
Abstract
Metastatic prostate cancer is a lethal disease that remains incurable despite the recent approval of new drugs, thus making the development of alternative treatment approaches urgently needed. A more precise understanding of the molecular mechanisms underlying prostate cancer dissemination could lead to the identification of novel therapeutic targets for the design of efficient anti-metastatic strategies. MicroRNA (miRNAs) are endogenous, small non-coding RNA molecules acting as key regulators of gene expression at post-transcriptional level. It has been clearly established that altered miRNA expression is a common hallmark of cancer. In addition, emerging evidence suggests their direct involvement in the metastatic cascade. In this review, we present a comprehensive overview of the data generated in experimental tumor models indicating that specific miRNAs may impinge on the different stages of prostate cancer metastasis, including (i) the regulation of epithelial-to-mesenchymal transition and cell migration/invasion, (ii) the interplay between cancer cells and the surrounding stroma, (iii) the control of angiogenesis, (iv) the regulation of anoikis, and (v) the colonization of distant organs. Moreover, we show preliminary evidence of the clinical relevance of some of these miRNAs, in terms of association with tumor aggressiveness/dissemination and clinical outcome, as emerged from translation studies carried out in prostate cancer patient cohorts. We also discuss the potential and the current limitations of manipulating metastasis-related miRNAs, by mimicking or inhibiting them, as a strategy for the development of novel therapeutic approaches for the advanced disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prostate cancer (PCa) is the second most commonly diagnosed malignancy and the fifth leading cause of cancer-related death in men [1]. PCa diagnosis is based on histopathological inspection of prostate core biopsies, in the presence of elevated serum prostate-specific antigen (PSA) levels and/or suspect digital rectal examination. Although PSA-based screening has resulted in enhanced detection at earlier stages of the disease and reduction of PCa-related mortality, it remains controversial because of adverse effects such as overdiagnosis [2]. In addition, the inability of currently available prognostic factors, including PSA, Gleason score and tumor stage, to properly discriminate low-risk from aggressive disease at the time of diagnosis has led to the overtreatment of patients suitable for conservative management.
Current treatment options for localized PCa include prostatectomy, external radiotherapy and brachytherapy, while active surveillance has evolved as an alternative to radical treatment for very low-risk PCa [3]. In case of relapse after radical treatment or in patients with locally advanced or metastatic disease, androgen deprivation therapy represents the standard treatment. Unfortunately, the efficacy of androgen deprivation is temporary and most patients develop a castration-resistant disease. Metastatic castration-resistant PCa is a lethal disease which remains incurable despite the recent approval of new drugs, including androgen pathway inhibitors (enzalutamide, abiraterone acetate), immunotherapeutic agents (sipuleucel-T), cytotoxic chemotherapeutics (cabazitaxel) and radiopharmaceuticals (radium-223), making the development of alternative treatment approaches urgently needed. In this context, a deeper understanding of the molecular mechanisms that underly PCa dissemination could lead the identification of novel therapeutic targets for the development of efficient anti-metastatic strategies [4].
Metastasis is the product of a multi-step process during which cancer cells, responding to intrinsic and extrinsic stimuli, detach from the primary tumor, invade the contiguous stroma, migrate over a long distance, and colonize other organs [5, 6]. In a classical view, tumor cells acquire the metastatic competence through the stochastic accumulation of serial genomic alterations, as exemplified by the colorectal cancer paradigm. However, recent next generation sequencing analyses have been elusive in identifying metastasis-specific alterations [5]. Rather, the same oncogenic forces driving tumor development seem to be clonally selected and emerge in metastases at secondary sites. Consistently, gene expression profiling studies had already shown that specific gene signatures found in primary lesions could predict the propensity of giving rise to metastases, again suggesting that clones with potential to disseminate are already present in the primary tumor [7]. The detection of circulating or bone marrow disseminated tumor cells in patients with clinically localized cancer suggests that cancer cell dissemination may begin when tumor is still at an early stage [6]. At diagnosis, a tumor may have already shed a huge number of cells in circulation but only a small percentage proves to efficiently colonize distant organs. Metastasis is indeed a very inefficient process and most of the cells die in circulation due to anoikis or at secondary sites due to hostile tissue microenvironment. A successful outcome relies on the proficient cross-talk between tumor cells and the microenvironment of the organ where cancer is initiated as well as the interaction with the stroma of the metastasis host tissue [8]. The fruitful combination of predisposing genetic alterations and epigenetic modifications induced by permissive microenvironmental signals can indeed provide a tumor cell with the plasticity needed to go through the different steps of the metastatic process. Among distorsions in the epigenome, miRNA alterations have been shown to importantly contribute to cancer metastasis as reviewed by Fenderico and colleagues [9].
miRNA biogenesis and functions
miRNAs are single stranded, endogenous, evolutionary conserved, non-coding RNA molecules acting as post-transcriptional regulators of gene expression [10]. Human miRNA biogenesis is a multi-step process that, starting from the transcription of a primary miRNA transcript (pri-miRNA) leads to the generation of a single-strand mature miRNA and the star miRNA (miRNA*). The mature miRNA, by interacting with argonaute proteins (AGO) within the multi-protein RNA-induced silencing complex (RISC), guides the complex onto complementary sequences present in the 3′- or 5′-untranslated regions (UTR) or coding regions of its target mRNAs [10]. According to the degree of complementarity, the recognition can lead to cleavage of the mRNA or to protein translation inhibition, respectively. A single miRNA can potentially regulate the expression of several transcripts, and each transcript can be targeted by more than one miRNA. Such intrinsic feature allows miRNAs to play roles in a wide range of biological processes, including development, differentiation, metabolism, proliferation, cell cycle, and apoptosis [10]. Although post-transcriptional repression remains the principal mechanism of action, it has been reported that miRNAs may also target specific sequences in gene promoters, thus exerting a function in transcriptional induction [11].
Deregulated miRNA expression and/or function have been causatively linked to the pathogenesis of several human diseases, including cancer [12]. Depending on their expression levels, cellular context and targets’ functions, miRNAs can act as oncogenes or tumor suppressors. For example, the down-regulation of a miRNA, the function of which is to control the abundance of a given oncogene, may result in aberrant overexpression of the target, thus ultimately promoting cancer [12]. In this regard, well-documented evidence suggests that miRNAs can take part in pathways sustaining all the diverse hallmarks of cancer and therefore actively contribute to tumor development and progression [12].
Involvement of miRNAs in the multi-step metastasis process of prostate cancer
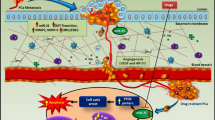
The metastatic program includes multiple sequential and interrelated steps by which cancer cells detach from the primary site, acquire cell motility and the ability to degrade the extracellular matrix (ECM), invade the contiguous host tissue and enter the systemic circulation through the lymph and blood vessels (intravasion). Circulating tumor cells survive until reaching the metastatic site, where they exit the blood stream (extravasation), colonize the foreign microenvironment and start proliferation to establish a secondary tumor [5, 6]. Here, we focus on a series of recent preclinical studies demonstrating that specific miRNAs may impinge on the different steps of PCa metastasis and summarize the current knowledge on the molecular mechanisms through which they regulate the process (Table 1; Fig. 1).

Overview of miRNAs involved in the metastatic cascade. miRNAs shown to play a role in promoting or preventing different steps of the PCa metastatic process, including the regulation of epithelial-to-mesenchymal transition and cell migration/invasion, the interplay between cancer cells and the surrounding stroma, the control of angiogenesis, the regulation of anoikis, and the colonization of distant organs, are reported
miRNAs regulating epithelial-to-mesenchymal transition and cell migration
Epithelial-to-mesenchymal transition (EMT) is an early key step of the metastatic process and consists of a series of events by which epithelial cells lose most of their epithelial features and gain properties that are typical of mesenchymal cells [13]. Specifically, epithelial cells break contact with their neighbors, by losing adherens and tight junctions, and change their cytoskeleton architecture assuming a spindle-shaped form that allows them to break through the basal membrane of epithelium and migrate over a long distance. Increased expression of mesenchymal markers, such as vimentin and N-cadherin, overexpression of E-cadherin transcriptional repressors, including zinc finger proteins SNAI1 and SNAI2, twist-related protein 1 (TWIST1) and zinc finger E-box-binding homeobox 1 and 2 (ZEB1, ZEB2), and membrane-to-nuclear localization of β-catenin are EMT hallmarks. Current knowledge indicates that changes in the expression levels of EMT-related genes are not only ascribable to genetic mutations but also to complicated networks between signaling pathways and miRNAs [14]. In this context, it has been reported that ectopic expression of miR-205, a miRNA that is down-regulated in PCa, in DU145 and PC-3 cells reversed their mesenchymal phenotype by inducing E-cadherin up-modulation, morphological changes, cytoskeleton rearrangements as well as reduction of cell locomotion and invasion. The concomitant repression of ZEB1/2 and protein kinase Cε (PKCε) by miR-205 has been suggested to drive such events [15]. More recently, it has been found that miR-205-induced inhibition of PCa cell migration and invasion was also mediated by direct regulation of centromere protein F [16]. Similarly, ectopic expression of miR-29b, another down-regulated miRNA in PCa, in PC-3 cells resulted in up-regulation of E-cadherin and concomitant decrease of N-cadherin and SNAI1, inhibition of wound healing capacity, invasiveness and ability to colonize the lungs and liver of severe combined immunodeficiency (SCID) mice following intravenous injection [17]. SNAI2 has been identified as one of the phylogenetically conserved targets of miR-1 and miR-200, two miRNAs whose expression is reduced during PCa progression. Forced expression of miR-1 or miR-200 in PCa cells induced an epithelial phenotype, as shown by increased E-cadherin and decreased vimentin protein levels [18]. The overexpression of miR-203, a miRNA down-regulated in PCa, was found to counteract EMT and inhibit PCa cell motility and invasion by repressing a cohort of pro-metastatic genes [19]. More recently, it has been shown that miR-203 exerts a dual function in PCa cells since its reconstitution counteracted EMT but also allowed growth factor-independent proliferation via repressing SNAI2 [20]. Reconstitution of miR-34b, which is silenced in PCa through CpG hypermethylation of the MIR34B/C locus, counteracted EMT in PCa cells and inhibited cell proliferation, migration/invasion, and growth in nude mice by directly targeting AKT and its downstream proliferative genes [21]. miR-145 was reported to be a strong repressor of EMT by targeting ZEB2, which in turn directly represses miR-145 transcription. Indeed, miRNA reconstitution in DU145 and C4-2B cells counteracted EMT and also inhibited cell migration and invasion [22]. Reconstitution of miR-573, a miRNA whose expression is lower in metastatic compared to matched primary PCa, was found to impair tumor cell migration and invasion as well as TGF-β1-induced EMT [23]. Finally, ectopic overexpression of miR-223 inhibited PC-3 cell growth, migration and invasion by targeting the integrin α3 (ITGA3)/integrin β1 (ITGB1) signaling pathway [24]. Both ITGA3 and ITGB1 proteins are known to be involved in the induction of EMT in cancer cells [25, 26].
A number of miRNAs have been shown to regulate pathways relevant to cell motility, which are at least in part independent of the EMT program, such as the Ras homolog gene family, member A (RhoA)/Rho-associated protein kinase 1 (ROCK1) axis [27]. In this context, stable transfection of PC-3 cells with miR-146a, which is profoundly down-regulated in castration-resistant PCa, was found to suppress the expression of the target protein-coding gene ROCK1, and markedly reduce cell proliferation and invasion [28].
Chemokines are master regulators of cell migration [29]. Specifically, it has been shown that binding of the C-X-C motif chemokine receptor type 4 (CXCR4) to its ligand stromal cell-derived factor 1/C-X-C motif chemokine 12 (SDF-1/CXCL12) mediates migration and metastatic spread in different types of cancer [30, 31]. In this regard, it has been reported that miR-494-3p can regulate CXCR4 expression post-transcriptionally and that ectopic overexpression of the miRNA inhibits migration and invasion of PC-3 and DU145 cells [32].
miRNAs involved in the interaction between PCa cells and tumor microenvironment
Compelling evidence supports the notion that tumor progression is strongly influenced by microenvironmental cues, including hypoxia, acidity, ECM composition, and host stromal cells, collectively called “reactive stroma” [8]. During tumor progression, cancer and its surrounding stroma co-evolve and contribute equally to the aggressive phenotype [8]. Recent results have offered understanding into how miRNAs are directly involved in the cross-talk between cancer cells and the microenvironment, giving rise to heterotypic cell signals aimed at preventing or fostering tumor development [33].
miRNAs and extracellular matrix
Loss of ECM structure by overproduction of metalloproteases (MMPs) is required for cancer cells to infiltrate adjacent tissue [5, 6]. Different studies demonstrated that MMPs production may be regulated at the post-transcriptional level by miRNAs. In this context, ectopic expression of miR-29b [17], miR-146a [30], miR-130b [31] and miR-205 [15] in PCa cells significantly inhibited the expression/proteolytic activity of MMP-2. Conversely, miR-21 was found to positively regulate PCa cell invasiveness through translational repression of the MMP inhibitor reversion-inducing-cysteine-rich protein with kazal motifs (RECK) [36].
miR-25 was reported to directly regulates pro-invasive αv- and α6-integrin expression. Indeed, ectopic overexpression of miR-25 in PCa cell lines and selected subpopulations of highly metastatic and tumorigenic cells, strongly affected the invasive cytoskeleton, causing reduced migration and metastasis formation via attenuation of extravasation [37].
The basement membrane (BM) is a thin sheet of ECM, which surrounds normal prostate gland and not only provides a structural support but also influences cell–cell and cell–protein interactions [38]. Lack or discontinuity of the BM is a prerequisite for tumor cell invasion into interstitial spaces, thus favoring metastasis. In this regard, it has been demonstrated that miR-205 participates in a network involving ΔNp63α, which is essential for maintenance of the BM in prostate epithelium. Functionally, miR-205 is able to control the deposition of laminin-332 and its receptor integrin-β4. Hence, pathological loss of miR-205, as widely observed in PCa, may favor tumor dissemination by creating discontinuities in the BM. Interestingly, replacement of miR-205 in PCa cells was able to restore BM deposition and three dimensional organization into normal-like acinar structures, thus hampering cancer progression [39].
miRNAs and cancer-associated fibroblasts
Among stromal cells, cancer-associated fibroblasts (CAFs) have been reported to play a key role in malignant progression, through secretion of soluble growth factors, such as vascular endothelial growth factor (VEGF), and inflammatory cytokines, including transforming growth factor beta (TGF-β), interleukin 6 (IL-6) and interleukin 10 (IL-10), production of ECM proteins, and release of matrix MMPs [40]. Musumeci and colleagues first demonstrated that down-regulation of miR-15 and miR-16 in PCa clinical samples was not confined to the cancer cell population but also involved fibroblasts surrounding the tumors. Reconstitution of miR-15 and miR-16 in CAFs was found to impair their tumor-supportive capability both in vitro and in vivo by suppressing fibroblast growth factor 2 (FGF2) and its receptor fibroblast growth factor receptor 1 (FGFR1) [41]. miR-205 was found to be the most down-modulated miRNA in PCa cells upon CAF stimulation, due to direct transcriptional repression by hypoxia inducible factor 1 (HIF-1). Rescue experiments demonstrated that ectopic miR-205 overexpression in PCa cells counteracted CAF-induced EMT, thus impairing enhancement of cell invasion, acquisition of stem cell traits, tumorigenicity, and metastatic dissemination. In addition, miR-205 blocked tumor-driven activation of surrounding fibroblasts by reducing the secretion of pro-inflammatory cytokines, such as IL-6 [42].
It has been demonstrated that overexpression of the hypoxia-induced miR-210 in young prostate fibroblasts increased their senescence-associated features and converted them into CAF-like cells, able to promote EMT in PCa cells, to support angiogenesis and to recruit endothelial precursor cells and monocytes/macrophages [43]. Ectopic expression of miR-409—a miRNA which is highly abundant in prostate CAFs—in normal prostate fibroblasts conferred a cancer-associated stroma-like phenotype and led to the release of the miRNA via extracellular vesicles to promote tumorigenesis through repression of tumor suppressor genes such as Ras suppressor 1 (RSU1) and stromal antigen 2 (STAG2) [44]. Finally, a comparative miRNA expression analysis aimed at identifying specific miRNAs consistently involved in prostate fibroblast activation by different stimuli revealed a role for miR-133b as a soluble factor secreted by activated fibroblasts to support paracrine activation of non-activated fibroblast and promote tumor progression [45].
miRNAs regulating angiogenesis
When tumor reaches a certain critical diameter, essential nutrients and oxygen become scarce. To face the problem, new blood vessels are originated through the sprouting of preexisting vessels. Indeed, a hallmark of cancer is the capability of tumor cells to induce angiogenesis, a finely tuned process, based on the balance between pro-angiogenic and anti-angiogenic factors [46]. Cancer cells also hijack the blood vasculature to infiltrate it and initiate their journey toward a distant site. Experimental evidence indicates that miRNAs may determine the quiescent or angiogenic state of endothelial cells by inducing changes in the levels of angiogenic activators or inhibitors. In this context, it was shown that overexpression of miR-21 in DU145 cells induces tumor angiogenesis by targeting phosphatase and tensin homolog (PTEN), which in turn activates AKT and extracellular-signal-regulated kinases 1/2 (ERK1/2) signaling pathways and finally enhances HIF-1α and VEGF expression, two of the strongest angiogenesis inducers [47]. Conversely, restoration of miR-146a in DU145 cells was found to reduce tumorigenicity and angiogenesis, as assessed by microvessel density in subcutaneous xenografts, by down-regulating epidermal growth factor receptor (EGFR) [34].
miRNAs and resistance to anoikis
Anoikis is the process through which epithelial cells undergo apoptosis after they lose the contacts with neighboring cells and ECM. Metastatic cancer cells are insensitive to anoikis and do not require adhesion to the ECM to survive the trip through the lymph and the blood circulation [48]. Although molecular mechanisms supporting anoikis resistance are poorly understood, it has been suggested that miRNAs may play a role. Specifically, miR-132, a miRNA down-regulated in PCa due to CpG island hypermethylation, has been reported to regulate anoikis in PCa cells [49]. Indeed, after miR-132 restoration in PC-3 cells, in vitro anoikis assay revealed cell detachment followed by cell death. This effect was partially explained by miR-132 targeting of two pro-survival factors, heparin-binding epidermal growth factor (HB-EGF) and Talin2 (TLN2), a protein that connects integrins to the actin cytoskeleton [49].
miRNAs and colonization to distant organs
Out of millions of cancer cells that infiltrate blood circulation, only a few survive and even less (estimated to be <0.01 %) ultimately develop into macroscopic metastases [6]. Experimental evidence suggests that miRNAs can influence the survival of tumor-propagating cells as well as the dissemination of tumor cells at bone, which represent the primary site of PCa metastasis. Specifically, it was shown that forced expression of miR-224, which is frequently down-regulated in metastatic PCa, was able to abrogate PCa cell proliferation, migration and invasion by inhibiting the expression of tribbles homolog 1 (TRIB1) [50]. Interestingly, TRIB1 appears an essential factor for prostate tumorigenesis and tumor-propagating cell survival through the regulation of the endoplasmic reticulum chaperone expression [51].
In an experimental bone metastasis model, the intravenous injection of atelocollagen-conjugated miR-16 was found to significantly inhibit the occurrence of PCa bone metastasis as a consequence of down-regulation of genes associated to cell cycle control and proliferation, such as cyclin-dependent kinases 1 (CDK1) and 2 (CDK2) [52]. Similarly, the ectopic overexpression of miR-145 and miR-143, two miRNAs found to be down-regulated in PCa bone metastases, was able to reduce bone invasion by PC-3 cells in an intra-tibial injection mouse model as a consequence of EMT repression [53]. Again, the ectopic expression of miR-203 was reported to inhibit bone metastasis and induce sensitivity to tyrosine kinase inhibitors (TKIs) in a PCa xenograft model. Functionally, it was demonstrated that the induction of bone metastasis and TKI resistance requires miR-203 down-regulation and activation of EGFR pathway via altered expression of EGFR ligands, epiregulin (EREG) and transforming growth factor alpha (TGF-α). Interestingly, in PCa clinical samples, miR-203 levels were inversely correlated with the expression of EREG and TGF-α, thus supporting the existence of a miR-203, EGFR, TKIs resistance regulatory network in PCa progression [54]. Ectopic expression of miR-34a was also found to inhibit bone metastasis in a Ras-dependent PCa xenograft model by directly targeting transcription factor 7 (TCF7), a Wnt signaling-related gene, which is considered a critical factor in bone metastasis, as well as the pro-survival gene baculoviral inhibitor of apoptosis repeat-containing 5 (BIRC5) [55]. Reconstitution of androgen-regulated miR-1, whose expression levels are progressively reduced in disseminated PCa, was reported to inhibit bone metastasis in a PCa xenograft model through the direct targeting of Src, a non-receptor tyrosine kinase that integrates numerous signaling pathways, including androgen receptor (AR). Specifically, Src interacts with and phosphorylates AR, resulting in enhanced activity. In addition, Src is downstream of receptors, such as EGFR and fibroblast growth factor receptor (FGFR), and upstream of signaling molecules, such as AKT and ERK, that have been implicated in PCa survival in the absence of sufficient AR signaling. Overall, results from the study propose that loss of miR-1 represents a mechanistic link between low canonical AR output and Src-promoted metastatic phenotypes [56]. In addition, it has been shown that activated nuclear EGFR acts as a transcriptional inhibitor of miR-1 and, in turn, promotes bone metastasis formation following intra-cardiac injection of PCa cells in nude mice as a consequence of the increased activity of the miR-1 target TWIST1 [57]. Consistent with these findings, a correlation between decreased miR-1 levels and enhanced expression of activated EGFR and TWIST1 was observed in PCa clinical specimens [57]. Interestingly, an independent study showed that miR-1 is significantly down-regulated in specimens from recurrent PCa patients compared to those from non-recurrent patients [58].
Concerning miRNAs potentially involved in the promotion of PCa metastatic spread, miR-96 was identified as a key factor in TGF-β-driven PCa bone metastasis. Based on the results obtained in murine and human PCa experimental models, a mechanism was proposed by which TGF-β stimulates mammalian target of rapamycin (mTOR) functions through the Smad-dependent canonical pathway to induce the expression of miR-96, which in turn targets and inhibits the expression of proline-rich AKT1 substrate 1 (AKT1S1), a negative regulator of mTOR [54]. Two additional reports indicate a role for specific components of the delta-like 1 homolog-deiodinase, iodothyronine 3 (DLK-1-DIO3) miRNA mega-cluster, which have been shown to be critical for embryonic development and EMT, as positive regulators of bone metastasis. The first study showed that intra-cardiac injection of miR-154* inhibitor-treated PCa cells in mice led to decreased bone metastasis and increased survival, and suggested that miRNA effects were mediated by down-regulation of its target STAG2, a component of the cohesin multi-protein complex [60]. The same research group reported that orthotopic delivery of miR-409-3p/-5p in the murine prostate gland induced tumors expressing EMT and stemness markers. In addition, intra-cardiac inoculation of miR-409-5p inhibitor-treated PCa cells in mice reduced bone metastasis and prolonged survival. The miRNA was found to target tumor suppressors, such as STAG2 and RSU1 [61].
A recent study elegantly demonstrated that loss of miR-15 and miR-16 cluster in cooperation with increased miR-21 expression promotes human PCa cell spreading and bone lesions in mouse models through the potentiation of TGF-β activity, which in turn leads to Hedgehog signaling activation, thus mediating local invasion, distant bone marrow colonization and osteolysis by tumor cells. These findings establish a new molecular circuitry for PCa metastasis spread and bone lesion formation [62].
Clinical relevance of miRNAs involved in the metastasis process in PCa experimental models
Results of preliminary studies aimed at evaluating in PCa clinical samples the expression of specific miRNAs identified as involved in the metastasis process in experimental models indicated that the expression levels of such miRNAs are progressively deregulated in aggressive disease and metastasis, and, in some cases, are correlated to clinical outcome (Table 2), thus supporting a possible role as novel therapeutic targets and/or prognostic biomarkers.
miR-205. Three independent studies consistently demonstrated that low miR-205 expression levels were associated to the metastatic disease [42, 63, 64]. Two of them also reported that miR-205 down-regulation was correlated to poor patient outcome in terms of increased probability of biochemical recurrence [51] and shortened overall survival [63].
miR-224. A significant association between miR-224 down-regulation and advanced clinical stage and metastasis was consistently shown in three studies [50, 65, 66]. Two of them also revealed that reduced/absent expression of miR-224 was significantly associated with shorter progression-free survival of PCa patients [50, 65].
miR-21. In a cohort of radical prostatectomy samples, high levels of miR-21 were found to be significantly associated with advanced pathological stage, high Gleason score, presence of lymph node metastasis and poor patient outcome. Specifically, multi-variate analysis indicated miR-21 expression as an independent predictor of 5-year biochemical recurrence [67].
miR-126. Sun and colleagues reported that the loss of miR-126 expression in PCa clinical samples was associated with aggressive clinico-pathological features, including advanced stage, positive angiolymphatic invasion and lymph node metastasis, and that patients with low miR-126 expression displayed shorter biochemical recurrence-free survival than those with high miR-126 expression [68].
miR-34b. Two studies consistently showed that reduced miR-34b levels were correlated with clinico-pathological progression of PCa, in terms of high Gleason score, perineural invasion and lymph node metastasis [21, 69]. In addition, patients with tumors expressing low miR-34b levels displayed a significantly higher risk of biochemical recurrence and a lower overall survival probability [21, 69].
miR-15 and miR-16. In a series of primary PCa and visceral/bone metastatic lesions, Bonci and colleagues showed that low expression of miR-15 and miR-16 was associated to the presence of bone metastasis and poor patient disease-free survival. In addition, experimental findings demonstrating that up-regulated miR-21 expression cooperates with miR-15 and miR-16 loss to promote cancer progression in murine PCa models were initially validated in the same sample cohort. Indeed a concomitant deregulation of miR-15/miR-16 and miR-21 was selectively observed in a fraction of PCa patients with progressive disease or distant lesions and was associated with a poor prognosis in terms of disease-free survival [62].
Potential of miRNA-based approaches for the development of anti-metastatic therapeutic approaches
The direct involvement of specific miRNAs in the regulation of the different steps of PCa dissemination program, together with the possibility to manipulate their expression, has aroused interest in the development of novel miRNA-based strategies for the prevention and treatment of metastasis [71]. Restoration of miRNA expression in tumor cells can be accomplished through the use of different types of molecules able to mimic native miRNAs. To reproduce a physiologic context, synthetic double-stranded RNA oligonucleotides that harbor chemical modifications to improve stability and cellular uptake are designed to allow the exclusive production of the mature miRNA of interest and to preserve the ability to interact with the natural targets thus minimizing the occurrence of off-target effects [72]. While the use of synthetic precursors is useful to induce a transient re-expression of the miRNA, cloning miRNA genes into viral vector systems represents a successful strategy to stably restore miRNA expression [73]. A main advantage of viral vectors in the context of a miRNA-based anti-metastatic therapy is represented by the possibility of injecting them directly into the tumor mass.
Mature miRNAs can be inhibited using antisense oligonucleotides, known as anti-miRNAs and miRNA sponges. Antisense oligonucleotides strongly bind a given miRNA and antagonize its interaction with the target mRNAs [72]. Locked nucleic acids (LNAs), i.e., bicyclic nucleic acid analogs bearing RNA bases with an extra bridge connecting the 2′ oxygen and 4′ carbon, exhibit the highest affinity towards complementary RNA and now represent the most widely used anti-miRNA molecules [72]. miRNA sponges are expression vectors that allow overexpression of RNA molecules containing multiple tandem binding sites to the miRNA of interest and block its function through competition with target transcripts [72]. A different inhibitory strategy is based on miR-masks, 22nt-long single stranded, chemically modified oligonucleotides complementary to the miRNA binding site in the 3′ UTR of a given protein-coding RNA messenger. Conversely to anti-miRNAs and miRNA sponges, the miR-mask approach is aimed at masking the interaction sites between the miRNA and its target mRNA [74].
Preclinical research conducted in mice is encouraging towards translating miRNA-based strategies into human cancer therapy. However, although these studies failed to reveal adverse events associated with miRNA manipulation and suggested that treatment with miRNA modulators is well tolerated, further improvements in terms of efficacy and targeted delivery to the tumor are still necessary. Indeed, the design of strategies to guarantee a more precise delivery to the tissue/organ of interest is a crucial issue due to the ubiquitous expression pattern reported for many miRNAs, which could increase the risk of off-target effects. In this context, strategies based on the conjugation of the therapeutic oligonucleotide with targeting molecules, such as peptides, antibodies or other active molecules, which may promote homing of the miRNA modulator to tumor cells, are currently being investigated [72]. In addition, a possible issue dealing with miRNA replacement approaches is related to the need of properly restoring the activity of the down-regulated/lost miRNA while preventing the introduction of supra-physiological levels of the miRNA [72]. Notably, the applicability of strategies aimed at modulating specific miRNA expression in the clinical setting are currently under investigation. Specifically, the first liposome-formulated mimic of the tumor suppressor miR-34a (MRX34) is completing a Phase I dose-escalation study in patients with unresectable primary liver cancer and advanced or metastatic cancer with liver involvement (http://www.clinicaltrials.gov; Identifier: NCT01829971). Moreover, in a Phase II clinical trial, the use of an LNA-modified anti-miRNA against miR-122, which is vital for the replication cycle of hepatitis C virus (HCV), has demonstrated to be an effective and safe treatment strategy for patients with chronic HCV genotype 1 infection, without evidence of viral resistance [75]. Although the approach has been developed to treat a non-oncologic disease, available results contribute to demonstrate that miRNA-inhibiting therapies may have a clinical interest also for cancer treatment.
Conclusions and future prospects
Results obtained in experimental models have demonstrated a direct involvement of specific miRNAs in the PCa dissemination process, by exerting either pro- or anti-metastatic functions, depending on their target genes. miRNAs, such as miR-205, miR-21, miR-15/miR-16 and miR-203, each involved in at least two steps of the metastatic cascade, could prove to be particularly useful targets for the development of novel therapeutic strategies for the advanced disease. In addition, results of translational studies carried out in PCa clinical specimens support the relevance of such miRNAs in tumor progression and patients’ outcome. However, before novel therapeutics based on modulators of such miRNAs can be translated into the clinic, the actual role of these miRNAs and the precise identification of the key targets relevant to the metastatic process need to be accurately defined in preclinical models of PCa spontaneous metastasis, which properly reproduce the clinical setting. Moreover, given miRNA redundancy, it should be carefully assessed whether targeting an individual miRNA may be sufficient to get a therapeutic effect.
Overall, although important questions dealing with the development of enhanced systems for the tumor specific delivery of miRNA-based molecules and the improvement of currently available information concerning their pharmacokinetics and safety profile are still open, a major role for novel therapeutic approaches based on the modulation of metastasis-related miRNAs in the management of advanced PCa patients can be envisaged in the not-too-distant future.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2014) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136(5):E359–E386. doi:10.1002/ijc.29210
Schröder FH, Hugosson J, Roobol MJ, Tammela TL et al (2014) ERSPC Investigators. Screening and prostate cancer mortality: results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow-up. Lancet 384(9959):2027–2035. doi:10.1016/S0140-6736(14)60525-0
Bangma CH, Valdagni R, Carroll PR, van Poppel H, Klotz L, Hugosson J (2015) Active surveillance for low-risk prostate cancer: developments to date. Eur Urol 67(4):646–648. doi:10.1016/j.eururo.2014.11.004
Hathaway AR, Baker MK, Sonpavde G (2015) Emerging agents for the therapy of advanced prostate cancer. Future Oncol 11(20):2775–2787. doi:10.2217/fon.15.224
Vanharanta S, Massagué J (2013) Origins of metastatic traits. Cancer Cell 24(4):410–421. doi:10.1016/j.ccr.2013.09.007
Valastyan S, Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147(2):275–292. doi:10.1016/j.cell.2011.09.024
Mittempergher L, Saghatchian M, Wolf DM, Michiels S, Canisius S, Dessen P, Delaloge S, Lazar V, Benz SC, Tursz T, Bernards R, van’t Veer LJ (2013) A gene signature for late distant metastasis in breast cancer identifies a potential mechanism of late recurrences. Mol Oncol 7(5):987–999. doi:10.1016/j.molonc.2013.07.006
Tlsty TD, Coussens LM (2006) Tumor stroma and regulation of cancer development. Annu Rev Pathol 1:119–150
Fenderico N, Casamichele A, Profumo V, Zaffaroni N, Gandellini P (2013) MicroRNA-mediated control of prostate cancer metastasis: implications for the identification of novel biomarkers and therapeutic targets. Curr Med Chem 20(12):1566–1584
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136(2):215–233. doi:10.1016/j.cell.2009.01.00219167326
Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R (2008) MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA 105(5):1608–1613. doi:10.1073/pnas.0707594105
Jansson MD, Lund AH (2012) MicroRNA and cancer. Mol Oncol 6(6):590–610. doi:10.1016/j.molonc.2012.09.006
Acloque H, Thiery JP, Nieto MA (2008) The physiology and pathology of the EMT. Meeting on the epithelial–mesenchymal transition. EMBO Rep 9(4):322–326. doi:10.1038/embor.2008.30
Miska EA (2008) MicroRNAs—keeping cells in formation. Nat Cell Biol 10(5):501–502. doi:10.1038/ncb0508-501
Gandellini P, Folini M, Longoni N, Pennati M, Binda M, Colecchia M, Salvioni R, Supino R, Moretti R, Limonta P, Valdagni R, Daidone MG, Zaffaroni N (2009) miR-205 exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cepsilon. Cancer Res 69(6):2287–2295. doi:10.1158/0008-5472.CAN-08-2894
Nishikawa R, Goto Y, Kurozumi A, Matsushita R, Enokida H, Kojima S, Naya Y, Nakagawa M, Ichikawa T, Seki N (2015) MicroRNA-205 inhibits cancer cell migration and invasion via modulation of centromere protein F regulating pathways in prostate cancer. Int J Urol 22(9):867–877. doi:10.1111/iju.12829
Ru P, Steele R, Newhall P, Phillips NJ, Toth K, Ray RB (2012) miRNA-29b suppresses prostate cancer metastasis by regulating epithelial–mesenchymal transition signaling. Mol Cancer Ther 11(5):1166–1173. doi:10.1158/1535-7163.MCT-12-0100
Liu YN, Yin JJ, Abou-Kheir W, Hynes PG, Casey OM, Fang L, Yi M, Stephens RM, Seng V, Sheppard-Tillman H, Martin P, Kelly K (2013) MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene 32(3):296–306. doi:10.1038/onc.2012.58
Viticchiè G, Lena AM, Latina A, Formosa A, Gregersen LH, Lund AH, Bernardini S, Mauriello A, Miano R, Spagnoli LG, Knight RA, Candi E, Melino G (2011) MiR-203 controls proliferation, migration and invasive potential of prostate cancer cell lines. Cell Cycle 10(7):1121–1131
Qu Y, Li WC, Hellem MR, Rostad K, Popa M, McCormack E, Oyan AM, Kalland KH, Ke XS (2013) MiR-182 and miR-203 induce mesenchymal to epithelial transition and self-sufficiency of growth signals via repressing SNAI2 in prostate cells. Int J Cancer 133(3):544–555. doi:10.1002/ijc.28056
Majid S, Dar AA, Saini S, Shahryari V, Arora S, Zaman MS, Chang I, Yamamura S, Tanaka Y, Chiyomaru T, Deng G, Dahiya R (2013) miRNA-34b inhibits prostate cancer through demethylation, active chromatin modifications, and AKT pathways. Clin Cancer Res 19(1):73–84. doi:10.1158/1078-0432.CCR-12-2952
Ren D, Wang M, Guo W, Huang S, Wang Z, Zhao X, Du H, Song L, Peng X (2014) Double-negative feedback loop between ZEB2 and miR-145 regulates epithelial–mesenchymal transition and stem cell properties in prostate cancer cells. Cell Tissue Res 358(3):763–778. doi:10.1007/s00441-014-2001-y
Wang L, Song G, Tan W, Qi M, Zhang L, Chan J, Yu J, Han J, Han B (2015) miR-573 inhibits prostate cancer metastasis by regulating epithelial–mesenchymal transition. Oncotarget 6(34):35978–35990. doi:10.18632/oncotarget.5427
Kurozumi A, Goto Y, Matsushita R, Fukumoto I, Kato M, Nishikawa R, Sakamoto S, Enokida H, Nakagawa M, Ichikawa T, Seki N (2015) Tumor-suppressive microRNA-223 inhibits cancer cell migration and invasion by targeting ITGA3/ITGB1 signaling in prostate cancer. Cancer Sci. doi:10.1111/cas.12842
Shirakihara T, Kawasaki T, Fukagawa A, Semba K, Sakai R, Miyazono K, Miyazawa K, Saitoh M (2013) Identification of integrin α3 as a molecular marker of cells undergoing epithelial–mesenchymal transition and of cancer cells with aggressive phenotypes. Cancer Sci 104(9):1189–1197. doi:10.1111/cas.12220
Yang J, Hou Y, Zhou M, Wen S, Zhou J, Xu L, Tang X, Du YE, Hu P, Liu M (2015) Twist induces epithelial–mesenchymal transition and cell motility in breast cancer via ITGB1-FAK/ILK signaling axis and its associated downstream network. Int J Biochem Cell Biol 71:62–71. doi:10.1016/j.biocel.2015.12.004
Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9(9):690–701. doi:10.1038/nrm2476
Lin SL, Chiang A, Chang D, Ying SY (2008) Loss of mir-146a function in hormone-refractory prostate cancer. RNA 14(3):417–424. doi:10.1261/rna.874808
Moser B, Wolf M, Walz A, Loetscher P (2004) Chemokines: multiple levels of leukocyte migration control. Trends Immunol 25(2):75–84
Dillenburg-Pilla P, Patel V, Mikelis CM, Zárate-Bladés CR, Doçi CL, Amornphimoltham P, Wang Z, Martin D, Leelahavanichkul K, Dorsam RT, Masedunskas A, Weigert R, Molinolo AA, Gutkind JS (2015) SDF-1/CXCL12 induces directional cell migration and spontaneous metastasis via a CXCR4/Gαi/mTORC1 axis. FASEB J 29(3):1056–1068. doi:10.1096/fj.14-260083
Ma N, Pang H, Shen W, Zhang F, Cui Z, Wang J, Wang J, Liu L, Zhang H (2015) Downregulation of CXCR4 by SDF-KDEL in SBC-5 cells inhibits their migration in vitro and organ metastasis in vivo. Int J Mol Med 35(2):425–432. doi:10.3892/ijmm.2014.2033
Shen PF, Chen XQ, Liao YC, Chen N, Zhou Q, Wei Q, Li X, Wang J, Zeng H (2014) MicroRNA-494-3p targets CXCR4 to suppress the proliferation, invasion, and migration of prostate cancer. Prostate 74(7):756–767. doi:10.1002/pros.22795
Kohlhapp FJ, Mitra AK, Lengyel E, Peter ME (2015) MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 34(48):5857–5868. doi:10.1038/onc.2015.89
Xu B, Wang N, Wang X, Tong N, Shao N, Tao J, Li P, Niu X, Feng N, Zhang L, Hua L, Wang Z, Chen M (2012) MiR-146a suppresses tumor growth and progression by targeting EGFR pathway and in a p-ERK-dependent manner in castration-resistant prostate cancer. Prostate 72(11):1171–1178. doi:10.1002/pros.22466
Chen Q, Zhao X, Zhang H, Yuan H, Zhu M, Sun Q, Lai X, Wang Y, Huang J, Yan J, Yu J (2015) MiR-130b suppresses prostate cancer metastasis through down-regulation of MMP2. Mol Carcinog 54(11):1292–1300. doi:10.1002/mc.22204
Reis ST, Pontes-Junior J, Antunes AA, Dall’Oglio MF, Dip N, Passerotti CC, Rossini GA, Morais DR, Nesrallah AJ, Piantino C, Srougi M, Leite KR (2012) miR-21 may acts as an oncomir by targeting RECK, a matrix metalloproteinase regulator, in prostate cancer. BMC Urol 12:14. doi:10.1186/1471-2490-12-14
Zoni E, van der Horst G, van de Merbel AF, Chen L, Rane JK, Pelger RC, Collins AT, Visakorpi T, Snaar-Jagalska BE, Maitland NJ, van der Pluijm G (2015) miR-25 modulates invasiveness and dissemination of human prostate cancer cells via regulation of αv- and α6-integrin expression. Cancer Res 75(11):2326–2336. doi:10.1158/0008-5472.CAN-14-2155
LeBleu VS, Macdonald B, Kalluri R (2007) Structure and function of basement membranes. Exp Biol Med (Maywood) 232(9):1121–1129
Gandellini P, Profumo V, Casamichele A, Fenderico N, Borrelli S, Petrovich G, Santilli G, Callari M, Colecchia M, Pozzi S, De Cesare M, Folini M, Valdagni R, Mantovani R, Zaffaroni N (2012) miR-205 regulates basement membrane deposition in human prostate: implications for cancer development. Cell Death Differ 19(11):1750–1760. doi:10.1038/cdd.2012.56
Cirri P, Chiarugi P (2012) Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastasis Rev 31(1–2):195–208. doi:10.1007/s10555-011-9340-x
Musumeci M, Coppola V, Addario A, Patrizii M, Maugeri-Saccà M, Memeo L, Colarossi C, Francescangeli F, Biffoni M, Collura D, Giacobbe A, D’Urso L, Falchi M, Venneri MA, Muto G, De Maria R, Bonci D (2011) Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene 30(41):4231–4242. doi:10.1038/onc.2011.140
Gandellini P, Giannoni E, Casamichele A, Taddei ML, Callari M, Piovan C, Valdagni R, Pierotti MA, Zaffaroni N, Chiarugi P (2014) miR-205 hinders the malignant interplay between prostate cancer cells and associated fibroblasts. Antioxid Redox Signal 20(7):1045–1059. doi:10.1089/ars.2013.5292
Taddei ML, Cavallini L, Comito G, Giannoni E, Folini M, Marini A, Gandellini P, Morandi A, Pintus G, Raspollini MR, Zaffaroni N, Chiarugi P (2014) Senescent stroma promotes prostate cancer progression: the role of miR-210. Mol Oncol 8(8):1729–1746. doi:10.1016/j.molonc.2014.07.009
Josson S, Gururajan M, Sung SY, Hu P, Shao C, Zhau HE, Liu C, Lichterman J, Duan P, Li Q, Rogatko A, Posadas EM, Haga CL, Chung LW (2015) Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 34(21):2690–2699. doi:10.1038/onc.2014.212
Doldi V, Callari M, Giannoni E, D’Aiuto F, Maffezzini M, Valdagni R, Chiarugi P, Gandellini P, Zaffaroni N (2015) Integrated gene and miRNA expression analysis of prostate cancer associated fibroblasts supports a prominent role for interleukin-6 in fibroblast activation. Oncotarget 6(31):31441–31460. doi:10.18632/oncotarget.5056
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3(6):401–410
Liu LZ, Li C, Chen Q, Jing Y, Carpenter R, Jiang Y, Kung HF, Lai L, Jiang BH (2011) MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1α expression. PLoS One 6(4):e19139. doi:10.1371/journal.pone.0019139
Taddei ML, Giannoni E, Fiaschi T, Chiarugi P (2012) Anoikis: an emerging hallmark in health and diseases. J Pathol 226(2):380–393. doi:10.1002/path.3000
Formosa A, Lena AM, Markert EK, Cortelli S, Miano R, Mauriello A, Croce N, Vandesompele J, Mestdagh P, Finazzi-Agrò E, Levine AJ, Melino G, Bernardini S, Candi E (2013) DNA methylation silences miR-132 in prostate cancer. Oncogene 32(1):127–134. doi:10.1038/onc.2012.14
Lin ZY, Huang YQ, Zhang YQ, Han ZD, He HC, Ling XH, Fu X, Dai QS, Cai C, Chen JH, Liang YX, Jiang FN, Zhong WD, Wang F, Wu CL (2014) MicroRNA-224 inhibits progression of human prostate cancer by downregulating TRIB1. Int J Cancer 135(3):541–550. doi:10.1002/ijc.28707
Mashima T, Soma-Nagae T, Migita T, Kinoshita R, Iwamoto A, Yuasa T, Yonese J, Ishikawa Y, Seimiya H (2014) TRIB1 supports prostate tumorigenesis and tumor-propagating cell survival by regulation of endoplasmic reticulum chaperone expression. Cancer Res 74(17):4888–4897. doi:10.1158/0008-5472.CAN-13-3718
Takeshita F, Patrawala L, Osaki M, Takahashi RU, Yamamoto Y, Kosaka N, Kawamata M, Kelnar K, Bader AG, Brown D, Ochiya T (2010) Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther 18(1):181–187. doi:10.1038/mt.2009.207
Peng X, Guo W, Liu T, Wang X, Tu X, Xiong D, Chen S, Lai Y, Du H, Chen G, Liu G, Tang Y, Huang S, Zou X (2011) Identification of miRs-143 and -145 that is associated with bone metastasis of prostate cancer and involved in the regulation of EMT. PLoS One 6(5):e20341. doi:10.1371/journal.pone.0020341
Siu MK, Abou-Kheir W, Yin JJ, Chang YS, Barrett B, Suau F, Casey O, Chen WY, Fang L, Hynes P, Hsieh YY, Liu YN, Huang J, Kelly K (2014) Loss of EGFR signaling regulated miR-203 promotes prostate cancer bone metastasis and tyrosine kinase inhibitors resistance. Oncotarget 5(11):3770–3784
Chen WY, Liu SY, Chang YS, Yin JJ, Yeh HL, Mouhieddine TH, Hadadeh O, Abou-Kheir W, Liu YN (2015) MicroRNA-34a regulates WNT/TCF7 signaling and inhibits bone metastasis in Ras-activated prostate cancer. Oncotarget 6(1):441–457
Liu YN, Yin J, Barrett B, Sheppard-Tillman H, Li D, Casey OM, Fang L, Hynes PG, Ameri AH, Kelly K (2015) Loss of androgen-regulated microRNA 1 activates SRC and promotes prostate cancer bone metastasis. Mol Cell Biol 35(11):1940–1951. doi:10.1128/MCB.00008-15
Chang YS, Chen WY, Yin JJ, Sheppard-Tillman H, Huang J, Liu YN (2015) EGF receptor promotes prostate cancer bone metastasis by downregulating miR-1 and activating TWIST1. Cancer Res 75(15):3077–3086. doi:10.1158/0008-5472.CAN-14-3380
Karatas OF, Guzel E, Suer I, Ekici ID, Caskurlu T, Creighton CJ, Ittmann M, Ozen M (2014) miR-1 and miR-133b are differentially expressed in patients with recurrent prostate cancer. PLoS One 9(6):e98675. doi:10.1371/journal.pone.0098675
Siu MK, Tsai YC, Chang YS, Yin JJ, Suau F, Chen WY, Liu YN (2015) Transforming growth factor-β promotes prostate bone metastasis through induction of microRNA-96 and activation of the mTOR pathway. Oncogene 34(36):4767–4776. doi:10.1038/onc.2014.414
Gururajan M, Josson S, Chu GC, Lu CL, Lu YT, Haga CL, Zhau HE, Liu C, Lichterman J, Duan P, Posadas EM, Chung LW (2014) miR-154* and miR-379 in the DLK1-DIO3 microRNA mega-cluster regulate epithelial to mesenchymal transition and bone metastasis of prostate cancer. Clin Cancer Res 20(24):6559–6569. doi:10.1158/1078-0432.CCR-14-1784
Josson S, Gururajan M, Hu P, Shao C, Chu GY, Zhau HE, Liu C, Lao K, Lu CL, Lu YT, Lichterman J, Nandana S, Li Q, Rogatko A, Berel D, Posadas EM, Fazli L, Sareen D, Chung LW (2014) miR-409-3p/-5p promotes tumorigenesis, epithelial-to-mesenchymal transition, and bone metastasis of human prostate cancer. Clin Cancer Res 20(17):4636–4646. doi:10.1158/1078-0432.CCR-14-0305
Bonci D, Coppola V, Patrizii M, Addario A, Cannistraci A, Francescangeli F, Pecci R, Muto G, Collura D, Bedini R, Zeuner A, Valtieri M, Sentinelli S, Benassi MS, Gallucci M, Carlini P, Piccolo S, De Maria R (2015) A microRNA code for prostate cancer metastasis. Oncogene. doi:10.1038/onc.2015.176
Hagman Z, Haflidadóttir BS, Ceder JA, Larne O, Bjartell A, Lilja H, Edsjö A, Ceder Y (2013) miR-205 negatively regulates the androgen receptor and is associated with adverse outcome of prostate cancer patients. Br J Cancer 108(8):1668–1676. doi:10.1038/bjc.2013.131
Kalogirou C, Spahn M, Krebs M, Joniau S, Lerut E, Burger M, Scholz CJ, Kneitz S, Riedmiller H, Kneitz B (2013) MiR-205 is progressively down-regulated in lymph node metastasis but fails as a prognostic biomarker in high-risk prostate cancer. Int J Mol Sci 14(11):21414–21434. doi:10.3390/ijms141121414
Mavridis K, Stravodimos K, Scorilas A (2013) Downregulation and prognostic performance of microRNA 224 expression in prostate cancer. Clin Chem 9(1):261–269. doi:10.1373/clinchem.2012.191502
Wan Y, Zeng ZC, Xi M, Wan S, Hua W, Liu YL, Zhou YL, Luo HW, Jiang FN, Zhong WD (2015) Dysregulated microRNA-224/apelin axis associated with aggressive progression and poor prognosis in patients with prostate cancer. Hum Pathol 46(2):295–303. doi:10.1016/j.humpath.2014.10.027
Li T, Li RS, Li YH, Zhong S, Chen YY, Zhang CM, Hu MM, Shen ZJ (2012) miR-21 as an independent biochemical recurrence predictor and potential therapeutic target for prostate cancer. J Urol 187(4):1466–1472. doi:10.1016/j.juro.2011.11.082
Sun X, Liu Z, Yang Z, Xiao L, Wang F, He Y, Su P, Wang J, Jing B (2013) Association of microRNA-126 expression with clinicopathological features and the risk of biochemical recurrence in prostate cancer patients undergoing radical prostatectomy. Diagn Pathol 8:208. doi:10.1186/1746-1596-8-208
Forno I, Ferrero S, Russo MV, Gazzano G, Giangiobbe S, Montanari E, Del Nero A, Rocco B, Albo G, Languino LR, Altieri DC, Vaira V, Bosari S (2015) Deregulation of MiR-34b/Sox2 predicts prostate cancer progression. PLoS One 10(6):e0130060. doi:10.1371/journal.pone.0130060
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18(1):11–22. doi:10.1016/j.ccr.2010.05.026
van Rooij E, Kauppinen S (2014) Development of microRNA therapeutics is coming of age. EMBO Mol Med 6(7):851–864. doi:10.15252/emmm.201100899
Wen D, Danquah M, Chaudhary AK, Mahato RI (2015) Small molecules targeting microRNA for cancer therapy: promises and obstacles. J Control Release 219:237–247. doi:10.1016/j.jconrel.2015.08.011
Liu YP (1809) Berkhout B (2011) miRNA cassettes in viral vectors: problems and solutions. Biochim Biophys Acta 11–12:732–745. doi:10.1016/j.bbagrm.2011.05.014
Wang Z (2011) The principles of MiRNA-masking antisense oligonucleotides technology. Methods Mol Biol 676:43–49. doi:10.1007/978-1-60761-863-8_3
Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR (2013) Treatment of HCV infection by targeting microRNA. N Engl J Med 368(18):1685–1694. doi:10.1056/NEJMoa1209026
Acknowledgments
The work in the authors’ laboratory was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC), Projects #11542 (PG) and #12162 (NZ), and the I. Monzino Foundation (NZ).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Doldi, V., Pennati, M., Forte, B. et al. Dissecting the role of microRNAs in prostate cancer metastasis: implications for the design of novel therapeutic approaches. Cell. Mol. Life Sci. 73, 2531–2542 (2016). https://doi.org/10.1007/s00018-016-2176-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2176-3