynaptic potential (fEPSP) slopes using varying stimulus intensities 0, 10, 20, 40, 60, 80, and 100 μA. For the measurement of paired-pulse facilitation (PPF), inter-stimulus intervals of 25, 50, 100, 200, 400, 1000, and 2000 ms were used. LTP was induced using a theta-burst stimulation [TBS; four trains, each consisting of 10–100-Hz bursts (four pulses) given at 5 Hz, repeated at 10-s intervals (4 × TBS)]. fEPSP slopes or FV amplitudes as specified in the graphs were measured, and data are expressed as the mean ± SEM.
Primary neuronal cultures
Hippocampal and cortical cultures obtained from Wistar rat embryos (E18–19) were cultured on poly-l-lysine (Sigma-Aldrich) pre-coated coverslips in Neurobasal medium (GIBCO) containing B-27 supplement (GIBCO), l-glutamine (Sigma), and a penicillin–streptomycin antibiotic mixture (GIBCO) at 37 °C in a 5 % CO2 humidified atmosphere for a maximum of 19 days in vitro (DIV19), as previously described [12]. Isolated primary neurons were plated at low density (52 cell/mm2) for immunofluorescence and high density (1500 cell/mm2) for Rac and RhoA activation assays, Western blotting, and BDNF assay.
shRNA-miR AAV viral vector, shRNA lentiviral particles, and transduction in vitro
shRNA-miR AAV viral vectors, shRNA lentiviral particles, and transduction-Scr (scrambled), or CDK5 shRNA-miR (short hairpin RNA in a microRNA backbone) were designed and validated in our previous studies [12, 34]. AAV particles were obtained from the Davidson Laboratory (University of Iowa Viral Vector Core). Briefly, Scr or CDK5 shRNA-miR expression (mouse U6 promoter) was cloned into pAAV.CMV.hrGFP, which contains AAV serotype 2/5-inverted terminal repeats and a CMV-humanized Renilla GFP (hrGFP) simian virus 40 poly (A) reporter cassette [35, 36]. The primary cultures at DIV7 were transduced with 2 μL of AAV 2/5 (10 genomes per mL) at a 1012 titer for 3 h at 37 °C. The transductions were kept for 12 days before administering glutamate and inhibitor treatments.
Treatments and inhibitors in primary cultures
The cultures were treated with 0.01, 0.05, 0.1, 1, 2.5, 5, 10, 25, 50, and 100 µM of NSC23766 (Rac inhibitor, Calbiochem) mixed in medium and prepared in milli-Q water to choose the lowest concentration that inhibits Rac. After 24 h, LDH released from culture media was used to evaluate potential cytotoxicity induced by Rac inhibition. NSC23766 (1 and 5 µM) treatment was performed for 30 min at DIV19 to test Rac-dependent neuroplasticity. Subsequently, the proteins were collected for Rho GTPase activation assay, or the cells were fixed for immunofluorescence analyses. DIV19 neurons were treated with 125-μM glutamate (Sigma) prepared in glutamate buffer, as described previously [37]. After this treatment, the proteins were collected at 5, 10, 15, 20, 25, and 30 min for analysis with Western blot assay. Twenty-four hours after glutamate treatment, the total lysates and media were collected for the Western blot BDNF assay.
Immunofluorescence
The cultures were fixed in 4 % paraformaldehyde prepared in cytoskeleton buffer for 20 min. Autofluorescence was blocked using 50-mM NH4Cl. The cells were permeabilized and blocked with PBS + 0.1 % Triton X-100 and 1 % fetal bovine serum for 1 h. The cultures were incubated overnight at 4 °C with the following primary antibodies: mouse PSD95 (1:250, Calbiochem) and Alexa 594 secondary fluorescent antibody were used (1:1000, Molecular Probes). The nuclei were stained with Hoechst 33258 (1:5000, Molecular probes) and phalloidin conjugated with Alexa 594 (1:1000, Molecular probes) was used for 1 h. The neurons were coverslipped using Gel Mount (Fluorsave, Calbiochem). PSD95 cluster quantification was performed according to [12] that relies on determining the eGFP and PSD95 thresholds. Once the eGFP-sized areas were quantified, the PSD95 cluster density was obtained after calculating the number of clusters/100 µm2 of eGFP. The average cluster size was obtained by summing the total area of each cluster and dividing by the number of fused clusters. In addition, mouse brains were cut in 50-μm coronal sections using a vibratome (Leica 1000), and the sections were treated with 50-mM NH4Cl for 10 min at room temperature. The slices were pre-incubated for 1 h in 1 % BSA with 0.3 % Triton X-100 in 0.1 M PB. The coverslips and slices were observed using a DSU spinning disc confocal microscope and analyzed as individual images for eGFP expression. No staining was observed when the primary antibodies were omitted. XY images were collected using a 10× (NA, 0.4), 40× (NA, 1.3), or 60× (NA, 1.42) objective. Z-stack (6–10 optical sections) images were collected at 0.4-μm intervals encompassing the partial dendritic arbor with a 60× oil-immersion objective (NA, 1.42). The image stacks were deconvolved using a 3D-deconvolution algorithm following maximum intensity projection using the CellM imaging software (Olympus).
Dendritic spine 3D reconstructions and quantitative image analysis
Images for dendritic spine 3D reconstructions were acquired with a Zeiss LSM 510 confocal microscope using a Plan-Apo-chromat 63×, NA 1.4, oil-immersion objective. Z-stacks consisting of approximately ten optical sections of voxel size Δx/Δy/Δz = 50/50/200 nm were taken. Images were deconvolved with the Huygens Professional 3.7 software (Scientific Volume Imaging, Hilversum, The Netherlands) and processed with image analysis routines based on Interactive Data Language IDL (ITT, Boulder, CO, USA). For the segmentation of spines, we used the following image processing steps: (1) Gaussian kernel, (2) histogram equalization, and (3) threshold. Dendritic spine types were classified according to Tortosa et al. [38]. Spine volumes were quantified in dendritic sections >100-μm length in ten cells per culture from at least five different cultures for each experimental condition.
Calcium imaging
Cytosolic calcium was registered in primary neuronal cultures using the calcium probe Rhod-3 (Molecular probes). Neurons were treated with the loading buffer in 1× PBS containing 2.5-mM probenecid acid, PowerLoad™ 1× and 10-μM Rhod-3 AM (acetoxymethyl) ester for 30 min at room temperature. Then, the cultures were treated during the same period at room temperature with incubation buffer containing 2.5-mM probenecid acid to allow complete cleavage of AM esters. Following incubation with the calcium indicator, the cultures were treated with 125-µM glutamate and cell imaging was performed using the 60× (NA, 1.42) water-immersion objective of the DSU spinning disk confocal microscope (Olympus). The estimated change in fluorescence intensity over time was determined as a pseudo-ratio (ΔF/F 0) using the ImageJ software (NIH). This ratio was calculated using the following equation: ΔF/F 0 = (F − F baseline)/(F baseline), where F is the indicator fluorescence intensity and F baseline or F 0 is the fluorescence intensity before stimulation with glutamate. For each image analysis, the ROI of somas and dendrites was determined separately using a manually set ROI in Image J.
Western blotting
After treatment, the cultures were homogenized in lysis buffer and protease inhibitor cocktail mixture (Cytoskeleton). Electrophoresis was performed with 30 μg of protein, and the proteins were transferred to nitrocellulose membranes (Amersham), as described previously [34]. The membranes were incubated overnight at 4 °C with the following primary antibodies: rabbit p35 1:500 (C-19, Santa Cruz), mouse CDK5 1:500 (Millipore), mouse TrkB 1:1000 (Millipore), rabbit CREB 1:500 (Cell Signaling Technology), mouse phospho-CREB (ser133) 1:500 (Cell Signaling Technology), mouse ERK1/2 1:1000 (Thermo-scientific), mouse phospho-ERK1/2 (Thr202, Tyr204) 1:1000 (Thermo-Scientific), and rabbit phosphor-CAMKII (Thr286) 1:500 (Cell Signaling Technology). βIII-tubulin 1:5000 (Promega) was used as a loading control. IRDye 800CW goat anti-mouse or IRDye 680 goat anti-rabbit 1:15,000 (LI-COR) were used as secondary probes. The blots were developed using the Odyssey infrared imaging system. The fluorescent blot images were analyzed using the Odyssey infrared imaging system application software version 3.0 (LI-COR). The quantification of each blot was determined in relative units with respect to the loading control values. The samples from all of the experimental groups were processed in parallel to minimize inter-assay variation.
Rac and RhoA activation assay
The Rho GTPases activity was measured using G-Lisa activation assay Biochem kit (Cytoskeleton). Briefly, the active Rac, Cdc42, or RhoA linked to the PAK-binding domain or ROCK-binding domain was maintained to the well bottom, whereas inactive Rac- or RhoA-GDP was removed during washing. The bound Rac or RhoA was detected with an antibody specific for Rac or RhoA (1:200). The immune-complex was reveled with horseradish peroxidase secondary antibody (HRP, 1:100). The colorimetric signal was measured at 490 nm using a spectrophotometer (ELISA BioRad reader, Roche). Four independent experiments were conducted.
BDNF assay
BDNF release from co-cultures was measured using a mouse anti-BDNF ELISA kit (Promega), as was described previously [12]. The supernatant solution from each treatment was used as a sample. The colorimetric signal was measured at 490 nm using spectrophotometer (ELISA BioRad reader, Roche). Three independent experiments were conducted.
LDH release
Cytotoxicity induced by Rac inhibition was assessed by measuring the lactate dehydrogenase (LDH) release to the culture media using an LDH kit (Roche Molecular Biochemicals, Indianapolis, IN, USA) as described previously [37].
Statistical analyses
The values were expressed as the mean ± SEM. The results were considered significant at p < 0.05. All sample groups were processed in parallel to reduce inter-assay variation. The number of mice used was 3–5 for electrophysiological recordings and in vitro assays. The parametric data were compared using a multivariable 2-way ANOVA followed by Bonferroni’s (2 groups) or Tukey’s post-hoc test for comparisons between groups. The non-parametric data were analyzed using the Kruskal–Wallis test and Dunn’s Multiple Comparison Test. Comparisons between two groups were performed using Student’s t test for parametric data or the Mann–Whitney test for non-parametric data. All sample groups were processed in parallel to reduce inter-assay variation. The data are expressed as the mean ± SEM. SPSS 17.0 and PRISM software were used. The results were considered significant at p < 0.05, p < 0.01, p < 0.001, or p < 0.0001.
Results
CDK5 shRNA-miR increases LTP in WT mice and recovers LTP in APPswe/PS1Δ9 mice
First of all, we validated the CDK5 silencing, where CDK5 shRNA-miR-transduced WT mice showed a decrease in CDK5 levels (Fig. 1a, b). To examine the synaptic basis of the CDK5 reduction, we evaluated the strength and plasticity of CA3–CA1 synapses in hippocampal slices in mice injected with saline, Scr, or CDK5 shRNA-miR for 3 weeks. The ratio between the slope of the field excitatory postsynaptic potentials (fEPSP) and the fiber volley (FV), a measure of synaptic strength, was similar among the groups (Fig. 1c; repeated-measures ANOVA, p > 0.05). We also evaluated paired-pulse facilitation (PPF), a form of short-term plasticity commonly used to detect changes in presynaptic function. We did not observe changes among the treatment groups for PPF (Fig. 1d; repeated-measures ANOVA, p > 0.05). These results indicate that silencing CDK5 does not affect basal synaptic transmission. Next, we evaluated whether LTP, a form of synaptic plasticity, was affected. To that end, we used theta-burst stimulation (4× TBS), a protocol highly effective in inducing LTP [39]. Mice that were injected with CDK5 shRNA-miR showed a significantly increased amplitude of LTP at 60-min post-TBS (average of 10 last minutes) compared with non-injected mice and Scr mice (Fig. 1e; *p < 0.05, repeated-measures ANOVA, with Tukey’s test), which indicates that CDK5 silencing promoted LTP as a form of synaptic plasticity in young adult and healthy mice.

Slices of young adult mice treated with CDK5 show an increase in LTP without changes in input strength or presynaptic function. a Expression of the viral vector (Scr or CDK5 ShRNA-miR) in the regions of dentate gyrus and CA1 of the hippocampus. eGFP represents the transduced areas with Scr or CDK5 shRNA-miR in the hippocampus. Magnification ×20. Scale bar 100 μm. b WT mice transduced with Scr or CDK5 shRNA-miR, the CDK5 protein levels were measured by western blot and were normalized each to tubulin by fluorescence intensity analysis on the bar graph as arbitrary units (RU), n = 4. *p < 0.05; ANOVA Tukey’s test. c Example eEPSP traces recorded with increasing stimulation intensity; eEPSP slope, FV, and eEPSP slope/FV amplitude ratio plotted against stimulation intensity; and eEPSP slope Vs FV amplitude. Note there is no difference in the maximum eEPSP slope and FV amplitude between treatments (repeated-measures ANOVA [F(5, 10) = 4.321, p = 0.235]). No significant difference in input–output functions when eEPSP slope changes were normalized to FV amplitudes. d Representative eEPSP traces taken at a 50-ms inter-stimulus interval (ISI). No change in PPF ratio across different ISIs (repeated-measures ANOVA [F(6, 12) = 100.8, p = 0.377]). e An increase in the magnitude of LTP following a 4× TBS protocol in 2-month-old CDK5 shRNA-miR mice (no injection, 133.82 ± 6.03 % of baseline at 1 h after 4× TBS, Scr shRNA-miR, 129.95 ± 5.21 %, CDK5 shRNA-miR, 153.59 ± 9.40 %. Repeated-measures ANOVA [F(81, 162) = 37.91, ****p < 0.0001], followed by the Tukey’s post-hoc test (*p < 0.05) in the last 10 min. Insets show representative eEPSP traces (black, gray and blue lines represent baselines of each treatment; red line (indicated by 2), 1-h post-induction stimulation). The values in parentheses indicate: the number of hippocampal slices and the number of animals used
Recently, it was demonstrated that six-month-old APPswe/PS1Δ9 mice showed alterations in synaptic transmission and LTP [31]. Hence, to evaluate the possible effect of silencing CDK5 in this Alzheimer’s disease model, littermate mice (control) and 5-month-old APPswe/PS1Δ9 mice were injected with Scr and CDK5 shRNA-miR. Synaptic activity was registered 3 weeks after injection. APPswe/PS1Δ9 mice injected with Scr shRNA-miR showed an altered basal excitatory synaptic transmission compared to controls. APPswe/PS1Δ9 mice with Scr showed a decreased ratio of fEPSP slopes and FV amplitude compared with controls. Interestingly, CDK5 shRNA-miR rescued the basal excitatory synaptic transmission in the APPswe/PS1Δ9 mice, resulting in values similar to those in controls (Fig. 2a; *p < 0.05, repeated-measures ANOVA, Tukey’s test). Such changes in transmission could be due to changes in the probability of neurotransmitter release. To confirm this, we used a PPF protocol to evaluate short-term plasticity and presynaptic functioning. We found that APPswe/PS1Δ9 Scr mice showed a decrease in the facilitation of the inter-stimulus intervals of 25, 50, and 100 ms. In addition, APPswe/PS1Δ9 mice treated with CDK5 shRNA-miR showed a recovery of facilitation of altered inter-stimulus intervals similar to those observed in control values (Fig. 2b; *p < 0.05 or **p < 0.01, repeated-measures ANOVA, Tukey’s test). Finally, we evaluated whether CDK5 shRNA-miR could modify LTP in these affected mice. APPswe/PS1Δ9 mice treated with Scr showed a decrease in LTP compared with controls. Interestingly, the APPswe/PS1Δ9 mice that were injected with CDK5 shRNA-miR recovered the LTP amplitude at 60-min post-TBS compared with the APPswe/PS1Δ9 mice with Scr (Fig. 2c; *p < 0.05, repeated-measures ANOVA, Tukey’s test). This suggests that treatment with CDK5 shRNA-miR prevents synaptic dysfunction and alterations in plasticity in APPswe/PS1Δ9 mice.

CDK5 shRNA-miR recovers PPF and LTP in adult APPswe/PS1Δ9 mice. a Example eEPSP traces recorded with increasing stimulation intensity, eEPSP slope, FV, and eEPSP slope/FV amplitude ratio plotted against stimulation intensity, and eEPSP slope vs FV amplitude. Note the recovery of the eEPSP slope/FV amplitude ratio in Tg-CDK5 shRNA-miR compared with Tg-Scr shRNA-miR. Repeated-measures ANOVA [F(5, 15) = 4.552, **p < 0.01], followed by the Tukey’s post-hoc test (*p < 0.05). b Representative eEPSP traces taken at 50 ms ISI. Reduced PPF ratio at shorter (25, 50, 100 ms) ISIs in Tg-Scr shRNA-miR, which is recovered in Tg-CDK5 shRNA-miR treatment to control values. Repeated-measures ANOVA [F(6, 18) = 125.4, ****p < 0.0001], followed by the Tukey’s post-hoc test (*p < 0.05 or **p < 0.01). c A decrease in the magnitude of LTP following a 4× TBS protocol in 6-month-old Tg-Scr shRNA-miR mice and a recovery of magnitude of LTP in Tg-CDK5 shRNA-miR mice (WT-Scr shRNA-miR, 158.68 ± 8.79 % of baseline at 1 h after 4× TBS; Tg-Scr shRNA-miR, 135.15 ± 7.69 %; WT-CDK5 shRNA-miR, 155.82 ± 5.78 % Tg-CDK5 shRNA-miR, 155.46 ± 7.20 %. Repeated-measures ANOVA [F(81, 243) = 86.65, ****p < 0.0001], followed by the Tukey’s post-hoc test (*p < 0.05) in the last ten min. Insets show representative eEPSP traces (gray, black, blue, and red lines represent baselines of each treatment; red line (indicated by 2), 1-h post-induction stimulation). The values in parentheses indicate: the number of hippocampal slices and the number of animals used
CDK5 shRNA-miR enhances spine morphogenesis
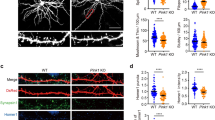
Dendritic spine morphogenesis and LTP have been considered as essential processes of synaptic plasticity. To determine whether silencing of CDK5 affected structural plasticity, we evaluated the effect of CDK5 shRNA-miR on dendritic spine formation in mature neuronal cultures. Neurons at DIV7 were transduced with AAV viral vector carrying either Scr or CDK5 shRNA-miR for 12 days. CDK5 shRNA-miR-transduced neurons showed a decrease in CDK5 levels (Fig. 3A) and an increase in the number of dendritic protrusions to 66 ± 3 per 100-μm segments compared with 52 ± 5 in neurons transduced with the Scr control (Fig. 3B, C; p < 0.05, t test). After CDK5 silencing, the average protrusion length was substantially higher than the control Scr shRNA-miR-transduced neurons, in which the average length was approximately 1.5 ± 0.15 and 2.8 ± 0.23 μm to CDK5 shRNA-miR (Fig. 3B, D; p < 0.001, t test). Moreover, after CDK5 silencing, the protrusion lengths were highly variable and the spines had an average length that was greater than 2.0 μm. Neurons with CDK5 shRNA-miR showed lengths ranging from 0.5 to 14 μm, and 50 % of the spines had an average length greater than 2.0 μm. The remaining 50 % of the spines retained an average length of less than 2 μm (Fig. 3F). Interestingly, silencing of CDK5 with shRNA-miR increased the area of PSD95-immunoreactivity in neurons (Fig. 3E, *p < 0.05, t test). In addition, some of the dendritic protrusions showed PSD95 puncta (Fig. 3Bc–Bf;). Complementarily, three-dimensional reconstructions were performed to classify the different dendritic protrusions. Protrusions from control neurons predominantly exhibited the morphological characteristics of mature mushroom-shaped, stubby, and thin spines, which were represented by protrusions between 0.01 and 0.4 μm3 (Fig. 4a, b; ***p < 0.001, t test). In contrast, CDK5 shRNAmiR-treated neurons displayed an increase for filopodia, or long-thin protrusions without a distinguishable head (Fig. 4a–c), which included protrusions between 0.5 and 1 μm3 (Fig. 4b, c; ***p < 0.001, t test). Interestingly, mushroom-shaped, stubby, and thin spines remained unchanged in CDK5 shRNAmiR-treated neurons (Fig. 4c). Taken together, these findings suggest that CDK5 shRNA-miR increased the total number of protrusions, which was represented by increased filopodia content, suggest an increase of synaptic contacts [40]. Therefore, the silencing of CDK5 could to induce dynamic filopodia contacts in mature neurons.

CDK5 silencing effect on spine morphogenesis. A Primary hippocampal neurons were transduced with SCR or CDK5 shRNA-miR and CDK5 protein levels were detected by western blotting. βIII-tubulin was used as the loading control. Relative units = RU, n = 4 (***p < 0.001). B Hippocampal neurons transduced with AAV vector viral eGFP-tagged Scr ShRNA-miR and CDK5 shRNA-miR at DIV7, with eGFP fluorescence visualized at day 19. Scr shRNA-miR is shown in a, with magnification shown in b; the magnification of neurons immunostained with PSD95 is shown in c. There was a morphological difference in the neurons transduced with CDK5 shRNA-miR, which show a greater extent of dendritic protrusions compared with Scr shRNA-miR (d–f). Scale bars 20 μm. Insets are showing the proximal dendritic shaft for each treatment. Scale bars 2 μm. The arrow and arrowhead show PSD95 clusters on elongated protrusions and mature spines, respectively. The pictures are representative of at least 90 % of the transduced neurons from n = 5. c The number of dendritic protrusions per 100 μm dendritic length quantified from 15 randomly selected DIV19-transduced neurons. CDK5 shRNA-miR-transduced neurons displayed an increase in the number of protrusions compared with Scr shRNA-miR (*p < 0.05). D Mean protrusion length is shown as the average of the length from the total dendritic spines quantified in each treatment (*p < 0.001). e Quantification of PSD95 clusters (red, B f) on dendritic protrusions (*p < 0.05). f Protrusion length distribution for each condition. The number of protrusions per 100-μm dendritic length was quantified from 15 randomly selected 19 DIV transduced neurons. Each experiment was performed in duplicate (n = 3)

CDK5 silencing increases filopodia protrusion. a 3D rendering of dendrites is shown for Scr and CDK5 shRNA-miR neurons. Scale bar 2 μm. The dendrites from Scr neurons have different types of spines, including mushroom (arrows), stubby (dotted arrow), and thin (dashed arrow), constituting the population of mature spines. In contrast, although CDK5 shRNA-miR neurons have different types of mature spines, they generate long protrusions without a distinguishable head corresponding to filopodia protrusions (arrowhead). b Protrusion volume distribution for each condition shows protrusion size categories. CDK5 shRNA-miR increased protrusions with a larger size than that in controls, corresponding to filopodia or intermediates (arrowhead). c The graph shows the different types of spines found in 100-μm dendritic lengths (ten neurons from three independent experiments per condition). Representative data for b and c are presented as the mean ± SEM of n = 4. *p < 0.05; ***p < 0.001; Student’s t test
Rac inhibition affects CDK5 shRNA-miR-induced spine morphogenesis
The Rho GTPases family regulates the actin cytoskeleton and plays a major role in dendritic spine morphogenesis, synapse formation, and nervous system plasticity [22, 41]. To evaluate whether Rac1 signaling contributed to CDK5 shRNA-miR-induced spine morphogenesis, we first determined the concentration range, at which the small molecule inhibitor NSC23766 inhibits Rac1 without an overt cytotoxic effect in neurons. We found that neurons treated with concentrations below 10 µM showed less than 10 % cytotoxicity (Fig. 5a), even after a 24-h exposure. Therefore, neurons were treated for 30 min with 1- and 5-µM NSC23766, which decreased the 30–50 % of Rac activity (Fig. 5b). Interestingly, neurons treated with 5-µM NSC23766 showed decrease of RhoA activity, possibly to a negative feedback between Rac-GTP and RhoA-GTP (Fig. 5b). Notably, CDK5 silencing led to an increase of active Rac1, and RhoA activity remained unchanged (Fig. 5b; *p < 0.05, ANOVA, Tukey’s test). However, Rac1 activation by CDK5 shRNA-miR was down-regulated by NSC23766 at 1 and 5 µM (Fig. 5b; *p < 0.05 and **p < 0.01, ANOVA, Tukey’s test). In addition, Rac inhibition (1 or 5 µM NSC23766) decreased dendritic protrusions in both experimental groups without nuclear fragmentation or condensation (Fig. 5c). Thus, our data suggest that CDK5 shRNA-miR-induced spine morphogenesis relies on the Rac GTPases-dependent regulation of actin cytoskeleton dynamics.

Rac inhibition affected CDK5 shRNA-miR-induced spine morphogenesis in mature neurons. a Primary hippocampal neurons were treated with 0.01-, 0.05-, 0.1-, 1-, 2.5-, 5-, 10-, 25-, 50-, and 100-μM NSC23766 for 24 h. After treatment, the LDH released from medium was measured to determine cytotoxicity. b Then, neurons were transduced with Scr or CDK5 shRNA-miR and DIV19 treated with 1- or 5-μM NSC23766 for 30 min to analyze spine morphogenesis. Rac and RhoA activities were quantified for the 30-min NSC23766 treatment as the amount of Rac-GTP and RhoA-GTP corresponding to the optical density observed using ELISA (λ = 490 nm). The data are presented as the mean ± SEM from n = 4 per duplicate. c Morphological characters show: AAV vector viral eGFP-tagged Scr and CDK5 shRNA-miR (green), nuclei visualized with Hoechst staining (blue), and F-actin cytoskeleton visualized with Alexa 594 Phalloidin dye (red). Magnification ×60. Scale bar 20 µm, n = 4. Insets are showing the proximal dendritic shaft for each treatment. Scale bars 2 μm. *p < 0.05; **p < 0.01; ANOVA with Tukey’s test for b
pCREB/BDNF signaling enhanced by CDK5 shRNA-miR
The activation of CREB transcription is a crucial process during the induction of synaptic plasticity and is sensitive to the activation of calcium-dependent MAPK and CaMKII pathways [42, 43]. Thus, we evaluated whether CDK5 shRNA-miR played a role in the plasticity signaling BDNF/CaMKII/ERK/CREB. We performed transduction of CDK5 shRNA-miR on DIV 7 for 12 days. All biochemical analyses were performed on DIV 19 at 24 h after glutamate treatment. Neurons transduced with CDK5 shRNA-miR increased BDNF following buffer or glutamate treatment (Fig. 6a; # p < 0.05, Kruskal–Wallis test with Dunn’s post-hoc test). Neurons transduced with CDK5 shRNA-miR showed a reduction of approximately 50 % of the total levels of CDK5 and increased p35 (Fig. 6b, c; *p < 0.05 Kruskal–Wallis test with Dunn’s post hoc test), as previously we reported in CDK5 silencing-induced neuroprotection [12]. In addition, CDK5 shRNA-miR enhanced the phosphorylation of ERK (Thr202/Tyr204), CaMKII (Thr286), and CREB (Ser133) in basal conditions (without glutamate) (Fig. 5b, c; *p < 0.05, **p < 0.01 and ***p < 0.001, Kruskal–Wallis test with Dunn’s post-hoc test). In addition, we evaluated the levels of TrKB (full-length = FL and truncated = T), because this neurotrophin receptor is involved in pCREB activation, plasticity, and the survival processes [44, 45]. Compared with Scr shRNA-miR, the CDK5 silencing increased TrkB-FL, specifically in the basal condition (Fig. 6b, c; *p < 0.05, Kruskal–Wallis test with Dunn’s post hoc test). In another hand, CDK5 shRNA-miR neurons treated with glutamate showed a recovery of p35 and an increase of pERK and pCaMKII (Fig. 6b, c; *p < 0.05, **p < 0.01 and ***p < 0.001, Kruskal–Wallis test with Dunn’s post-hoc test). Interestingly, CDK5 shRNA-miR-treated neurons showed an increase of TrkB-T concomitant to an increase in BDNF (Fig. 6; *p < 0.05, Kruskal–Wallis test with Dunn’s post-hoc test). These findings show that silencing of CDK5 enhances pCREB/BDNF signaling involved in plasticity and pERK/pCaMKII/BDNF also implied in neuroprotection.

BDNF/pCREB signaling enhanced by CDK5 shRNA-miR. Primary hippocampal neurons were transduced with Scr (black bars) or CDK5 shRNA-miR at DIV7 (gray bars). DIV18 neurons were treated with 125-μM glutamate for 20 min, and evaluated 24-h post-glutamate. a BDNF protein levels for each treatment were measured through BDNF Emax immunoassay system. The data are presented as the mean ± SEM from n = 5, performed in duplicate. The values were normalized to the control. b Representative blots and c quantification of TrkB (FL and T), pERK1/2 (ratio to ERK), pCAMKII and pCREB (ratio to CREB), and p35 and CDK5 protein levels were measured by western blot, and were normalized each βIII-tubulin by fluorescence intensity analysis on the bar graph as arbitrary units (RU), n = 4; *p < 0.05, **p < 0.01, ***p < 0.001; Kruskal–Wallis’ test with Dunn’s for a and c
CDK5 shRNA-miR promotes early activation of pCREB/BDNF pathway
We evaluated whether silencing of CDK5 induced an early activation of ERK, CaMKII, and CREB at 30 min after glutamate exposure. Neurons with CDK5 shRNA-miR, which were treated with glutamate, revealed a greater increase in the phosphorylation of ERK, CaMKII, and CREB between 5 and 30 min compared with the Scr glutamate-treated neurons (Fig. 7; *p < 0.05, **p < 0.01, two-way ANOVA, Bonferroni’s test). Compared with CDK5 shRNA-miR, the Scr neurons did not show changes in TrkB-T levels; however, CDK5 shRNA-miR prevented a decrease in TrkB-FL compared with the Scr, specifically at 30 min after glutamate treatment (Fig. 7; *p < 0.05, **p < 0.01, ***p < 0.001, two-way ANOVA, Bonferroni’s test). This early effect induced by CDK5 shRNA-miR supports the increased levels of BDNF compared with neurons with Scr shRNA-miR at 24 h after glutamate treatment (Fig. 6a; *p < 0.05, Kruskal–Wallis’ test with Dunn’s post-hoc test). These results suggest that CDK5 downregulation promotes an early activation of ERK/CaMKII/CREB pathway maybe involving a late BDNF production-induced neuroprotection.

Kinetics of pCREB/BDNF signaling induced by CDK5 shRNA-miR. Primary hippocampal neurons were transduced with Scr or CDK5 shRNA-miR at DIV7 and DIV18 neurons were treated with 125-μM glutamate for 2, 5, 10, 15, 25, and 30 min. A Representative blots and B quantification of temporal course of a TrkB (FL and T), b pERK1/2, c pCAMKII and d pCREB protein levels were measured by western blot and were normalized each to βIII-tubulin by fluorescence intensity analysis on the bar graph as arbitrary units (RU), n = 4. Two-way ANOVA [F(1, 48) = 33.96, ****P < 0.0001] for B a, [F(1, 48) = 37.37, ****p < 0.0001] for B b, [F(1, 48) = 34.73, ****p < 0.0001] for B c, [F(1, 48) = 80.76, ****p < 0.0001] for B d; followed by the Bonferroni’s post-hoc test (*p < 0.05, **p < 0.01, ***p < 0.01) in each time
CDK5 shRNA-miR facilitated calcium distribution in neurites
Synaptic changes during LTP require calcium for synaptic potentiation [46–48]. Thus, we determined the changes in intracellular calcium levels in the soma and neurites of hippocampal neurons treated with CDK5 shRNA-miR using Rhod3 DIV19 after glutamate exposure. Neurons transduced with Scr shRNA-miR revealed an increase in calcium concentration in the soma and neurites after glutamate treatment. In particular, the largest increase in calcium occurred in the soma at 20 min after glutamate (Fig. 8a, b). Similarly, neurons that were transduced with CDK5 shRNA-miR and treated with glutamate showed increased calcium concentration in the soma and neurites (Fig. 8a). In addition, we determined the average ΔF/F 0 at 5 min, because this is the time, at which the calcium levels had a peak. Neurons with Scr shRNA-miR showed increased calcium levels in the soma and neurites, whereas neurons with CDK5 shRNA-miR prevented the increased calcium levels in the neurites during the initial period (5 min) (Fig. 8b, c; *p < 0.05, ANOVA, Tukey’s test). However, CDK5 shRNA-miR maintained a gradual increase of calcium concentration in the neurites for 20 min compared with Scr neurons treated with glutamate (Fig. 8a, b, d). These findings suggest that CDK5 shRNA-miR protects in the acute phase of glutamate exposure and facilitates the calcium dynamics in neurites maybe associated with neuroprotection and plasticity in a late phase.

CDK5 shRNA-miR increases calcium levels into neurites. A Hippocampal neurons transduced with AAV vector viral eGFP-tagged Scr shRNA-miR and CDK5 shRNA-miR at DIV7, with eGFP and Rho3 fluorescence visualized at DIV 18. Scale bars 20 μm. The images represent the initial (0 min) and final frames (20 min) of Rhod3-loaded neurons (cytoplasmic calcium) registered every 15 s during a 20-min period. Neurons were recorded in buffer of glutamate (vehicle) for 3 min and treated 17 min later with glutamate. Representative pseudocolored images of Rho3-loaded neurons (a–d) and the respective surface intensity distribution (a′–d′) are shown immediately before (a, a′, c, c′) and 17 min after (b, b′, d, d′) the addition of glutamate. Magnification ×60. Scale bar 20 µm. B ΔF/F o fluorescence ratio was quantified in soma and neurites in neurons transduced with Scr and CDK5 shRNA-miR and exposed to buffer or glutamate. The graph shows the mean of three independent experiments. The red arrow indicates the addition of glutamate. Stimulus ΔF/F o graph represents the mean fluorescence ratio of the first 5 min (C) and 20 min (D) after glutamate treatment, quantified in soma and neurites in neurons transduced with Scr and CDK5 shRNA-miR. *p < 0.05; ANOVA with Tukey’s test
Discussion
These findings suggest that silencing CDK5 is a promising strategy to promote the neuroplasticity processes, which includes dendritic protrusion morphogenesis, enhancement of calcium signaling in neurites, paired-pulse facilitation, and LTP. Moreover, CREB activation and subsequent BDNF release underlying to silencing of CDK5. Several studies based on CDK5 suppression have shown increased synaptic plasticity, mainly in the models of the adult synapse [23, 24, 49]. Here, we demonstrate that silencing CDK5 increases LTP in young mice and recovers PPF and LTP in a mice model of AD.
We previously showed that CDK5 shRNA-miR exhibited a silencing of 50 %, supported by specific reduction of CDK5 protein and mRNA levels. In addition, CDK5 silencing also decreases the formation of CDK5/p35 and CDK5/p25 complexes and prevents the cleavage of p35 to p25, thus reducing the CDK5 activity [12, 34]. In this study, under basal conditions, CDK5 silencing increased the density of dendritic protrusions by modulating Rho GTPases, promoting the activity of Rac, but not affecting RhoA activity, which is related to the F-actin polymerization and stabilization that is necessary for the formation of dendritic spines and synaptic transmission. Recently, we confirmed that silencing of CDK5 in neurons from DIV 5 to DIV 7 (immature stage) resulted in a decrease in neurite arborization [12]. Thus, we evaluated the effect of silencing CDK5 on spine morphogenesis in DIV 19 mature cultures. These findings are controversial, because it was demonstrated that spine formation is altered, because of suppressed CDK5 phosphorylation of TrkB or CDK5 silencing mediated by artificial shRNAs [2, 26]. However, other models have demonstrated that the absence of CDK5-induced phosphorylation of δ-catenin, a homolog of p120-catenin, increases the density of dendritic protrusions [50]. We previously showed that Cdk5 silencing, knockout, or pharmacology inhibition increase p120-catenin in neurites when exposed to neurodegenerative insults [51], which is consistent with a previous report, where p120-catenin also induces Rac activation and RhoA inhibition, increasing actin polymerization rate, and the number of dendritic spines [52].
The fine regulation of Rho GTPases could result from CDK5 activity; hence, we suggest that CDK5 silencing could give way to a dynamic regulation of Rho GTPases activity, which could be supported by our previous findings, in which CDK5 knockdown resulted in improved learning and memory in transgenic AD mice that were associated with Rac1 activation [12]. The relationship between CDK5 and Rac1 in learning and memory was demonstrated during a process of contextual extinction in the hippocampus. Regulation of Rac1 activity by CDK5 mediated through the p35 relocation from the membrane to the cytosol and dissociation of CDK5 are involved in the suppression of extinction [13, 53]. This study shows that Rac1 inhibition suppresses F-actin polymerization in dendritic processes, and reverts the activity of Rac and RhoA. The regulation of Rho GTPases is determined by “cross-talk” pathways, where active Rac1 promotes RhoA activity. Rac1 inhibition eventually could generate a decrease in RhoA activity [17]. According to these reports and our results, we suggest that pharmacological inhibition of Rac1 blocks the actin polymerization during dendritic protrusion morphogenesis induced by CDK5 reduction.
Accordingly, one of the effects associated with CDK5 silencing was an increase in dendritic spine length, thus resembling filopodia morphology. High-resolution studies have shown a functional relationship among the various spine morphologies: “thin”, “stubby”, and “mushroom”. Therefore, through the observation of live cells, it is known that a mature “mushroom” spine passes or changes over time to an undifferentiated filopodia-type state. With these findings, it was suggested that an undifferentiated spine or filopodia is in a state of transition, where it can search for new presynaptic targets to return to the “mushroom” type to promote mechanisms of synaptic plasticity and new contacts, which could have positive effects on memory and learning [54, 55]. This view is consistent with our observations that dendritic protrusions presented PSD95 puncta and intermediate-filopodia characteristics. In fact, Trommald and Hulleberg recognized that some spines had intermediate forms and were difficult to classify according to common criteria [56]. Also, Arellano and colleagues suggested that dendritic protrusions exhibited a continuum variability of shapes, that correlated with PSD area, suggesting a large variability in the synaptic strength [40]. In addition, CDK5 silencing induced a normal mature proportion of mature spines and an increased PSD95 clusters, which supports our previous study, in which CDK5 RNAi may enhance the PSD95 protein levels on spines associated with learning and spatial memory function improvement in 3xTgAD mice [12]. Furthermore, the function and localization of PSD95 are regulated by CDK5 phosphorylation at its N-terminal [57]. This phosphorylation inhibits the multimerization of PSD95 and the anchoring of the PSD95 interacting potassium channels. The genetic ablation of CDK5 or its pharmacological inhibition promotes the formation of large PSD95 complexes [57, 58]. This increase in PSD95 favors the recruitment of other proteins, which are involved in the dynamics of the spines, allowing an active state during neurotransmission.
Importantly, our findings showed that young, healthy mice treated with CDK5 shRNA-miR increased LTP as a form of synaptic plasticity. Protocols that induce LTP and LTD can vary widely [59]. Nevertheless, diverse experimental approaches involving CDK5 and synaptic plasticity have also used pharmacological methods, such as roscovitine or olomoucine, to reduce CDK5 activity. In contrast, using a prion promoter system in CDK5, conditional knockout animals that were temporarily regulated by the transgene of the estrogen receptor demonstrated that the 50 % decrease of total CDK5 could induce an increase in hippocampal-dependent spatial memory in mice evaluated with the Morris water maze test and contextual fear conditioning [23]. In CDK5 conditional knockout mice, high-frequency LTP was associated with an increase in spatial memory. Specifically, LTP associated with an increase in spatial memory observed in CDK5 conditional knockout mice was attributed to an increase in NR2B availability in the membrane and a reduction in the degradation of total NMDA levels [23, 24]. Moreover, pharmacological inhibition of CDK5 with roscovitine resulted in an increase in EPSCs in striated neurons, which promotes the neuronal plasticity necessary for the relay of information between the cortex and basal ganglia [60], and increased LTP induction in corticostriatal synapses through the PKA pathway and activation of both NMDA receptors and L-type calcium channels [61].
Likewise, we have suggested that CDK5 silencing recovers cognitive dysfunction in an aged 3xTg-AD model and reduced neurofibrillary tangles and β-amyloid [12, 34, 62, 63]; however, reduction of these two neuropathological markers do not fully explain the cognitive recovery in the AD pathology. Here, we showed that silencing CDK5 increases synaptic plasticity potentiation. Using APPswe/PS1Δ9 mice as AD models, we showed that treatment with CDK5 shRNA-miR improved the synaptic changes. These 6-month-old animals demonstrated synaptic dysfunction and reduction of dendritic spines [31]. Consistent with this concept, it was suggested that the over-activity of CDK5 (CDK5/p25 complex) is an initial trigger for the synaptic damage in the Alzheimer-type pathology [49]. Disruption of presynaptic function in APPswe/PS1Δ9 mice results from altered calcium dynamics caused by mutations in PS1, which increase the release of calcium from the endoplasmic reticulum (ER) [64–67]. In AD murine models, abnormally high calcium signaling increases neuronal death and supports the hypothesis that the release of intracellular calcium is affected in AD [64]. Neurotoxic calcium transfer from the ER to mitochondria is triggered by increased CDK5 activity [68, 69], and inhibition of CDK5 can increase calcium storage in the ER [70]. A transient increase in presynaptic residual calcium levels underlies PPF [71], and we showed that CDK5 silencing improved PPF. Thus, we suggest that CDK5 silencing improves changes in calcium dynamics and it is possible that the resulting presynaptic functions are supported by its redistribution to neurites under neuroprotection.
In addition, it is well known that the membrane depolarization induced by high KCl has been tested as a strategy to induce synaptic activity [72]. In in vitro membrane, depolarization experiments have been shown that inhibition of CDK5 increases the influx of Ca2+, increases the release of Ca2+ from the ER to the cytoplasm, and decreases the rate of removal of Ca2+ [70]. PPF is known as a form of synaptic plasticity to short term that evaluates the Ca2+ dependent-presynaptic function. PPF performing a protocol in APPswe/PS1Δ9 mice has shown a decreased presynaptic dysfunction concerning the facilitation of the postsynaptic response [31]. Interestingly, silencing of CDK5 recovered the presynaptic dysfunction in APPswe/PS1Δ9 mice. Although Ca2+ levels were not determined in the in vivo tissue slices model, neurons in culture expressing CDK5 shRNA-miR showed an increase of Ca2+ in neurites, which we hypothesize could be linked to the facilitation of Ca2+ residual needed for presynaptic function; however, this needs to be tested in future experiments.
In in vivo and in vitro models, we have shown that silencing of CDK5 promotes plasticity through an increase in BDNF levels [29, 30]. In this study, we demonstrated that treatment with glutamate restricted an increase in calcium levels, especially in the soma, whereas CDK5 silencing modulated and directed it to neurites. We also observed an increase in BDNF/pCREB within the first minutes and 24 h after glutamate treatment. It has been suggested that the phosphorylation of CaMKII and ERK is required during LTP for an early activation of CREB and memory formation [27, 42, 43, 73]. In addition, downregulation of CDK5 following NMDA treatment facilitated CaMKII activation, leading to an increase in LTP [74]. The formation of the BDNF/TrkB-FL complex is required for cell survival; however, in an excitotoxic context, TrkB-FL is converted to TrkB-T, TrkB-T without BDNF is correlated to cell death; however, TrkB-T/BDNF promotes survival and plasticity, as was showed by CDK5 silencing [44]. The direct involvement of p35 and CDK5 in this signaling pathway must be clarified in future studies. Nevertheless, we suggest that silencing half of the CDK5 is sufficient to increase p35 and an efficient modulation of BDNF/pCREB, as neuroplasticity targets. Interestingly, we recently reported that neuroprotection and plasticity induced by CDK5 knockdown depend on p35 [12] and BDNF receptor, TRKB [77]. BDNF-dependent ERK activation not only induces pro-plasticity and survival pathways, but also promotes p35 expression [75, 76]. In this study, we found that CDK5 knockdown induced a modest early ERK phosphorylation, which was increased after 24-h post-glutamate; at the same time, we also found increased CREB phosphorylation likely dependent on ERK activation [42]. Together, this evidence suggests a positive feedback, where ERK1/2 and CREB phosphorylation are implicated on p35 and BDNF production, favoring neuroplasticity.
In conclusion, our data suggest an effect on dendritic protrusion morphogenesis and synaptic plasticity conferred by CDK5 silencing, and this effect supports the facilitation of LTP and the notion of enhanced neuroplasticity. Our findings validate pCREB activation and BDNF release, as cell targets for inducing neuroplasticity in CDK5 knockdown condition. The potential therapeutic effects of CDK5 silencing in neurodegenerative pathologies might involve multiple mechanisms; this study provides evidence of silencing by means of the functional recovery of the synapse.
Abbreviations
- CDK5:
-
Cyclin-dependent kinase 5
- Glu:
-
Glutamate
- AD:
-
Alzheimer’s disease
- DIV:
-
Day in vitro
- MAP-2:
-
Microtubule-associated protein-2
- ROCK:
-
Rho kinase
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- DTT:
-
Dithiothreitol
- G-LISA:
-
GTP enzyme-linked immunosorbent assay
- IFU:
-
Infectious units of virus
- PSD95:
-
Postsynaptic density-95
- NMDAR:
-
NMDA receptor
- 3xTg-AD:
-
Triple transgenic Alzheimer’s disease mice
- AAV:
-
Adeno-associated virus
- shRNA-miR:
-
Short hairpin RNA in a microRNA backbone
- PAK:
-
p21-activated kinase
- Scr:
-
Scrambled
References
Dhavan R, Tsai L-H (2001) A decade of CDK5. Nat Rev Mol Cell Biol 2(10):749–759
Lai KO, Ip NY (2009) Recent advances in understanding the roles of Cdk5 in synaptic plasticity. Biochim Biophys Acta 1792(8):741–745. doi:10.1016/j.bbadis.2009.05.001
Grant P, Sharma P, Pant HC (2001) Cyclin-dependent protein kinase 5 (Cdk5) and the regulation of neurofilament metabolism. Eur J Biochem 268(6):1534–1546. doi:10.1046/j.1432-1327.2001.02025.x
Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu C-L, Lanier LM, Gertler FB, Vidal M, Van Etten RA, Tsai L-H (2000) Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 26(3):633–646. doi:10.1016/s0896-6273(00)81200-3
Cheung ZH, Fu AK, Ip NY (2006) Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron 50(1):13–18. doi:10.1016/j.neuron.2006.02.024
Cheung ZH, Chin WH, Chen Y, Ng YP, Ip NY (2007) Cdk5 is involved in BDNF-stimulated dendritic growth in hippocampal neurons. PLoS Biol 5(4):e63. doi:10.1371/journal.pbio.0050063
Cheung ZH, Ip NY (2007) The roles of cyclin-dependent kinase 5 in dendrite and synapse development. Biotechnol J 2(8):949–957. doi:10.1002/biot.200700056
Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402(6762):615–622. doi:10.1038/45159
Bhatt DH, Zhang S, Gan WB (2009) Dendritic spine dynamics. Annu Rev Physiol 71:261–282. doi:10.1146/annurev.physiol.010908.163140
Bourne JN, Harris KM (2008) Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci 31(1):47–67. doi:10.1146/annurev.neuro.31.060407.125646
Collins MO, Yu L, Coba MP, Husi H, Campuzano I, Blackstock WP, Choudhary JS, Grant SG (2005) Proteomic analysis of in vivo phosphorylated synaptic proteins. J Biol Chem 280(7):5972–5982. doi:10.1074/jbc.M411220200
Posada-Duque RA, López-Tobón A, Piedrahita D, González-Billault C, Cardona-Gomez GP (2015) p35 and Rac1 underlie the neuroprotection and cognitive improvement induced by CDK5 silencing. J Neurochem 134(2):354–370. doi:10.1111/jnc.13127
Sananbenesi F, Fischer A, Wang X, Schrick C, Neve R, Radulovic J, Tsai LH (2007) A hippocampal Cdk5 pathway regulates extinction of contextual fear. Nat Neurosci 10(8):1012–1019. doi:10.1038/nn1943
Xin X, Ferraro F, Back N, Eipper BA, Mains RE (2004) Cdk5 and Trio modulate endocrine cell exocytosis. J Cell Sci 117(Pt 20):4739–4748. doi:10.1242/jcs.01333
Tolias KF, Duman JG, Um K (2011) Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol 94(2):133–148. doi:10.1016/j.pneurobio.2011.04.011
Li Z, Van Aelst L, Cline HT (2000) Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat Neurosci 3(3):217–225. doi:10.1038/72920
Li Z, Aizenman CD, Cline HT (2002) Regulation of rho GTPases by crosstalk and neuronal activity in vivo. Neuron 33(5):741–750
Govek EE, Newey SE, Van Aelst L (2005) The role of the Rho GTPases in neuronal development. Genes Dev 19(1):1–49. doi:10.1101/gad.1256405
Tada T, Sheng M (2006) Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol 16(1):95–101. doi:10.1016/j.conb.2005.12.001
Lee T, Winter C, Marticke SS, Lee A, Luo L (2000) Essential roles of Drosophila RhoA in the regulation of neuroblast proliferation and dendritic but not axonal morphogenesis. Neuron 25(2):307–316
Ruchhoeft ML, S-i Ohnuma, McNeill L, Holt CE, Harris WA (1999) The neuronal architecture of xenopus retinal ganglion cells is sculpted by rho-family GTPases in vivo. J Neurosci 19(19):8454–8463
Murakoshi H, Wang H, Yasuda R (2011) Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472(7341):100–104. doi:10.1038/nature09823
Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM, Cooper DC, Bibb JA (2007) Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci 10(7):880–886. doi:10.1038/nn1914
Plattner F, Hernández A, Kistler TM, Pozo K, Zhong P, Yuen EY, Tan C, Hawasli AH, Cooke SF, Nishi A, Guo A, Wiederhold T, Yan Z, Bibb JA (2014) Memory enhancement by targeting Cdk5 regulation of NR2B. Neuron 81(5):1070–1083. doi:10.1016/j.neuron.2014.01.022
Li BS, Sun MK, Zhang L, Takahashi S, Ma W, Vinade L, Kulkarni AB, Brady RO, Pant HC (2001) Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc Natl Acad Sci USA 98(22):12742–12747. doi:10.1073/pnas.211428098
Lai K-O, Wong ASL, Cheung M-C, Xu P, Liang Z, Lok K-C, Xie H, Palko ME, Yung W-H, Tessarollo L, Cheung ZH, Ip NY (2012) TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat Neurosci 15(11):1506–1515. http://www.nature.com/neuro/journal/v15/n11/abs/nn.3237.html
Kida S (2012) A functional role for CREB as a positive regulator of memory formation and LTP. Exp Neurobiol 21(4):136–140
Yiu AP, Rashid AJ, Josselyn SA (2011) Increasing CREB Function in the CA1 region of dorsal hippocampus rescues the spatial memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 36(11):2169–2186. http://www.nature.com/npp/journal/v36/n11/suppinfo/npp2011107s1.html
Posada-Duque RA, Palacio-Castañeda V, Cardona-Gómez GP (2015) CDK5 knockdown in astrocytes provide neuroprotection as a trophic source via Rac1. Mol Cell Neurosci 68:151–166. doi:10.1016/j.mcn.2015.07.001
Gutierrez-Vargas JA, Munera A, Cardona-Gomez GP (2015) CDK5 knockdown prevents hippocampal degeneration and cognitive dysfunction produced by cerebral ischemia. J Cereb Blood Flow Metab. doi:10.1038/jcbfm.2015.150
Megill A, Tran T, Eldred K, Lee NJ, Wong PC, Hoe HS, Kirkwood A, Lee HK (2015) Defective age-dependent metaplasticity in a mouse model of Alzheimer’s disease. J Neurosci 35(32):11346–11357. doi:10.1523/JNEUROSCI.5289-14.2015
Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzales V, Wong MPF, Price DL, Tang F, Markowska AL, Borchelt DR (2005) Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: relationships to β-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis 18(3):602–617. doi:10.1016/j.nbd.2004.10.022
Boric K, Munoz P, Gallagher M, Kirkwood A (2008) Potential adaptive function for altered long-term potentiation mechanisms in aging hippocampus. J Neurosci 28(32):8034–8039. doi:10.1523/JNEUROSCI.2036-08.2008
Piedrahita D, Hernández I, López-Tobón A, Fedorov D, Obara B, Manjunath BS, Boudreau RL, Davidson B, LaFerla F, Gallego-Gómez JC, Kosik KS, Cardona-Gómez GP (2010) Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s mice. J Neurosci 30(42):13966–13976. doi:10.1523/jneurosci.3637-10.2010
Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, Davidson BL (2009) Nonallele-specific silencing of mutant and wild-type Huntingtin demonstrates therapeutic efficacy in huntington’s disease mice. Mol Ther 17(6):1053–1063. http://www.nature.com/mt/journal/v17/n6/suppinfo/mt200917s1.html
Urabe M, Ding C, Kotin RM (2002) Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther 13(16):1935–1943
Posada-Duque RA, Velasquez-Carvajal D, Eckert GP, Cardona-Gomez GP (2013) Atorvastatin requires geranylgeranyl transferase-I and Rac1 activation to exert neuronal protection and induce plasticity. Neurochem Int 62(4):433–445. doi:10.1016/j.neuint.2013.01.026
Tortosa E, Montenegro-Venegas C, Benoist M, Härtel S, González-Billault C, Esteban JA, Avila J (2011) Microtubule-associated protein 1B (MAP1B) is required for dendritic spine development and synaptic maturation. J Biol Chem 286(47):40638–40648. doi:10.1074/jbc.M111.271320
Albensi BC, Oliver DR, Toupin J, Odero G (2007) Electrical stimulation protocols for hippocampal synaptic plasticity and neuronal hyper-excitability: are they effective or relevant? Exp Neurol 204(1):1–13. doi:10.1016/j.expneurol.2006.12.009
Arellano JI, Benavides-Piccione R, DeFelipe J, Yuste R (2007) Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front Neurosci. doi:10.3389/neuro.01.1.1.010.2007
Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G (2009) Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol 186(1):85–97. doi:10.1083/jcb.200901084
Wu G-Y, Deisseroth K, Tsien RW (2001) Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci 98(5):2808–2813. doi:10.1073/pnas.051634198
Wayman GA, Tokumitsu H, Davare MA, Soderling TR (2011) Analysis of CaM-kinase signaling in cells. Cell Calcium 50(1):1–8. doi:10.1016/j.ceca.2011.02.007
Gomes JoR, Costa JoT, Melo CV, Felizzi F, Monteiro PC, Pinto MJ, Inácio AR, Wieloch T, Almeida RD, Graos MR, Duarte CB (2012) Excitotoxicity downregulates TrkB.FL signaling and upregulates the neuroprotective truncated TrkB receptors in cultured hippocampal and striatal neurons. J Neurosci 32(13):4610–4622. doi:10.1523/jneurosci.0374-12.2012
Fenner BM (2012) Truncated TrkB: beyond a dominant negative receptor. Cytokine Growth Factor Rev 23(1–2):15–24. doi:10.1016/j.cytogfr.2012.01.002
Stevens CF, Tonegawa S, Wang Y (1994) The role of calcium, calmodulin kinase II in three forms of synaptic plasticity. Curr Biol 4(8):687–693. doi:10.1016/S0960-9822(00)00153-6
Murakoshi H, Wang H, Yasuda R (2011) Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472(7341):100–104. http://www.nature.com/nature/journal/v472/n7341/abs/10.1038-nature09823-unlocked.html
Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3(3):175–190. http://www.nature.com/nrn/journal/v3/n3/suppinfo/nrn753_S1.html
Sheng Y, Zhang L, Su SC, Tsai L-H, Julius Zhu J (2015) Cdk5 is a new rapid synaptic homeostasis regulator capable of initiating the early alzheimer-like pathology. Cerebral Cortex. doi:10.1093/cercor/bhv032
Poore CP, Sundaram JR, Pareek TK, Fu A, Amin N, Mohamed NE, Zheng Y-L, Goh AXH, Lai MK, Ip NY, Pant HC, Kesavapany S (2010) Cdk5-mediated phosphorylation of β-catenin regulates its localization and GluR2-mediated synaptic activity. J Neurosci 30(25):8457–8467. doi:10.1523/jneurosci.6062-09.2010
Uribe-Arias A, Posada-Duque RA, González-Billault C, Villegas A, Lopera F, Cardona-Gómez GP (2016) p120-catenin is necessary for neuroprotection induced by CDK5 silencing in models of Alzheimer’s disease. J Neurochem. doi:10.1111/jnc.13697
Elia LP, Yamamoto M, Zang K, Reichardt LF (2006) p120 catenin regulates dendritic spine and synapse development through Rho-family GTPases and cadherins. Neuron 51(1):43–56
Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A (2008) Switching on and off fear by distinct neuronal circuits. Nature 454(7204):600–606. doi:10.1038/nature07166
Zagrebelsky M, Holz A, Dechant G, Barde YA, Bonhoeffer T, Korte M (2005) The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J Neurosci 25(43):9989–9999. doi:10.1523/JNEUROSCI.2492-05.2005
Hotulainen P, Llano O, Smirnov S, Tanhuanpaa K, Faix J, Rivera C, Lappalainen P (2009) Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol 185(2):323–339. doi:10.1083/jcb.200809046
Trommald M, Hulleberg G (1997) Dimensions and density of dendritic spines from rat dentate granule cells based on reconstructions from serial electron micrographs. J Comp Neurol 377(1):15–28. doi:10.1002/(sici)1096-9861(19970106)377:1<15:aid-cne3>3.0.co;2-m
Morabito MA, Sheng M, Tsai LH (2004) Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J Neurosci 24(4):865–876. doi:10.1523/JNEUROSCI.4582-03.2004
Bianchetta MJ, Lam TT, Jones SN, Morabito MA (2011) Cyclin-dependent kinase 5 regulates PSD-95 ubiquitination in neurons. J Neurosci 31(33):12029–12035. doi:10.1523/JNEUROSCI.2388-11.2011
Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361(6407):31–39. doi:10.1038/361031a0
Chergui K, Svenningsson P, Greengard P (2004) Cyclin-dependent kinase 5 regulates dopaminergic and glutamatergic transmission in the striatum. Proc Natl Acad Sci USA 101(7):2191–2196. doi:10.1073/pnas.0308652100
Miranda-Barrientos J, Nieto-Mendoza E, Hernandez-Echeagaray E (2014) The Cdk5 inhibitor Roscovitine increases LTP induction in corticostriatal synapses. ASN Neurol 6(2). doi:10.1042/AN20140006
Castro-Alvarez JF, Uribe-Arias A, Cardona-Gómez GP (2015) Cyclin-dependent kinase 5 targeting prevents β-amyloid aggregation involving glycogen synthase kinase 3β and phosphatases. J Neurosci Res 93(8):1258–1266. doi:10.1002/jnr.23576
Castro-Alvarez JF, Uribe-Arias SA, Kosik KS, Cardona-Gomez GP (2014) Long- and short-term CDK5 knockdown prevents spatial memory dysfunction and tau pathology of triple transgenic Alzheimer’s mice. Front Aging Neurosci 6:243. doi:10.3389/fnagi.2014.00243
Cheung K-H, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VMY, Foskett JK (2008) Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58(6):871–883. doi:10.1016/j.neuron.2008.04.015
Stutzmann GE, Smith I, Caccamo A, Oddo S, LaFerla FM, Parker I (2006) Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci 26(19):5180–5189. doi:10.1523/jneurosci.0739-06.2006
Stutzmann GE, Caccamo A, LaFerla FM, Parker I (2004) Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci 24(2):508–513. doi:10.1523/jneurosci.4386-03.2004
Müller M, Cárdenas C, Mei L, Cheung K-H, Foskett JK (2011) Constitutive cAMP response element binding protein (CREB) activation by Alzheimer’s disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling. Proc Natl Acad Sci 108(32):13293–13298. doi:10.1073/pnas.1109297108
Fdr Darios, Muriel M-P, Khondiker ME, Brice A, Ruberg M (2005) Neurotoxic calcium transfer from endoplasmic reticulum to mitochondria is regulated by cyclin-dependent kinase 5-dependent phosphorylation of Tau. J Neurosci 25(16):4159–4168. doi:10.1523/jneurosci.0060-05.2005
Sun K-H, De Pablo Y, Vincent F, Shah K (2008) Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J Neurochem 107(1):265–278. doi:10.1111/j.1471-4159.2008.05616.x
Choi HS, Chung SH (2010) Roscovitine increases intracellular calcium release and capacitative calcium entry in PC12 cells. Neurosci Lett 469(1):141–144. doi:10.1016/j.neulet.2009.11.061
Wu L, Saggau P (1994) Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. J Neurosci 14(2):645–654
Liang Z, Ye T, Zhou X, Lai KO, Fu AK, Ip NY (2015) Cdk5 regulates activity-dependent gene expression and dendrite development. J Neurosci 35(45):15127–15134. doi:10.1523/JNEUROSCI.1443-15.2015
Takemoto-Kimura S, Suzuki K, Kamijo S, Ageta-Ishihara N, Fujii H, Okuno H, Bito H (2010) Differential roles for CaM kinases in mediating excitation–morphogenesis coupling during formation and maturation of neuronal circuits. Eur J Neurosci 32(2):224–230. doi:10.1111/j.1460-9568.2010.07353.x
Hosokawa T, Saito T, Asada A, Ohshima T, Itakura M, Takahashi M, Fukunaga K, S-i Hisanaga (2006) Enhanced activation of Ca2+/calmodulin-dependent protein kinase II upon downregulation of cyclin-dependent kinase 5-p35. J Neurosci Res 84(4):747–754. doi:10.1002/jnr.20975
Li B-S, Ma W, Jaffe H, Zheng Y, Takahashi S, Zhang L, Kulkarni AB, Pant HC (2003) Cyclin-dependent kinase-5 is involved in neuregulin-dependent activation of phosphatidylinositol 3-kinase and Akt activity mediating neuronal survival. J Biol Chem 278(37):35702–35709. doi:10.1074/jbc.M302004200
Zheng YL, Li BS, Kanungo J, Kesavapany S, Amin N, Grant P, Pant HC (2007) Cdk5 modulation of mitogen-activated protein kinase signaling regulates neuronal survival. Mol Biol Cell 18(2):404–413. doi:10.1091/mbc.E06-09-0851
Gutierrez-Vargas JA, Moreno H, Cardona-Gómez GP (2016) Targeting CDK5 post-stroke provides long-term neuroprotection and rescues synaptic plasticity. J Cereb Blood Flow Metab. doi:10.1177/0271678X16662476
Acknowledgments
The authors thank American Journal Experts for editing the English language in this manuscript. This research was supported by a grant from the Colciencias, projects # 111545921503 and #111554531400 (GPC-G), Mobility’s Project Colombia-Chile 576-2011, and the PROLAB Collaboration in Latin America/IBRO (CG-B and GPC-G). In addition, the Fogarty International Center and the NIA of NIH under Award Number RO1-AG029802-01 supported this research (GPC-G). R.A.P.-D. was sponsored by ENLAZA mundos fellowship. F.B. and C.G.-B. were supported by FONDAP Grant 15150012, and OR and SH were supported by FONDECYT 1151029, CONICYT PIA ACT1402, DAAD and ICM P09-015-F (Chile). The authors would like to thank the Advanced Microscopy Unit and Viral Vector and Gene Therapy Cores of the Group of Neuroscience of the University of Antioquia. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work presents new findings that CDK5 RNAi induces neuroplastic changes in mature neurons. Interestingly, we show that CDK5 downregulation induces dendritic protrusion morphogenesis and increases LTP in both normal and dysfunctional synapses. CDK5 RNAi neuroplasticity was confirmed by calcium in neurites and enhancement of BDNF/CREB activation.
Rights and permissions
About this article

Cite this article
Posada-Duque, R.A., Ramirez, O., Härtel, S. et al. CDK5 downregulation enhances synaptic plasticity. Cell. Mol. Life Sci. 74, 153–172 (2017). https://doi.org/10.1007/s00018-016-2333-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2333-8