
Abstract
Marfan syndrome (MFS) is a connective tissue disorder with multiple organ manifestations. The genetic cause of this syndrome is the mutation of the FBN1 gene, encoding the extracellular matrix (ECM) protein fibrillin-1. This genetic alteration leads to the degeneration of microfibril structures and ECM integrity in the tunica media of the aorta. Indeed, thoracic aortic aneurysm and dissection represent the leading cause of death in MFS patients. To date, the most effective treatment option for this pathology is the surgical substitution of the damaged aorta. To highlight novel therapeutic targets, we review the molecular mechanisms related to MFS etiology in vascular smooth muscle cells, the foremost cellular type involved in MFS pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Marfan syndrome (MFS) is a connective tissue disease with an autosomal-dominant inheritance and an estimated incidence of 2–3 in 10,000 individuals. The main typical manifestations of this syndrome are seen in multiple organ systems, such as the cardiovascular, skeletal and ocular systems [1].
Dietz et al. [2] made a major breakthrough in MFS by discovering the genetic cause of the disease, mutations in the FBN1 gene which encodes the extracellular glycoprotein fibrillin-1. There are close to 3000 known FBN1 mutations, varying from point mutations to large rearrangements [3–5]. The hereditary component has been diagnosed in approximately 80 % of MFS cases, whereas the remaining cases are due to de novo mutations in FBN1 without gender and ethnicity discrimination. Fibrillins are large glycoproteins that form complex extracellular structures called microfibrils [6]. These molecules provide elasticity and structural support to tissues modulating elastic fiber biogenesis and homeostasis, and regulating the bioavailability/activity of different growth factors. FBN1 mutations lead to impaired fibrillin-1 protein synthesis, secretion and/or incorporation in extracellular matrix (ECM) [7–9] which determines the degeneration of the microfibrillar architecture, loss of tissue homeostasis and subsequent destruction of the ECM integrity [10, 11]. These alterations drive the fibrillin-1 intracellular accumulation and the reduction of extracellular distribution of other ECM proteins, including fibronectin and tenascin [12]. Furthermore, mutant fibrillin-1 is incapable of maintaining transforming growth factor-β (TGF-β) in an inactive form [13]. Consequently, MFS patients have increased levels of active TGF-β [14].
In MFS, the main affected vessel is the thoracic aorta. Indeed, progressive aortic root aneurysm and dissection are the leading cause of death in MFS patients [15]. Thoracic aortic aneurysm (TAA) is characterized by aortic dilatation with thinning of the medial lamina. Furthermore, in many MFS cases, a typical histopathologic finding of the aortic wall is cystic medial degeneration (CMD) characterized by the accumulation of basophilic ground substance with cyst-like lesions, disruption of the lamellar organization of elastic fibers, degradation of ECM, and apoptosis of vascular smooth muscle cells (VSMCs) [16]. VSMCs are the major cell type in blood vessels and their main function is to regulate blood flow and pressure through vessel wall contraction and relaxation. Unlike many other mature cell types in the adult body, VSMCs hold a remarkable plasticity. They have the distinctive ability to alternate between a differentiated and quiescent “contractile” state and a highly proliferative and migratory “synthetic” phenotype [17]. A variety of studies demonstrate that changes in VSMC phenotype underlie MFS as well as many vascular occlusive diseases [18]. Specifically, it was reported that in MFS, there is a conversion of normally contractile aortic VSMCs into a less differentiated state of proliferation, migration, and copious ECM secretion, a process generally referred as phenotypic modulation or plasticity [19]. It is important to note that, contrary to the cells of the abdominal aorta which are derived from embryonic mesoderm progenitors, the VSMCs of thoracic aorta differentiate from cells of the neural crest [20]. The different embryonic origins render VSMCs more susceptible to the action of the TGF-β which is one of the key proteins involved in MFS alterations. The effects of TGF-β on neural crest-derived VSMCs result in a significant increase in cell proliferation and collagen deposition, whereas mesoderm-derived VSMCs do not display the same effects [21]. This explains, at least in part, the reason why aneurysms in MFS patients develop prevalently in the thoracic aorta.
This review is focused on the essential role of VSMCs in MFS pathogenesis and highlights how they can be considered, not only as a cellular model, but also as putative targets for the development of therapeutic strategies for patients with MFS.
Molecular mechanisms and signaling pathways
TGF-β signaling
The TGF-β signaling pathway has been designated as the major pathway involved in MFS. This molecule modulates several VSMC functions, such as proliferation, differentiation and ECM protein deposition [22].
TGF-β is primarily secreted and stored in the ECM as a latent complex composed by the N-terminal pro-domain of TGF-β, also called latency-associated peptide (LAP), and the fibrillin-like latent TGF-β binding protein (LTBP) [23]. It is important to point out that the TGF-β latent complex is attached to microfibrils by LTBP, thus fibrillin-1 plays a key role in maintaining TGF-β in an inactive form [24].
TGF-β activation relies on integrin-dependent and independent mechanisms. The former mechanism is mediated by structural alterations of the latency complex governed by the binding of specific integrins with LAP [25] and appears to be more active with increased ECM and microfibrillar complex stiffness [26]. On the other hand, the latter integrin-independent mechanisms involved in TGF-β activation are (1) plasmin [27] and matrix metalloproteinase (MMP) cleavage activity [25], which are able to perform fibrinolysis and ECM protein degradation, respectively, (2) changes in pH [27] and production of reactive oxygen species (ROS) [28], which can lead to disassociation of the latency protein from active TGF-β, and (3) thrombospondin-1 activity [29] which induces a conformational rearrangement of the LAP.
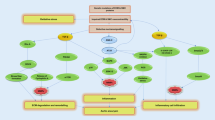
The mutation in fibrillin-1 associated with MFS leads to an uncontrolled release of TGF-β from the ECM. Once active, TGF-β can exert its effects by binding to its receptor TGF-β receptor II (TGFBR2), which, in turn, binds and transphosphorylates TGFBR1 (also known as ALK5). The excessive release of TGF-β is concomitant with overactive TGF-β signaling via canonical (by Smad2/3) [30] and/or non-canonical (by Erk1/2) [13] pathways (Fig. 1).

Molecular events occurring in MFS thoracic aorta aneurysm. The main mechanisms at the basis of dilatation and dissection of the thoracic aorta in MFS are: (1) the direct binding of AngII to its receptor AT1R and/or activation of AT1R by mechanical forces, (2) the uncontrolled release of TGF-β from the alterated ECM, (3) the activation of several MMPs, mediated both by miR-29b upregulation and by the direct binding of RGD motif of fragmented fibrillin to integrins, (4) the upregulation of miR-29b and the subsequent activation and translocation of caspases to cell membrane, and (5) the activation of Erk signaling by MMP-2. AngII angiotensin II, AT1R angiotensin II receptor 1, β-Arr2 β-Arrestin2, Smads small mothers against decapentaplegic, Erk extracellular signal-regulated kinase, TGF-β transforming growth factor-β, TGFβR1/2 transforming growth factor-β receptor 1 and 2, MMP matrix metalloproteinase, miR-29b microRNA-29b
For this reason, increased fragmentation of elastin fibers associated with aneurysm formation and overactivation of TGF-β signaling was detected in the same aortic region in the hypomorphic fibrillin-1 mouse model (Fbn1mgR/mgR) of Marfan-like syndrome [31]. Zilberberg et al,. in 2015, generated an MFS mice model with Ltbp3 gene deletion (Fbn1mgR/mgR; Ltbp3−/−). This model, lacking the fibrillin-like protein involved in the latent complex of TGF-β, showed an improved survival, no aneurysm formation, a diminished disruption and fragmentation of medial elastic fibers, and a decreased Smad2/3 and Erk1/2 activation in aorta when compared with Fbn1mgR/mgR [32].
Intriguingly, the overexpression of TGF-β in MFS is associated with altered hyaluronan synthesis, increased apoptosis, and impaired CD34-positive progenitor cell recruitment [33]. Activation of canonical signaling mediated by Smad2 plays a crucial role in the development TAA in MFS since its increased activity correlates with histologically evident aortic modifications [30]. Moreover, Carta et al. found in fibrillin-1-deficient mice (Fbn1mgN/mgN) an improper activation of Smad2/3 signaling, that was specific for VSMCs. Also, in an attempt to understand which mediator lies upstream of impaired Smad signaling in MFS mice, they showed an unwarranted p38 mitogen-activated protein kinase (MAPK) activity leading to a constitutive activation of Smad2/3 signaling in the aortic wall [34]. It has been proposed that the loss in fibrillin-1 deposition promotes ROS production resulting in the increased detrimental activity of TGF-β-activated kinase 1 (TAK1), p38 MAPK, and Smad2 [34].
Cumulatively, these results have led to the general consensus that suppressing TGF-β signaling in VSMCs could improve aortic complications associated with MFS. About 10 years ago, this assumption was demonstrated to be likely since attenuation of this signaling by means of TGF-β-neutralizing antibodies prevented and reversed multiple disease manifestations in MFS mice, including TAA [35] and abnormal mitral valve phenotype [36]. Recently, these very promising results were hampered by the demonstration that transgenic mice with conditional inactivation of TGFBR2 in VSMCs increased MAPK signaling and caused thickening, dilatation, and dissection of thoracic aorta [37, 38]. However, a step forward in understanding the role of TGF-β in MFS was achieved by Cook et al. [39] who demonstrated that TGF-β antagonism at post-natal day 16 (before TAA formation) accelerates aortic disease, while treatment at day 45 (after TAA formation) mitigated aneurysm development and dissection. These results further suggest a dimorphic role for TGF-β whereby beneficial or detrimental effects of TGF-β signaling depend on the developmental stage. In fact, it is well known that TGF-β is critical before birth for the development of the cardiovascular system [40]. However, in later life, TGF-β is expressed in reaction to injury, leading to the fibrotic response [41].
The frontier in understanding the molecular mechanisms underlying TGF-β signaling and Smad2 overactivation in MFS has now advanced to the level of epigenetics. Gomez et al. [30] correlated VSMC phenotype with respect to TGF-β signaling in different forms of TAA: monogenic (mainly MFS), degenerative, and bicuspid aortic valve (BAV)-associated forms. All types of TAA shared a complex dysregulation of Smad2 signaling which was VSMC-specific and heritable; however, this study surprisingly highlighted that this effect was independent of TGF-β. Also, it was found that Smad2 overexpression in TAA is due to increased H3K9/14 acetylation and H3K4 methylation in cell- and transcription start site-specific manner. To deeply understand the mechanism regulating the constitutive Smad2 overexpression in VSMCs, the same authors studied specific histone-modifying enzymes, transcription factors, and cofactors [42]. Interestingly, p53 and the complex composed of p300 and p300/CBP-associated proteins were shown to be critical determinants in Smad2 activation in human aneurysmal VSMCs.
Nonetheless, a prognostic role of circulating TGF-β in MFS patients has been shown as increased serum TGF-β levels in MFS patients correlate with larger aortic root diameters and faster aortic root growth [43].
Although more studies are necessary to fully understand all the molecular mechanisms involved in TAA formation, the literature to date suggests that dysregulated TGF-β signaling might be a result and not a cause of the aortic injury observed in MFS patients.
Angiotensin II signaling
To date, there are no published data highlighting specific mechanistic insights between mutated fibrillin-1 and angiotensin II (AngII) both in MFS models and patients. However, since fibrillin-1 mutations determine a perturbation of ECM that protects cells from blood pressure stress, it is conceivable that, in MFS, cells sense an altered shear stress which directly activates AngII receptor 1 (AT1R), independently of AngII. In fact, in different pathological conditions, such as cardiac hypertrophy, it has been shown that AT1R can be activated mechanically by shear stress [44]. Nevertheless, Nishimoto, in 2002, showed increased levels of local angiotensin-converting enzyme (ACE) and chymase in human aneurysmatic aorta samples, suggesting that also a local increase of AngII activates AT1R [45]. Consistently, Daugherty and collaborators discovered that chronic infusion of AngII in mice promotes the development of aortic aneurysms [46]. Thus, it is also logical to propose that in the context of MFS, fibrillin-1 mutations promote an increase of local AngII that directly activates AT1R. On the other hand, it is important to note that fibrillin microfibrils are a niche for growth factors. Therefore, it is possible that mutated fibrillin-1 determines the release of factors that indirectly activate AngII signaling (Fig. 1).
Furthermore, a strict connection between AngII and TGF-β was elucidated [47]. More importantly, AngII has been proposed as the molecule responsible for increased levels of TGF-β and phospho-Smad2. Specifically, AngII can activate the Smad2 signaling pathway directly through AT1R [48, 49] and/or indirectly by enhancing TGF-β expression [50]. All these observations converge on the possible involvement of AngII/AT1R and AT2R axis in MFS.
Nagashima et al. [51] were the first to evaluate whether this pathway is involved in VSMC apoptosis associated with CMD. As expected, AngII concentration was significantly higher in CMD than in controls. However, while AT1R expression was significantly decreased, AT2R expression was significantly increased in MFS aortas compared to controls. Furthermore, an ACE inhibitor (ACEI), as well as an AT2R blocker significantly inhibited serum deprivation-induced VSMC apoptosis. Conversely, this effect was not evident using the AT1R blocker. This study demonstrated that VSMC apoptosis is determined by ACE-dependent AngII formation and signaling via upregulated AT2R. Thus, ACEI could be a therapeutic strategy in the prevention and treatment of CMD in MFS.
Recently, more direct evidence that AngII signaling is involved in MFS was reported by Franken et al. [52]. In particular, it was demonstrated that the early molecular response in Fbn1mgR/mgR mice includes the constitutive overactivation of AT1R and, at the same time, an initial TGF-β production. However, it was proposed that the continuous constitutive AT1R activity leads to improper TGF-β signaling which drives medial degeneration [39]. Thus, the concept of TGF-β as the leading mediator responsible for aortic dilatation in MFS patients should be nuanced, since it is evident that also AngII/AT1R and AT2R axis synergistically acts with TGF-β, affecting several pathways that capitulate in aneurysmal formation.
Consistently, in a murine MFS model, β-arrestin2 (βarr2), a known mediator of AT1R-dependent MAPK activation, was found to be involved in the Erk1/2-dependent expression of pro-aneurysmal genes and proteins downstream of AT1R [53]. Thus, these data further confirm that AT1R contributes to TAA formation in MFS and indicate that this effect is mediated by βarr2.
The pivotal role of AngII/AT1R axis was also confirmed by the effectiveness of treatment with AngII receptor blockers (ARBs, i.e., sartans) in reducing the aortic root dilatation rate in MFS mice [35]. These interesting preclinical studies encouraged the initiation of several clinical trials aimed to prove whether AT1R blockage could reduce the progression of aortic dilatation in MFS patients [54–56]. To date, the major clinical trials are the COMPARE trial [57], the Marfan Sartan trial [54], and the Pediatric Heart Network study [56] which includes the Ghent Marfan Trial patients. Following analysis of the partial results from these studies, it is possible to conclude that a specific ARB (i.e., losartan) could be a valid alternative drug to β-blockers, although a double treatment of losartan together with β-blockers seems to be more effective than a low dose of β-blockers [52]. Nevertheless, the scientific community is still debating the efficacy of ARBs and β-blockers in MFS treatment. Intriguingly, recent data provided by Franken et al. have shown different response to losartan therapy in MFS patients displaying two different FBN1 mutations. Specifically, patients with haploinsufficient FBN1 mutations showed a higher inhibition rate in aortic root dilatation when compared to patients with dominant negative FBN1 mutation [58]. The authors hypothesized that the enhanced efficacy of losartan therapy in haploinsufficient patients may be related to: (1) the higher aortic stretch, due to thinner fibrillin-1 network, directly activating AT1R, and (2) the higher levels of locally produced AngII. Thus, future studies are necessary to discover the most effective treatment option to limit the development and progression of TAA in patients showing different type of mutation in FBN1 gene.
Proteases
MMPs
MMPs are enzymes that hydrolyze components of the ECM. Since MFS is characterized by ECM proteolysis, many investigators evaluated the involvement of MMPs in this disease. In 1998, Segura et al. [59] reported the upregulation of MMP-2 and MMP-9 in TAA and aortic valves of MFS patients. The authors suggested a possible link between FBN1 mutations and MMP activation. Specifically, it was described that defects in fibrillin-1 in MFS are accounted for the deposition of an abnormally aggregated elastin which is more prone to be degraded by MMPs in comparison with normal elastin. They also proposed that aberrant elastin per se leads to the upregulation of the MMP synthesis and the subsequent progressive impairment of ECM. Interestingly, it has also been shown that fibrillin-1 fragments containing the RGD motif are directly able to induce MMP-1 and -3 protein expression in vitro [60].
These results lead to the hypothesis for a vicious cycle by which MMPs are responsible for the generation of the fibrillin-1 fragments that further trigger MMP upregulation and the progressive destruction of connective tissue at the basis of TAA (Fig. 1).
Also, it was demonstrated that the increased expression profile of MMP-9 is a conditio sine qua non for the phenotypic changes observed in VSMCs isolated from the aorta of MFS patients [19], and that the upregulation of MMP-2 might play a role in MFS VSMC apoptosis [12]. When comparing TAA samples between MFS patients and non-MFS patients, Ikonomidis et al. [61] observed a very peculiar expression pattern of MMPs and TIMPs (endogenous MMP inhibitors). Specifically, decreased MMP-2 and TIMP-3 expression as well as increased MMP-12, MT1-MMP (a member of the membrane-type MMP) and TIMP-2 expression was found in MFS samples suggesting that each MMP family member in MFS aortas is specifically modulated. Conversely, an analysis performed in a mouse model of MFS demonstrated an increase in MMP-2 and MMP-9 expression in addition to a time-dependent increase in the MMP2:TIMP-2 ratio compared with controls [62]. These divergent data suggest that the MFS mouse model might not fully recapitulate all of the human MFS pathological mechanisms.
The importance of the MMPs in MFS was also confirmed by treating with doxycycline the Fbn1C1039G/+ mouse model of MFS [63], which expresses a gene mutation similar to that causing the classic manifestations of Marfan syndrome in humans (Cys1039Tyr). Strikingly, this non-specific inhibitor of MMPs enhanced aortic wall integrity by averting elastic fiber degeneration, normalizing vasomotor functions and inhibiting TGF-β activation. Notably, the beneficial effect of doxycycline was higher than atenolol, the β-blocker drug that has been previously reported to be a common component of Marfan patients’ treatment. Furthermore, the combination of doxycycline with losartan enhanced the survival of MFS mice by decreasing both Smad2 and Erk1/2 phosphorylation. These data suggest that MMP-2 acts both on canonical and non-canonical TGF-β signaling pathways to participate in the progression of MFS [64] (Fig. 1).
Calpains
Calpains are another class of proteases associated with MFS aortic aneurysm. In particular, Pilop et al. [65], by performing a proteomic analysis in aortic media of patients with MFS, found an increased activity of calpain-2. An increased amount of filamin A C-terminal fragment, which likely derives from the activity of the protease calpain-2, was observed. It is worth mentioning that calpains are calcium-dependent cysteine proteases that cleave not only structural proteins, but also many intracellular signaling mediators. For instance, it was reported that calpains, through their p53-degrading properties, counteract mechanically induced excessive VSMC apoptosis [66]. Taking these data together, the contribution of calpain-2 in aneurysm formation might be defined as a “double-edged sword” since its increased activity can lead to excessive elastolysis and limited VSMC apoptosis in dilated aorta of MFS patients.
Caspases
The deficiency of fibrillin-1 initiates the detachment of VSMCs from connective tissue leading to their apoptosis. A group of intracellular proteases called caspases plays a major role in this process. Thus, it was determined whether the expression/activity of this class of protein was modulated in MFS. The increased apoptosis in Fbn1C1039G/+ mouse aortas was strictly related to increased activity of caspase-3 and caspase-9 and decreased levels of the antiapoptotic proteins Mcl-1 and Bcl-2 [67]. Noticeably, caspases were expressed on the cell surface of ascending VSMCs, and inhibition of caspases with a pan inhibitor attenuated VSMC apoptosis, elastin degradation and aneurysm development in the Fbn1C1039G/+ Marfan mouse model [68] (Fig. 1).
Smooth muscle LDL receptor-related protein-1
LDL receptor-related protein-1 (LRP1) is a multifunctional receptor that controls expression, activity and trafficking of the platelet-derived growth factor (PDGF) receptor-β in VSMCs. Foremost, LRP1 was also reported to be identical to TGFβR(V), and LRP1/TGFβR(V) activity was required for the growth inhibitory response mediated by TGF-β and Smad2/3 signaling [69, 70].
Interestingly, Boucher et al. [71], using a transgenic mouse model, found that the deletion of Lrp1 in VSMCs causes a Marfan-like syndrome, characterized by nuclear accumulation of phospho-Smad2/3, disruption of elastic layers, tortuous aorta, and increased expression of the TGF-β target genes (TSP1 and PDGFRβ) in the vascular wall. A similar Marfan-like phenotype was observed by Basford et al. [72] who showed an age-dependent progression of aortic aneurysms in Lrp1-knockout mice. Also, this model showed secondary cardiomyopathy which was improved by treatment with an ACEI (captopril).
Further investigations on LRP1 molecular mechanisms revealed that this protein may be involved in preventing MFS complications by controlling PDGFRβ-dependent activation of phosphatidylinositol-3-kinase (PI3K) which, in turn, regulates actin organization and cell migration [73].
Proliferator-activated receptor-γ (PPARγ)
A large body of literature reported that the peroxisome proliferator-activated receptor-γ (PPARγ), a transcription factor of the nuclear receptor superfamily, determines anti-proliferative effects on VSMCs [74, 75]. Indeed, evidence was provided that two different PPARγ agonists induce apoptosis of cultured VSMCs [76, 77]. Thus, Sakomura et al. [78] evaluated whether PPARγ expression correlated with the aforementioned CMD. Peculiarly, PPARγ expression was upregulated in VSMCs during CMD and the severity of CMD correlated with the expression of PPARγ in medial VSMCs. Since PPARγ activation has anti-proliferative effects in VSMCs and its expression correlates with CMD, it seems logical to speculate that its activation by agonists should further exacerbate the medial degeneration in MFS. Surprisingly, Boucher et al. described that the treatment of Marfan-like mice with rosiglitazone, a PPARγ agonist, markedly reduced nuclear phospho-Smad2/3 accumulation and fibrosis which reverted the pathological phenotype [71]. These arguable positive effects might be related to the established capacity of PPARγ agonists to potently inhibit TGF-β signaling [79–81].
Thus, although more studies are necessary to unravel the role of PPARγ in MFS pathogenesis, these results suggest that the modulation of PPARγ signaling might lead to a clinical approach in the treatment of MFS.
miR-29b
MicroRNAs (miR) are short, non-coding, single-stranded RNAs that regulate hundreds of genes. A number of studies reported dysregulated miR expression as key determinant in the pathogenesis of numerous cardiovascular diseases [82, 83].
Interestingly, Jones et al. reported both a significant downregulation of miR-1, -21, -29a, -133a, -486 expression levels in human TAA and an inverse correlation between their expression and aortic diameter [84]. One of these miRs, miR-29a, was found to target MMP-2. Thus, the upregulated expression of MMP-2 in TAA samples can be explained, at least in part, by the downregulated expression of miR-29a [84].
Inversely, Boon et al. found a strong association between the augmented expression of miR-29 family members and the downregulation of ECM components, contributing in aortic structure loss and, subsequently, to aneurysm development [85]. In the specific context of MFS, miR-29b was found to be upregulated in both ascending aorta and VSMCs of Fbn1C1039G/+ mice, and its inhibition prevented early aneurysm development, apoptosis of cells of the aortic wall, and ECM degradation [67].
In vitro studies confirmed the enhanced expression of miR-29b in VSMCs isolated from Fbn1C1039G/+ mice and established that these cells are more prone to respond to excessive TGF-β signaling compared to the wild-type cells. Furthermore, investigations into the molecular mechanisms underpinning miR-29b expression recognized that excessive TGF-β signaling, by decreasing the activation of nuclear factor-κB (NF-κB), upregulates miR-29b expression [85]. Therefore, it is possible to speculate that the increased miR-29b expression is the trigger for MMP modulation which, in turn, leads to ECM degradation and VSMC apoptosis (Fig. 1).
Mechanotransduction
The thoracic aorta is subjected to the largest cyclic circumferential stretch from distending blood pressure, and axial stretch from gross motions of the heart [86]. The normal thoracic aorta can adapt well to modest sustained changes in hemodynamics, mostly by the action of VSMCs which can sense their chemomechanical environment to ensure appropriate compliance and structural integrity. A seminal finding regarding the response of VSMCs to a mechanical stimulus showed their change in ECM production, including fibrillar collagens and glycosaminoglycans, after cyclic mechanical loading [87].
Subsequently, it was discovered that VSMCs, through integrins and cytoskeleton proteins (actin and myosin), transduce mechanical information to intracellular signaling pathways to control the synthesis of matrix components and, successively, alter their cytoskeleton in response to cycles of increased mechanical load [88–90]. A consequential response of these mechanical stimuli is the release of several factors by cells within the aortic wall, which contribute to mechanically regulated matrix remodeling. Among these factors, there are the above-mentioned TGF-β and AngII (Fig. 1). The former binds to the matrix in a latent form and is activated by proteases or integrins through mechanical-dependent processes [25], while the latter is released from VSMCs in response to cyclic loading [89]. PDGF is another factor released from the matrix through the action of MMPs, which, in turn, can be influenced by the mechanical state of the matrix [86].
Mutated fibrillin-1 protein may determine an uncontrolled and exaggerated release of factors by: (1) losing the ability to sequester certain factors in the ECM, i.e., TGF-β and bone morphogenic proteins (BMP), and (2) affecting the compliance of ECM which may stimulate the release of factors from aortic cells [86]. Along with the crucial function of fibrillin-1 in the regulation of growth factors through their sequestration and/or deactivation, fibrillin-1 per se plays a relevant role in mechanotransduction from the endothelium and ECM to VSMCs. In addition, fibrillins contribute to the long-term stability of elastic fibers [91]. Indeed, in MFS, there is a loss of elastic fiber integrity which not only decreases VSMC functionality [92], but also reduces aortic resilience allowing aortic wall dilatation and aneurysm formation [93, 94]. Notably, in MFS, both mechanically damaged and proteolytically degraded matrices can alter the phenotype of VSMCs leading to mechanical and biological consequences.
Based on these findings, it is conceivable that in MFS, TAA arises mainly because of an inappropriate mechanosensing and mechanoregulation of the ECM by medial VSMCs that renders the aortic wall vulnerable to dilatation and dissection. Thus, strategies targeting mechanobiology of relevant MFS tissues will be essential for future progress in tackling this disorder.
Phenotypic changes and alteration of mechanical homeostasis
Almost all the molecular mechanisms outlined above drive the phenotypic changes in MFS VSMCs which lead to elastolysis in MFS. Specifically, Bunton et al. [19] found that MFS VSMCs lose their ECM attachments and connections that are normally mediated by fibrillin-1, and acquire morphological changes. This, in turn, promotes a synthetic state in VSMCs which increases the synthesis of MMP-9, a known mediator of elastolysis, contributing to the structural collapse of the vessel wall observed in MFS aortas.
Recently, an interesting work by Egea et al. [95] provided novel insights into the phenotypic alterations of MFS VSMCs. Specifically, overactive TGF-β signaling which was essential to orchestrate the phenotypic changes affecting contractile proteins (i.e., α-smooth muscle actin, smoothelin, smooth muscle protein 22 α, and calponin-1) and collagen I was found in MFS aorta and VSMCs isolated from MFS patients. These phenotypic changes were consistent with greater cellular and ECM stiffness which might contribute to the aortic rigidity observed in MFS aneurysms. This augmented rigidity might also result from aortic calcification. Indeed, although there are no data reporting a systemic dysregulation of calcium homeostasis in MFS, several authors have shown, both in animal models of MFS and patients, calcification deposits at the sites of breaks in elastic laminae [19, 91, 96–98]. Thus, it is possible to speculate that loss of elastic fiber connections in MFS might trigger calcium deposition which can negatively affect both phenotype and survival of VSMC [19]. However, it is worth noting that vessel rigidity in patients with MFS is not limited to the aorta. In fact, Eberth et al. [99] examined the common carotid arteries from the heterozygous Fbn1mgR/+ and the homozygous Fbn1mgR/mgR mice under biaxial loading. Carotids from Fbn1mgR/mgR mice were slightly stiffer in the circumferential direction and remarkably different in the axial direction compared to WT and Fbn1mgR/+ mice. The changes in the axial mechanical properties might be due to a compensatory mechanism triggered by the loss of an essential structural constituent to guarantee structural integrity and maintain mechanical homeostasis.
Furthermore, studies conducted on resistance-sized arteries revealed an increase in the stiffness of second-order mesenteric arteries in Fbn1C1039G/+ vs WT mice and an impairment in their vasomotor function [100].
Since Ca2+ waves are tightly associated with vasoconstriction, it was also evaluated if the decreased tonic contraction in MFS is due to aberrant Ca2+ wave signaling [101]. The measurement of the isometric force and intracellular Ca2+ in second-order mesenteric arteries from Fbn1C1039G/+ mice revealed a decreased frequency of Ca2+ waves and gives an explanation for the reduced tonic contraction in MFS.
Conclusions and future perspectives
The recent advances in surgical intervention have strongly extended the life expectancy of MFS patients. The pharmacological strategies employed to date are based on clinical and empirical practice and seems to be not supported by strong evidences grounded in a thorough knowledge of disease-specific molecular mechanisms. Thus, the therapy for TAA is purely surgical and, unfortunately, a definitive cure still remains unknown. Notably, current surgical guidelines are unable to predict the relative risk of aortic dissection in MFS patients, since they do not consider the aortic cellular component and extracellular milieu. In fact, from a microscopic viewpoint, cells populating the tunica intima, media and adventitia undergo critical aberrations dictated from the activation of well-studied pathways, including the AngII, TGF-β and MMP (Fig. 1). As reported earlier in this review, VSMCs are the main cell type affected in MFS. They undergo phenotypic alterations and apoptosis rendering the aortic wall fragile. Thus, VSMCs constitute the all-important “bricks” in the aortic wall and deserve an in-depth study to gain new insights into the molecular mechanisms underlying MFS. This new knowledge may provide the much needed rationale to develop innovative therapeutic strategies to contain as well as prevent aneurysm development in MFS patients.
References
Ammash NM, Sundt TM, Connolly HM (2008) Marfan syndrome-diagnosis and management. Curr Probl Cardiol 33(1):7–39. doi:10.1016/j.cpcardiol.2007.10.001
Dietz HC, Cutting GR, Pyeritz RE et al (1991) Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352(6333):337–339. doi:10.1038/352337a0
Loeys B, De Backer J, Van Acker P et al (2004) Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat 24(2):140–146. doi:10.1002/humu.20070
Collod-Beroud G, Le Bourdelles S, Ades L et al (2003) Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat 22(3):199–208. doi:10.1002/humu.10249
Baetens M, Van Laer L, De Leeneer K et al (2011) Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys-Dietz syndromes. Hum Mutat 32(9):1053–1062. doi:10.1002/humu.21525
Davis MR, Summers KM (2012) Structure and function of the mammalian fibrillin gene family: implications for human connective tissue diseases. Mol Genet Metab 107(4):635–647. doi:10.1016/j.ymgme.2012.07.023
Schrijver I, Liu W, Brenn T et al (1999) Cysteine substitutions in epidermal growth factor-like domains of fibrillin-1: distinct effects on biochemical and clinical phenotypes. Am J Hum Genet 65(4):1007–1020. doi:10.1086/302582
Aoyama T, Francke U, Dietz HC et al (1994) Quantitative differences in biosynthesis and extracellular deposition of fibrillin in cultured fibroblasts distinguish five groups of Marfan syndrome patients and suggest distinct pathogenetic mechanisms. J Clin Invest 94(1):130–137. doi:10.1172/JCI117298
Whiteman P, Handford PA (2003) Defective secretion of recombinant fragments of fibrillin-1: implications of protein misfolding for the pathogenesis of Marfan syndrome and related disorders. Hum Mol Genet 12(7):727–737
Pereira L, Andrikopoulos K, Tian J et al (1997) Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet 17(2):218–222. doi:10.1038/ng1097-218
Potter KA, Hoffman Y, Sakai LY et al (1993) Abnormal fibrillin metabolism in bovine Marfan syndrome. Am J Pathol 142(3):803–810
Nataatmadja M, West M, West J et al (2003) Abnormal extracellular matrix protein transport associated with increased apoptosis of vascular smooth muscle cells in marfan syndrome and bicuspid aortic valve thoracic aortic aneurysm. Circulation 108(Suppl 1):II329–II334. doi:10.1161/01.cir.0000087660.82721.15
Holm TM, Habashi JP, Doyle JJ et al (2011) Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 332(6027):358–361. doi:10.1126/science.1192149
Chaudhry SS, Cain SA, Morgan A et al (2007) Fibrillin-1 regulates the bioavailability of TGFbeta1. J Cell Biol 176(3):355–367. doi:10.1083/jcb.200608167
Milewicz DM, Dietz HC, Miller DC (2005) Treatment of aortic disease in patients with Marfan syndrome. Circulation 111(11):e150–e157. doi:10.1161/01.CIR.0000155243.70456.F4
Yuan SM, Jing H (2011) Cystic medial necrosis: pathological findings and clinical implications. Rev Bras Cir Cardiovasc 26(1):107–115
Li Z, Zhao X, Bai S et al (2008) Proteomics identification of cyclophilin A as a potential prognostic factor and therapeutic target in endometrial carcinoma. Mol Cell Proteomics 7(10):1810–1823. doi:10.1074/mcp.M700544-MCP200
Nguyen AT, Gomez D, Bell RD et al (2013) Smooth muscle cell plasticity: fact or fiction? Circ Res 112(1):17–22. doi:10.1161/CIRCRESAHA.112.281048
Bunton TE, Biery NJ, Myers L et al (2001) Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res 88(1):37–43
Thieszen SL, Dalton M, Gadson PF et al (1996) Embryonic lineage of vascular smooth muscle cells determines responses to collagen matrices and integrin receptor expression. Exp Cell Res 227(1):135–145. doi:10.1006/excr.1996.0258
Gadson PF Jr, Dalton ML, Patterson E et al (1997) Differential response of mesoderm- and neural crest-derived smooth muscle to TGF-beta1: regulation of c-myb and alpha1 (I) procollagen genes. Exp Cell Res 230(2):169–180. doi:10.1006/excr.1996.3398
Massague J, Wotton D (2000) Transcriptional control by the TGF-beta/Smad signaling system. EMBO J 19(8):1745–1754. doi:10.1093/emboj/19.8.1745
Saharinen J, Taipale J, Monni O et al (1998) Identification and characterization of a new latent transforming growth factor-beta-binding protein, LTBP-4. J Biol Chem 273(29):18459–18469
Dietz HC (2007) 2006 Curt Stern Award Address. Marfan syndrome: from molecules to medicines. Am J Hum Genet 81(4):662–667. doi:10.1086/521409
Annes JP, Munger JS, Rifkin DB (2003) Making sense of latent TGFbeta activation. J Cell Sci 116(Pt 2):217–224
Wipff PJ, Hinz B (2008) Integrins and the activation of latent transforming growth factor beta1—an intimate relationship. Eur J Cell Biol 87(8–9):601–615. doi:10.1016/j.ejcb.2008.01.012
Lyons RM, Keski-Oja J, Moses HL (1988) Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol 106(5):1659–1665
Barcellos-Hoff MH, Dix TA (1996) Redox-mediated activation of latent transforming growth factor-beta 1. Mol Endocrinol 10(9):1077–1083. doi:10.1210/mend.10.9.8885242
Schultz-Cherry S, Murphy-Ullrich JE (1993) Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J Cell Biol 122(4):923–932
Gomez D, Coyet A, Ollivier V et al (2011) Epigenetic control of vascular smooth muscle cells in Marfan and non-Marfan thoracic aortic aneurysms. Cardiovasc Res 89(2):446–456. doi:10.1093/cvr/cvq291
Howell DW, Popovic N, Metz RP et al (2014) Regional changes in elastic fiber organization and transforming growth factor beta signaling in aortas from a mouse model of marfan syndrome. Cell Tissue Res 358(3):807–819. doi:10.1007/s00441-014-1993-7
Zilberberg L, Phoon CK, Robertson I et al (2015) Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc Natl Acad Sci USA 112(45):14012–14017. doi:10.1073/pnas.1507652112
Nataatmadja M, West J, West M (2006) Overexpression of transforming growth factor-beta is associated with increased hyaluronan content and impairment of repair in Marfan syndrome aortic aneurysm. Circulation 114(1 Suppl):I371–I377. doi:10.1161/CIRCULATIONAHA.105.000927
Carta L, Smaldone S, Zilberberg L et al (2009) p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem 284(9):5630–5636. doi:10.1074/jbc.M806962200
Habashi JP, Judge DP, Holm TM et al (2006) Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312(5770):117–121. doi:10.1126/science.1124287
Ng CM, Cheng A, Myers LA et al (2004) TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest 114(11):1586–1592. doi:10.1172/JCI22715
Boileau C, Guo DC, Hanna N et al (2012) TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet 44(8):916–921. doi:10.1038/ng.2348
Lindsay ME, Schepers D, Bolar NA et al (2012) Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet 44(8):922–927. doi:10.1038/ng.2349
Cook JR, Clayton NP, Carta L et al (2015) Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol 35(4):911–917. doi:10.1161/ATVBAHA.114.305150
Sridurongrit S, Larsson J, Schwartz R et al (2008) Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev Biol 322(1):208–218. doi:10.1016/j.ydbio.2008.07.038
Ford CM, Li S, Pickering JG (1999) Angiotensin II stimulates collagen synthesis in human vascular smooth muscle cells. Involvement of the AT(1) receptor, transforming growth factor-beta, and tyrosine phosphorylation. Arterioscler Thromb Vasc Biol 19(8):1843–1851
Gomez D, Kessler K, Michel JB et al (2013) Modifications of chromatin dynamics control Smad2 pathway activation in aneurysmal smooth muscle cells. Circ Res 113(7):881–890. doi:10.1161/CIRCRESAHA.113.301989
Franken R, den Hartog AW, de Waard V et al (2013) Circulating transforming growth factor-beta as a prognostic biomarker in Marfan syndrome. Int J Cardiol 168(3):2441–2446. doi:10.1016/j.ijcard.2013.03.033
Zou Y, Akazawa H, Qin Y et al (2004) Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 6(6):499–506. doi:10.1038/ncb1137
Nishimoto M, Takai S, Fukumoto H et al (2002) Increased local angiotensin II formation in aneurysmal aorta. Life Sci 71(18):2195–2205
Daugherty A, Manning MW, Cassis LA (2000) Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 105(11):1605–1612. doi:10.1172/JCI7818
Chen X, Lu H, Rateri DL et al (2013) Conundrum of angiotensin II and TGF-beta interactions in aortic aneurysms. Curr Opin Pharmacol 13(2):180–185. doi:10.1016/j.coph.2013.01.002
Carvajal G, Rodriguez-Vita J, Rodrigues-Diez R et al (2008) Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int 74(5):585–595. doi:10.1038/ki.2008.213
Rodriguez-Vita J, Sanchez-Lopez E, Esteban V et al (2005) Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 111(19):2509–2517. doi:10.1161/01.CIR.0000165133.84978.E2
Franken R, Radonic T, den Hartog AW et al (2015) The revised role of TGF-beta in aortic aneurysms in Marfan syndrome. Neth Heart J 23(2):116–121. doi:10.1007/s12471-014-0622-0
Nagashima H, Sakomura Y, Aoka Y et al (2001) Angiotensin II type 2 receptor mediates vascular smooth muscle cell apoptosis in cystic medial degeneration associated with Marfan’s syndrome. Circulation 104(12 Suppl 1):I282–I287
Franken R, Mulder BJ (2015) Aortic disease: Losartan versus atenolol in the Marfan aorta-how to treat? Nat Rev Cardiol 12(8):447–448. doi:10.1038/nrcardio.2015.95
Wisler JW, Harris EM, Rasich MC et al (2015) The role of beta-arrestin2-dependent signaling in thoracic aortic aneurysm formation in a murine model of Marfan syndrome. Am J Physiol Heart Circ Physiol ajpheart 00291:2015. doi:10.1152/ajpheart.00291.2015
Milleron O, Arnoult F, Ropers J et al (2015) Marfan Sartan: a randomized, double-blind, placebo-controlled trial. Eur Heart J 36(32):2160–2166. doi:10.1093/eurheartj/ehv151
Chiu HH, Wu MH, Wang JK et al (2013) Losartan added to beta-blockade therapy for aortic root dilation in Marfan syndrome: a randomized, open-label pilot study. Mayo Clin Proc 88(3):271–276. doi:10.1016/j.mayocp.2012.11.005
Lacro RV, Dietz HC, Sleeper LA et al (2014) Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med 371(22):2061–2071. doi:10.1056/NEJMoa1404731
Groenink M, den Hartog AW, Franken R et al (2013) Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J 34(45):3491–3500. doi:10.1093/eurheartj/eht334
Franken R, den Hartog AW, Radonic T et al (2015) Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ Cardiovasc Genet 8(2):383–388. doi:10.1161/CIRCGENETICS.114.000950
Segura AM, Luna RE, Horiba K et al (1998) Immunohistochemistry of matrix metalloproteinases and their inhibitors in thoracic aortic aneurysms and aortic valves of patients with Marfan’s syndrome. Circulation 98(19 Suppl):II331–II337 (discussion II337–8)
Booms P, Pregla R, Ney A et al (2005) RGD-containing fibrillin-1 fragments upregulate matrix metalloproteinase expression in cell culture: a potential factor in the pathogenesis of the Marfan syndrome. Hum Genet 116(1–2):51–61. doi:10.1007/s00439-004-1194-7
Ikonomidis JS, Jones JA, Barbour JR et al (2006) Expression of matrix metalloproteinases and endogenous inhibitors within ascending aortic aneurysms of patients with Marfan syndrome. Circulation 114(1 Suppl):I365–I370. doi:10.1161/CIRCULATIONAHA.105.000810
Chung AW, Au Yeung K, Sandor GG et al (2007) Loss of elastic fiber integrity and reduction of vascular smooth muscle contraction resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in the thoracic aortic aneurysm in Marfan syndrome. Circ Res 101(5):512–522. doi:10.1161/CIRCRESAHA.107.157776
Chung AW, Yang HH, Radomski MW et al (2008) Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in marfan syndrome through the inhibition of matrix metalloproteinase-2 and -9. Circ Res 102(8):e73–e85. doi:10.1161/CIRCRESAHA.108.174367
Xiong W, Meisinger T, Knispel R et al (2012) MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ Res 110(12):e92–e101. doi:10.1161/CIRCRESAHA.112.268268
Pilop C, Aregger F, Gorman RC et al (2009) Proteomic analysis in aortic media of patients with Marfan syndrome reveals increased activity of calpain 2 in aortic aneurysms. Circulation 120(11):983–991. doi:10.1161/CIRCULATIONAHA.108.843516
Sedding DG, Homann M, Seay U et al (2008) Calpain counteracts mechanosensitive apoptosis of vascular smooth muscle cells in vitro and in vivo. FASEB J 22(2):579–589. doi:10.1096/fj.07-8853com
Merk DR, Chin JT, Dake BA et al (2012) miR-29b participates in early aneurysm development in Marfan syndrome. Circ Res 110(2):312–324. doi:10.1161/CIRCRESAHA.111.253740
Emrich FC, Okamura H, Dalal AR et al (2015) Enhanced caspase activity contributes to aortic wall remodeling and early aneurysm development in a murine model of Marfan syndrome. Arterioscler Thromb Vasc Biol 35(1):146–154. doi:10.1161/ATVBAHA.114.304364
Tseng WF, Huang SS, Huang JS (2004) LRP-1/TbetaR-V mediates TGF-beta1-induced growth inhibition in CHO cells. FEBS Lett 562(1–3):71–78. doi:10.1016/S0014-5793(04)00185-1
Huang SS, Ling TY, Tseng WF et al (2003) Cellular growth inhibition by IGFBP-3 and TGF-beta1 requires LRP-1. FASEB J 17(14):2068–2081. doi:10.1096/fj.03-0256com
Boucher P, Li WP, Matz RL et al (2007) LRP1 functions as an atheroprotective integrator of TGFbeta and PDFG signals in the vascular wall: implications for Marfan syndrome. PLoS One 2(5):e448. doi:10.1371/journal.pone.0000448
Basford JE, Koch S, Anjak A et al (2013) Smooth muscle LDL receptor-related protein-1 deletion induces aortic insufficiency and promotes vascular cardiomyopathy in mice. PLoS One 8(11):e82026. doi:10.1371/journal.pone.0082026
Zhou L, Takayama Y, Boucher P et al (2009) LRP1 regulates architecture of the vascular wall by controlling PDGFRbeta-dependent phosphatidylinositol 3-kinase activation. PLoS One 4(9):e6922. doi:10.1371/journal.pone.0006922
Bruemmer D, Berger JP, Liu J et al (2003) A non-thiazolidinedione partial peroxisome proliferator-activated receptor gamma ligand inhibits vascular smooth muscle cell growth. Eur J Pharmacol 466(3):225–234
Duan SZ, Usher MG, Mortensen RM (2008) Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ Res 102(3):283–294. doi:10.1161/CIRCRESAHA.107.164384
Okura T, Nakamura M, Takata Y et al (2000) Troglitazone induces apoptosis via the p53 and Gadd45 pathway in vascular smooth muscle cells. Eur J Pharmacol 407(3):227–235
Redondo S, Ruiz E, Santos-Gallego CG et al (2005) Pioglitazone induces vascular smooth muscle cell apoptosis through a peroxisome proliferator-activated receptor-gamma, transforming growth factor-beta1, and a Smad2-dependent mechanism. Diabetes 54(3):811–817
Sakomura Y, Nagashima H, Aoka Y et al (2002) Expression of peroxisome proliferator-activated receptor-gamma in vascular smooth muscle cells is upregulated in cystic medial degeneration of annuloaortic ectasia in Marfan syndrome. Circulation 106(12 Suppl 1):I259–I263
Fu M, Zhang J, Zhu X et al (2001) Peroxisome proliferator-activated receptor gamma inhibits transforming growth factor beta-induced connective tissue growth factor expression in human aortic smooth muscle cells by interfering with Smad3. J Biol Chem 276(49):45888–45894. doi:10.1074/jbc.M105490200
Law RE, Meehan WP, Xi XP et al (1996) Troglitazone inhibits vascular smooth muscle cell growth and intimal hyperplasia. J Clin Invest 98(8):1897–1905. doi:10.1172/JCI118991
Zirlik A, Leugers A, Lohrmann J et al (2004) Direct attenuation of plasminogen activator inhibitor type-1 expression in human adipose tissue by thiazolidinediones. Thromb Haemost 91(4):674–682. doi:10.1267/THRO04040674
Urbich C, Kuehbacher A, Dimmeler S (2008) Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res 79(4):581–588. doi:10.1093/cvr/cvn156
Maegdefessel L, Azuma J, Toh R et al (2012) Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest 122(2):497–506. doi:10.1172/JCI61598
Jones JA, Stroud RE, O’Quinn EC et al (2011) Selective microRNA suppression in human thoracic aneurysms: relationship of miR-29a to aortic size and proteolytic induction. Circ Cardiovasc Genet 4(6):605–613. doi:10.1161/CIRCGENETICS.111.960419
Boon RA, Seeger T, Heydt S et al (2011) MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res 109(10):1115–1119. doi:10.1161/CIRCRESAHA.111.255737
Humphrey JD, Milewicz DM, Tellides G et al (2014) Cell biology. Dysfunctional mechanosensing in aneurysms. Science 344(6183):477–479. doi:10.1126/science.1253026
Leung DY, Glagov S, Mathews MB (1976) Cyclic stretching stimulates synthesis of matrix components by arterial smooth muscle cells in vitro. Science 191(4226):475–477
Li S, Van Den Diepstraten C, D’Souza SJ et al (2003) Vascular smooth muscle cells orchestrate the assembly of type I collagen via alpha2beta1 integrin, RhoA, and fibronectin polymerization. Am J Pathol 163(3):1045–1056
Schwartz MA (2010) Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol 2(12):a005066. doi:10.1101/cshperspect.a005066
Dingemans KP, Teeling P, Lagendijk JH et al (2000) Extracellular matrix of the human aortic media: an ultrastructural histochemical and immunohistochemical study of the adult aortic media. Anat Rec 258(1):1–14
Pereira L, Lee SY, Gayraud B et al (1999) Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci USA 96(7):3819–3823
Li DY, Brooke B, Davis EC et al (1998) Elastin is an essential determinant of arterial morphogenesis. Nature 393(6682):276–280. doi:10.1038/30522
Valentin A, Humphrey JD, Holzapfel GA (2011) A multi-layered computational model of coupled elastin degradation, vasoactive dysfunction, and collagenous stiffening in aortic aging. Ann Biomed Eng 39(7):2027–2045. doi:10.1007/s10439-011-0287-4
Wilson JS, Baek S, Humphrey JD (2012) Importance of initial aortic properties on the evolving regional anisotropy, stiffness and wall thickness of human abdominal aortic aneurysms. J R Soc Interface 9(74):2047–2058. doi:10.1098/rsif.2012.0097
Crosas-Molist E, Meirelles T, Lopez-Luque J et al (2015) Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler Thromb Vasc Biol 35(4):960–972. doi:10.1161/ATVBAHA.114.304412
Takebayashi S, Kubota I, Takagi T (1973) Ultrastructural and histochemical studies of vascular lesions in Marfan’s syndrome, with report of 4 autopsy cases. Acta Pathol Jpn 23(4):847–866
Takebayashi S, Taguchi T, Kawamura K et al (1988) “Osmiophilic elastolysis” of peripheral organ arteries in patients with Marfan’s syndrome. Acta Pathol Jpn 38(11):1433–1443
Dingemans KP, Teeling P, van der Wal AC et al (2006) Ultrastructural pathology of aortic dissections in patients with Marfan syndrome: comparison with dissections in patients without Marfan syndrome. Cardiovasc Pathol 15(4):203–212. doi:10.1016/j.carpath.2006.03.004
Eberth JF, Taucer AI, Wilson E et al (2009) Mechanics of carotid arteries in a mouse model of Marfan Syndrome. Ann Biomed Eng 37(6):1093–1104. doi:10.1007/s10439-009-9686-1
Syyong HT, Chung AW, Yang HH et al (2009) Dysfunction of endothelial and smooth muscle cells in small arteries of a mouse model of Marfan syndrome. Br J Pharmacol 158(6):1597–1608. doi:10.1111/j.1476-5381.2009.00439.x
Syyong HT, Chung AW, van Breemen C (2011) Marfan syndrome decreases Ca2+ wave frequency and vasoconstriction in murine mesenteric resistance arteries without changing underlying mechanisms. J Vasc Res 48(2):150–162. doi:10.1159/000318804
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Perrucci, G.L., Rurali, E., Gowran, A. et al. Vascular smooth muscle cells in Marfan syndrome aneurysm: the broken bricks in the aortic wall. Cell. Mol. Life Sci. 74, 267–277 (2017). https://doi.org/10.1007/s00018-016-2324-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2324-9