
Abstract
Cancer stem cells (CSCs), are thought to be at the origin of tumor development and resistance to therapies. Thus, a better understanding of the molecular mechanisms involved in the control of CSC stemness is essential to the design of more effective therapies for cancer patients. Cancer cell stemness and the subsequent expansion of CSCs are regulated by micro-environmental signals including neurotrophins. Over the years, the roles of neurotrophins in tumor development have been well established and regularly reviewed. Especially, nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) are reported to stimulate tumor cell proliferation, survival, migration and/or invasion, and favors tumor angiogenesis. More recently, neurotrophins have been reported to regulate CSCs. This review briefly presents neurotrophins and their receptors, summarizes their roles in different cancers, and discusses the emerging evidence of neurotrophins-induced enrichment of CSCs as well as the involved signaling pathways.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer stem cells (CSCs), also known as tumor initiating cells, represent a rare population of tumor cells with the biological characteristics that are similar to normal stem cells: self-renewal and differentiation. CSCs are thought to be the fundamental driving force of tumor initiation and metastasis. They are resistant to conventional therapies and are proposed to be responsible for recurrence. Thus, a better understanding of the molecular mechanisms involved in the control of cancer cell stemness is essential to the design of more effective therapies for cancer patients. The stemness of cancer cells and their subsequent expansion are regulated by micro-environmental signals as well as genetic and epigenetic alterations. Neurotrophins are a family of structurally conserved growth factors including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3) and neurotrophin 4/5 (NT-4/5). Neurotrophins and their receptors are expressed by both tumor and microenvironmental cells, and are involved in the development of various tumors. Recently, neurotrophins have been demonstrated to enlarge CSC pool by influencing the behaviors of bo th CSCs and non-CSCs. Here, following an overview of cancer stem cell model, we briefly present neurotrophins and summarize their well-known roles in cancers. We then highlight the emerging evidence of neurotrophins-induced enrichment of CSCs and discuss the involved molecular mechanisms.
Cancer stem cell model
Two mutually non-exclusive models have been proposed to explain tumor development and intratumoral heterogeneity: the stochastic model and the cancer stem cell model. The stochastic model postulates that every cell within a tumor is equally likely to be the cell of origin. According to this model, cancer is raised from cells accumulating genetic mutations. The cancer stem cell model posits that cancers arise from, and are sustained by a unique subpopulation of cells that possess stem-like properties, the so-called tumor-initiating cells or cancer stem cells (CSCs). CSCs have the capacities of unrestricted self-renewal and differentiation, giving rise to progenitors and more differentiated cells with limited proliferation and tumorigenic potential. The cancer stem cell model for solid cancer was first introduced in breast cancer by the group of Clarke. This group identified a rare subpopulation having the phenotype of CD44+/CD24−/low/Lineage− as putative CSCs-enriched population. Indeed, as few as 100 CD44+/CD24−/low/Lineage− cells were sufficient to recapitulate the tumor, when injected into immunodeficient SCID mice [1]. Based upon serial transplant xenograft/limiting dilution assays and the use of tumor-specific CSC markers, studies in other solid tumors such as brain, ovarian and colon cancer constantly verified that CSCs exist at low frequencies within a tumor and are able to recapitulate some of the heterogeneity of the original tumors when injected into immunodeficient mice [2–4].
The importance of CSCs in tumor development was further reinforced by lineage tracing experiments in mouse models, which permit the follow-up of individual cells at different stages of tumor progression. In these experiments, CSCs are clearly shown to be at the origin of tumor formation, chemotherapy resistance and relapse of several cancers including intestinal adenoma, glioblastoma and squamous skin tumor [5–7]. The clinical relevance of CSCs is illustrated by findings showing that a stem cell-like gene expression signature is predictive of patient outcome in human leukemia, breast cancer, glioblastoma, and ovarian cancer [8–11].
Mounting evidences indicate that the stochastic model and the cancer stem cell model are not mutually exclusive and can be unified by cancer cell plasticity. CSCs within an established tumor are found to be heterogeneous [12–14]. It is hypothesized cancer may originate from the oncogenic activation of an original CSC, which can give rise to other CSCs that accumulate genetic and epigenetic modifications necessary for tumor initiation and progression. Each CSC subclone, derived from the initial CSC, has the capabilities of self-renewal and differentiating to intermediate transit-amplifying progenitors and more differentiated cells. A subset of these progenitors would be capable of bidirectional conversion between non-CSC and CSC states in response to microenvironment stimuli, including cytokines, chemokines and growth factors [15].
Neurotrophins and their receptors
Neurotrophins are a family of structurally conserved growth factors including NGF, BDNF, NT-3 and NT-4/5. Neurotrophin transcripts are first translated into pre-proneurotrophins. After signal peptide elimination, the proneurotrophins are cleaved at a dibasic amino acid site by intracellular proteases such as furin and proconvertases, or by extracellular proteases such as plasmin, MMP-3 and MMP-7, hence generating mature neurotrophins [16, 17]. Although neurotrophins have been initially studied for their role in nervous system development, they exert various effects on non-neuronal cells including cancer cells from different tissues.
Neurotrophins exert their biological functions mainly via two types of cell membrane receptors: the Trk tyrosine kinase receptors and the common neurotrophin receptor P75NTR. The tyrosine kinase receptors include TrkA, TrkB and TrkC, each of which exhibiting specificity for the different neurotrophins. TrkA preferentially binds to NGF, TrkB preferentially binds to BDNF and NT-4/5, TrkC preferentially binds to NT-3. Binding of Trks by their preferred neurotrophins activates their kinase domain to trigger downstream signaling pathways including MAPK, PI3 K and PLCγ-PKC [18]. Moreover, Trks can be activated by a number of receptors including steroid receptors, G-protein coupled receptors (GPCR) and CD44 [18, 19]. Other receptor tyrosine kinases such as c-MET can also transactivate Trks in the absence of neurotrophins [20]. On the other hand, P75NTR, binds to neurotrophins and proneurotropins with similar affinity. P75NTR does not have intrinsic enzymatic activity, and it owes its signaling to the recruitment of intracellular binding proteins or through regulated proteolysis signaling [21]. Although P75NTR has the ability to signal alone, many of its functions rely on its interaction with Trks and other co-receptors. For example, the formation of a P75NTR/Trk complex increases the affinity of each neurotrophin for its Trk receptor, most likely by the induction of conformational changes in its intracellular and extracellular domains [22]. More recently, a direct interaction between P75NTR and TrkA has been demonstrated, even in the absence of NGF [23]. P75NTR participates also in several signaling platforms by interacting with co-receptors such as sortilin, Nogo receptor and LINGO-1. Interactions with co-receptors seem to be dependent on P75NTR cellular localization, its post-translational modifications and the state of cellular differentiation [24].
Given the diverse (co-)receptors described above, the overall outcome of neurotrophin signaling is the consequence of the integration of distinct receptor signaling networks. This leads to divergent cellular responses including cell survival, apoptosis, proliferation, differentiation, migration, and invasion, depending on cell type and cell context.
Cancer promoting effects of neurotrophins
Over the years, accumulating data have shown the expression of neurotrophins and their receptors in different tumors. In most cases, neurotrophins have been shown to favor tumor development and progression. In this part, we will sum up major findings concerning NGF/TrkA, BDNF/TrkB, and neurotrophins/p75NTR signaling axes.
NGF/TrkA axis
Overexpression of NGF and/or TrkA has been correlated with perineural invasion in several cancers including pancreatic cancer [25, 26], oral squamous cell carcinoma [27] and adenoid cystic carcinoma [28]. The active form of TrkA (phospho-TrkA) has been associated with poor patient outcome in ovarian and breast carcinomas [29, 30] suggesting the involvement of the NGF/TrkA axis in tumor progression. NGF exhibits protumoral effects in several types of tumors including pancreatic, ovarian and breast cancers [29, 31, 32]. We have shown that NGF is overexpressed in the majority of breast cancers, and that NGF/TrkA inhibition reduces tumor growth in xenograft mouse model [32]. In breast cancer cell lines, ectopic overexpression of TrkA enhances anoikis resistance, invasion and metastasis [33, 34]. In addition to its involvement in promoting breast cancer cell proliferation, survival, migration and invasion, NGF promotes also angiogenesis by inducing the expression of proangiogenic factors such as vascular endothelial growth factor (VEGF) and transforming growth factor beta (TGFβ) [35, 36].
By contrast to most of the solid tumors, in neuroblastoma, NGF/TrkA may exert anti- or protumoral effects, depending on the expression of TrkA isoforms TrkAI or TrkAIII. Overexpression of TrkAI (exon 9 excluded) has been correlated with better prognosis in neuroblastoma [37–39]. NGF inhibits cell growth and induces terminal differentiation in neuroblastoma cell lines expressing high levels of TrkAI [40]. In contrast, TrkAIII (exons 6, 7 and 9 excluded) was described to be associated with neuroblastoma of poor prognosis [41, 42]. TrkAIII lacks the extracellular D4 Ig-like domain and related N-glycosylation sites required for cell surface localization [43]. TrkAIII is retained within the intracellular membrane, where it exerts protumoral activity through different mechanisms independent of NGF. These include constitutive PI3K/Akt/NF-kB signaling [41] and interaction with the centrosome, promoting centrosome amplification and genetic instability [44].
BDNF/TrkB axis
Mounting data show that the BDNF/TrkB axis is often associated with metastatic potential and poor prognosis in different cancers including neuroblastoma and cancers of non-neuronal origin such as head and neck, lung, breast, stomach and colon cancers [45–52]. For example, elevated levels of TrkB and BDNF predict a poor prognosis in neuroblastoma [50] and Wilm’s tumor [51]. TrkB-positive pancreatic tumors develop more rapidly liver metastasis than TrkB-negative tumors [52]. Increased BDNF expression at the invasive front of primary tumors is significantly correlated with poor prognosis in gastric cancer [48]. The co-expression of BDNF and TrkB mRNA is associated with liver and peritoneal metastasis in colorectal cancer [49]. Where studied, BDNF promotes cell proliferation, migration, invasion, and inhibits anoikis. Blockade of BDNF/TrkB signaling in different cancer cell lines significantly decreases their proliferative, migratory and metastatic ability in vitro and in vivo [46, 53]. Apart from direct action on tumor cells, BDNF exhibits also strong angiogenic property. BDNF increases the expression of HIF-1α which in turn up-regulates VEGF [54] and TrkB expression [55, 56]. Moreover, BDNF stimulates neovascularization via recruitment of TrkB-expressing endothelial progenitor cells [57], raising the possibility that any tumor cells secreting BDNF may be also able to induce angiogenesis through similar mechanisms.
Neurotrophins/P75NTR
P75NTR, the common receptor of neurotrophins and proneurotrophins, has been suggested to act as a tumor suppressor in gastric, bladder and prostate cancers by blocking cell cycle progression and inducing apoptosis [58–60]. In the majority of other tumors including melanoma, glioma, breast cancer and squamous cell carcinoma, P75NTR is proposed to favor tumor development [34, 47, 61, 62]. For example, NGF/P75NTR signaling is known to be implicated in melanoma cell proliferation and migration [63] and has been associated with increased brain metastases [64–66]. P75NTR expression is observed in high grade glioma [62] and is associated with poor prognoses and a risk of local recurrence of oral cancer [67]. P75NTR is correlated with perineural invasion of skin cancers [68]. More recently, P75NTR has been reported as a marker of CSCs in melanoma, esophageal and hypopharyngeal carcinomas [69].
The protumoral effects (i.e. prognostic value of expression and/or biological effects) of neurotrophins and their associated receptors in different solid tumors are summarized in Table 1.
Evidence for the role of neurotrophins in cancer stem cells
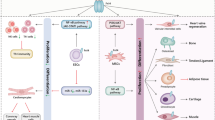
Neurotrophins have been reported to regulate CSCs in several types of cancers such as glioma, neuroblastoma, head and neck squamous cell carcinoma, melanoma and breast cancer. Neurotrophins can induce enrichment of CSCs through two major mechanisms: direct action on CSCs or indirect action through epithelial-to-mesenchymal transition (EMT) (Fig. 1).

Mechanisms of neurotrophins-induced enrichment of cancer stem cells (CSCs). Cancer cell plasticity designates the capacity of cancer cells to interconvert between differentiated and stem-like states, through a continuum of cell fate specifications. This phenotype shifting is modulated by microenvironmental signals and cellular interactions arising in the tumor niche. Among numerous factors from the microenvironment, neurotrophins are found to regulate cancer cell plasticity by acting on different types of cells. Particularly, neurotrophins can enlarge CSC pool: (1) by stimulating CSC self-renewal; (2) by inducing differentiated epithelial cells to epithelial-mesenchymal transition (EMT) and the conversion of mesenchymal like cancer cells (MLCCs) to CSCs (central frame). First, (pro)neurotrophin are found to increase CSC renewal in glioma and breast cancer (left frame). In glioma cells, BDNF, NT3 stimulate CSC proliferation through tyrosine kinase receptors TrkB-, TrkC-dependent activation of ERK and Akt pathways, while NGF stimulates CSC proliferation through the cleavage of P75NTR that gives rise to the soluble P75 intracellular domain (P75NTR-ICD). P75NTR-ICD is able to activate Akt pathway to stimulate CSC proliferation. In breast cancer cells, NGF induces P75NTR-mediated expression of the pluripotency transcription factors SOX2, NANOG and MYC. ProNGF increases also SOX2 expression in a p75NTR-independent manner. Moreover, neurotrophins are also described to induce EMT-linked enrichment of CSCs in lung and breast cancers (right frame). In lung carcinoma, BDNF and TrkB increase the expression of the master EMT transcription factors SLUG, TWIST and SNAIL. In breast cancer cells, NGF enhances p75NTR-dependent expression of SLUG. Thus, through the common transcription factors, neurotrophins activate signaling networks, allowing for the reprogramming of differentiated epithelial cancer cells to CSCs in a stepwise manner
In glioma, several cell lines named brain tumor initiating cells (BTICs) have been established by culturing cells, derived from patient tumors, on laminin-coated flasks in the presence of EGF and FGF2. The established cell lines exhibit cancer stem cell properties, as they express neural stem cell markers and are able to form neurospheres in vitro and tumors in xenograft mouse model. Forsyth et al. reported that neurotrophins (NGF, BDNF and NT-3) and their receptors (TrkA, TrkB, TrkC and P75NTR) are detected in several lines of BTICs [75]. Moreover, NGF, BDNF and NT3 are able to stimulate the proliferation of BTICs. The authors further showed that NGF stimulates BTIC proliferation through P75NTR cleavage. P75NTR cleavage is a highly regulated two-step process: P75NTR is firstly cleaved at the extracellular domain by the metalloproteases ADAM17 to generate a membrane-bound C-terminal fragment (P75NTR-CTF); the P75NTR-CTF is subsequently cleaved within the transmembrane domain by γ-Secretase and gives rise to the soluble P75 intracellular domain (P75NTR-ICD). Interestingly, ectopic expression of P75NTR-ICD is sufficient by itself to stimulate BTIC cell invasion and proliferation [73, 75]. Under physiological conditions, P75NTR cleavage implies the activation of tyrosine kinase receptors Trks, as inhibition of Trks by the pharmacological inhibitor K252a blocks accumulation of P75 fragments and prevents NGF-stimulated BTIC proliferation. Moreover, P75NTR-ICD is able to induce Akt activation in BTICs [75]. It is already reported that P75NTR cleavage is needed for Akt activation and neurotrophins-induced survival in PC12 and neurons [95–97]. Furthermore, Akt pathway is required for brain cancer stem cell growth [98, 99]. Thus P75NTR-ICD-induced Akt activation could be the key mechanism of neurotrophins-stimulated BTIC proliferation. How Akt is activated by P75NTR-ICD in the context of BTICs is still to be determined. By using another set of patients-derived BTICs, Lawn et al. reported that BDNF, NT3, TrkB, TrkC and P75NTR are frequently expressed [72]. In these cells, BDNF and NT3 promote BITC growth through the activation of tyrosine kinase receptors TrkB and TrkC and the downstream activation of ERK and Akt pathways. Taken together, it seems that neurotrophins and their receptors are widely expressed in BITCs to promote proliferation, invasion and survival through different pathways including ERK and Akt activation as well as P75NTR cleavage (Fig. 1, left frame). Although more detailed and complete activation of these pathways in the context of CSCs remains to be clarified, it is known that neurotrophin-dependent MAP kinase activation in neurons is mediated by SoS-Ras-MAP kinase and Frs2/ARMS-Crk pathways. Moreover, Akt activation in these models is mediated by SoS-Ras-PI3K [100]. MAP kinase and Akt activation regulates RSK kinase, CREB phosphorylation, and NFκB activation, which promote transcription of genes necessary for neuronal survival [101–103].
In neuroblastoma, works from the group of AR Mackay showed that in the SH-SY5Y neuroblastoma cell line, the NGF non responsive TrkAIII variant promotes the formation of larger spheres and the expression of stemness markers including Nanog, Nestin, SOX2 and CD117 through its tyrosine activity [104]. These data are consistent with previous findings demonstrating that expression TrkAIII in SH-SY5Y cells can induce an undifferentiated stem cell-like phenotype that exhibits increased tumorigenic and metastatic behavior [41]. However, whether this occurs in primary tumors remains to be determined.
In head and neck squamous cell carcinomas, it has been recently reported that P75NTR is a functional and targetable marker of CSCs. In these cells, loss of P75NTR inhibits cell proliferation and tumor formation. Moreover, targeting of P75NTR with a monoclonal antibody reduces NGF-induced Erk activation in head and neck squamous cell carcinoma [105]. In melanoma cells, knock down of P75NTR induces a change in morphology from spindled-shaped to epithelial-like cells with loss of expression of stemness markers including SOX10 and SOX2. Moreover, cells knocked-down for p75NTR did not form any tumors in xenograft mouse model [83].
In breast cancer, we have shown that NGF and proNGF enrich for a CSC subpopulation by regulating the dynamics between quiescence and proliferation and by increasing the frequency of symmetric divisions of CSCs [106]. NGF and proNGF lower the proportion of cells undergoing asymmetric division and decrease the expression of NUMB, a cell fate determinant involved in the asymmetric division of stem cells [107]. We observed an enrichment of P75NTR expressing cells in the ALDH1+ CSC population. In addition, P75NTR siRNA silencing abolishes the NGF/proNGF-enhanced sphere-forming capacity, indicating that NGF/proNGF-induced sphere formation is mediated by P75NTR. However, it seems that NGF and proNGF imply different molecular mechanisms to increase the CSC pool. For example, NGF is able to induce P75NTR-mediated expression of pluripotency transcription factors including sox2, nanog, and myc, which are involved the maintenance of stemness. In contrast, proNGF increases the expression of sox2 in a P75NTR-independent manner (Fig. 1, left frame). This may be explained by the different involved receptors, as NGF and proNGF can bind to both common and specific receptors. NGF exerts its biological effects via P75NTR and TrkA receptors, while proNGF, at least in neuronal cells, induces its effects through complexes often formed with P75NTR and sortilin [108] and less frequently with TrkA and sortilin [109]. In breast cancer cells, we showed that the pro-invasive effects of proNGF are mediated by TrkA and sortilin but not by P75NTR [110]. On the other hand, NGF binding to TrkA permits the recruitment of membrane CD44, which in turn activates Rho GTPase pathways to increase the aggressive phenotype of breast cancer cells [19]. This is particularly interesting, as CD44 is increasingly shown to be involved in the maintenance of stemness and survival of CSCs [111, 112]. Indeed, CD44 functions as a signaling platform by interacting with both extracellular matrix components (i.e. hyaluronan) and several types of membrane receptors including TrkA, c-MET, EGFR, PDGFR [113]. Clearly, NGF and proNGF signaling pathways through different (co-)receptors remain to be studied in the context of breast CSCs.
Yin et al. have shown the involvement of BDNF/TrkB in sustaining CSCs of recurrent triple-negative breast cancers (TNBC) [90]. TNBC express neither estrogen receptor, nor progesterone receptor and do not overexpress human epidermal growth factor receptor 2 (HER2). TNBC are clinically characterized as more aggressive with a poorer overall prognosis due to high recurrence rate. By developing an elegant post-chemotherapy relapse xenograft mouse model of TNBC, using cancer cells freshly isolated from patients with primary TNBC, Yin B et al. demonstrated that differentiated recurrent TNBC cells after paclitaxel treatment express and secrete BDNF, following activation of the JNK-CREB pathway [90]. BDNF acts then in a paracrine manner on ALDH1+/TrkB+ cells to induce the expression of KLF4, a zinc finger-type transcription factor of Krüppel-like factor family, already known to be involved in cell reprogramming and the maintenance of stemness [114, 115]. The BDNF-induced expression of KLF4 in ALDH1+/TrkB+ is found to be necessary for maintaining the stemness of ALDH1+/TrkB+ TNBC stem cells. Thus, differentiated recurrent TNBC cells constitute a specific microenvironment by providing BDNF to support the self-renewal capacity of CSCs. The subpopulation of ALDH1+ CSCs expressing TrkB is more resistant to chemotherapeutic agents both in vitro and in vivo. Moreover, using a genetically engineered mouse model of TNBC, the authors showed that ablation of the TrkB+ CSCs in the endogenous tumors prevents relapse of malignant tumors and prolongs survival of mice, further indicating that the TrkB+ CSCs represent the real source of TNBC recurrence [90].
Neurotrophins at the crossroad between epithelial-mesenchymal transition and cancer stem cells
Epithelial-to-mesenchymal transition (EMT) is a developmental process wherein epithelial cells transdifferentiate into mesenchymal cells. This process is characterized by molecular reprogramming including a decrease in the expression of proteins that enhance cell–cell contact such as E-cadherin and an increase in the expression of mesenchymal markers such as vimentin and fibronectin. Consequently, epithelial cells lose cellular junctions, reorganize cytoskeleton to gain the ability to migrate and invade adjacent tissue. EMT is coordinated by pleiotropic EMT transcription factors including zinc finger E-box binding homeobox members ZEB1 and ZEB2, the SNAIL zinc finger family, and the TWIST family of basic helix-loop-helix transcription factors [116]. Activation of EMT programs is also described to endow neoplastic epithelial cells with both mesenchymal phenotype and stemness traits. Chaffer et al. showed that ZEB1 can drive the conversion of breast neoplastic non stem cells (CD44-) into a stem-like state (CD44+) [117]. Moreover, interaction between EMT transcription factors and CSC transcription factors such as SOX and NANOG can promote stem cell self-renewal as well as commitment to either epithelial and/or mesenchymal lineage programs, depending on cellular context. For example, SOX2 binds directly to the promoters of the EMT transcription factors SLUG, SNAIL and TWIST1, leading to the loss of E-cadherin and the acquisition of stem cell features in pancreatic cancer cells [118]. Similarly, forced expression of SLUG with Sox9 in breast cancer cells can efficiently induce entrance into the CSC state [119]. More recently, by using the MMTV-PyMT transgenic model of mammary tumor development, the group of Robert A. Weinberg showed that SNAIL but not SLUG is tightly associated with a CSC phenotype [120].
Among numerous diffusible factors in tumor microenvironment, neurotrophins are increasingly described to be involved in the regulation of EMT and EMT-linked CSC enrichment (Fig. 1). Indeed, the BDNF/TrkB axis is constantly reported as an important promotor of EMT in a variety of cancers, including gastric [48], colon [49, 92], head and neck [82], lung [46, 84, 121], endometrial [122, 123], and breast cancers [90, 124]. BDNF/TrkB signaling activates Akt and MAP kinases, which in turn induce the expression of EMT transcription factors including TWIST, SNAIL, ZEB1, EZB2 [82, 121, 125, 126]. These EMT transcription factors can drive EMT by directly acting on target genes, and can also act in a stepwise manner. For example, in TrkB-transformed rat kidney epithelial cells, TrkB-induced EMT and metastasis is mediated by ZEB1, which acts downstream of a MAPK-dependent TWIST-SNAIL axis [126].
Ricci et al. clearly demonstrated the involvement of BDNF/TrkB in EMT-linked enrichment of CSCs of lung carcinoma by using as model system primary cell cultures derived from patients with malignant pleural effusions [127]. This system has been shown to reproduce the natural heterogeneity of non-small-cell lung cancer and to constitute a source of tumor-initiating cells as they can form tumors with histopathological features similar to those of original human tumors when propagated in immunodeficient mice [128, 129]. Using this model system, Ricci et al. showed that the couple of BDNF/TrkB is overexpressed in sphere culture conditions. This is associated with an increase of sphere formation and vimentin expression. Pharmacological inhibition of TrkB with K252a or silencing of TrkB by siRNA strongly reduces sphere formation and expression of EMT markers including vimentin, SLUG, TWIST and SNAIL (Fig. 1, right frame). Moreover, spheroids generated in the presence of siRNA against TrkB are not able to implant in immunodeficient mice, further supporting the importance of TrkB in tumor-initiating cells [84].
On the other hand, by using a mouse model system to mimic recurrent triple negative breast cancers as already mentioned above, Yin et al. showed that the ALDH1+/TrkB+ CSCs of recurrent triple negative breast cancers express higher levels of EMT markers including vimentin and TWIST [90]. ALDH1+/TrkB+ CSCs exhibit enhanced invasive capacity when compared to the corresponding ALDH1+/TrkB- and ALDH1- cells. These results suggest that in recurrent triple negative breast cancers, EMT and stemness maintenance may require the common signaling pathways of BDNF/TrkB.
We showed that NGF-treated luminal breast cancer cells, cultured under mammosphere conditions, exhibit an enhanced ability to generate tumors. Of note, the in vitro NGF pretreatment of breast cancer cells promotes EMT in tumors of SCID mice, as evidenced by the acquisition of migratory properties, a spindle-like cell morphology and the downregulation of epithelial markers, including E-cadherin, keratin 18 and keratin 19, but also the upregulation of mesenchymal markers such as vimentin and SLUG (Fig. 1, right frame). Moreover, the NGF-induced EMT yields cells with a CD44high/CD24−/low antigenic phenotype, which is widely used to identify breast CSCs [1, 117], thus linking NGF signaling to both EMT and CSCs. Interestingly, NGF increases expression of the SNAIL2, SNAIL1 and TWIST1 transcription factors in luminal breast cancer cells even under monolayer culture condition [106]. This suggests that NGF primes molecular changes in breast epithelial cancer cells, which may lead to EMT and CSC emergence from non-stem epithelial cells, depending on cellular context and tumor microenvironment.
Conclusion
In this review, we summarize the role of neurotrophins in cancer development and highlight the emerging evidence of neurotrophins in the regulation of CSCs. Accumulated data suggest that targeting the neurotrophin signaling pathways in CSCs may provide a new therapeutical option against treatment resistance and tumor relapse. Clearly, further study is needed to decipher the downstream signaling pathways of each neurotrophin receptor including TrkA, TrkB, P75NTR, and to identify key molecular events involved in neurotrophins-induced CSC enrichment. Moreover, given the plasticity of cancer cells and the dynamic interactions between CSCs and their microenvironment, it should be interesting to investigate the potential influence of neurotrophins and their receptors on the phenotype switching between CSCs and non CSCs.
References
Al-Hajj M, Wicha MS, Benito-Hernandez A et al (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 100:3983–3988. doi:10.1073/pnas.0530291100
Singh SK, Hawkins C, Clarke ID et al (2004) Identification of human brain tumour initiating cells. Nature 432:396–401. doi:10.1038/nature03128
Curley MD, Therrien VA, Cummings CL et al (2009) CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 27:2875–2883. doi:10.1002/stem.236
O’Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445:106–110. doi:10.1038/nature05372
Schepers AG, Snippert HJ, Stange DE et al (2012) Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 337:730–735. doi:10.1126/science.1224676
Chen J, Li Y, Yu T-SS et al (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488:522–526. doi:10.1038/nature11287
Driessens G, Beck B, Caauwe A et al (2012) Defining the mode of tumour growth by clonal analysis. Nature 488:527–530. doi:10.1038/nature11344
Eppert K, Takenaka K, Lechman ER et al (2011) Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 17:1086–1093. doi:10.1038/nm.2415
Liu S, Liu C, Min X et al (2013) Prognostic value of cancer stem cell marker aldehyde dehydrogenase in ovarian cancer: a meta-analysis. PLoS One 8:e81050. doi:10.1371/journal.pone.0081050
Balbous A, Cortes U, Guilloteau K et al (2014) A mesenchymal glioma stem cell profile is related to clinical outcome. Oncogenesis 3:e91. doi:10.1038/oncsis.2014.5
Wicha MS (2012) Migratory gene expression signature predicts poor patient outcome: are cancer stem cells to blame? Breast Cancer Res 14:114. doi:10.1186/bcr3338
Tang DG (2012) Understanding cancer stem cell heterogeneity and plasticity. Cell Res 22:457–472. doi:10.1038/cr.2012.13
Pece S, Tosoni D, Confalonieri S et al (2010) Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 140:62–73. doi:10.1016/j.cell.2009.12.007
Hope KJ, Jin L, Dick JE (2004) Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol 5:738–743. doi:10.1038/ni1080
Plaks V, Kong N, Werb Z (2015) The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 16:225–238. doi:10.1016/j.stem.2015.02.015
Bresnahan PA, Leduc R, Thomas L et al (1990) Human fur gene encodes a yeast KEX2-like endoprotease that cleaves pro-beta-NGF in vivo. J Cell Biol 111:2851–2859
Seidah NG, Benjannet S, Pareek S et al (1996) Cellular processing of the nerve growth factor precursor by the mammalian pro-protein convertases. Biochem J 314(Pt 3):951–960
Chao MV (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4:299–309. doi:10.1038/nrn1078
Aubert L, Guilbert M, Corbet C et al (2015) NGF-induced TrkA/CD44 association is involved in tumor aggressiveness and resistance to lestaurtinib. Oncotarget 6:9807–9819
Hecht M, Schulte JH, Eggert A et al (2005) The neurotrophin receptor TrkB cooperates with c-Met in enhancing neuroblastoma invasiveness. Carcinogenesis 26:2105–2115. doi:10.1093/carcin/bgi192
Skeldal S, Matusica D, Nykjaer A, Coulson EJ (2011) Proteolytic processing of the p75 neurotrophin receptor: a prerequisite for signalling?: Neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75(NTR). BioEssays 33:614–625. doi:10.1002/bies.201100036
Esposito D, Patel P, Stephens RM et al (2001) The cytoplasmic and transmembrane domains of the p75 and Trk A receptors regulate high affinity binding to nerve growth factor. J Biol Chem 276:32687–32695. doi:10.1074/jbc.M011674200
Iacaruso MFF, Galli S, Martí M et al (2011) Structural model for p75(NTR)-TrkA intracellular domain interaction: a combined FRET and bioinformatics study. J Mol Biol 414:681–698. doi:10.1016/j.jmb.2011.09.022
Lu B, Pang PT, Woo NH (2005) The yin and yang of neurotrophin action. Nat Rev Neurosci 6:603–614. doi:10.1038/nrn1726
Dang C, Zhang Y, Ma Q, Shimahara Y (2006) Expression of nerve growth factor receptors is correlated with progression and prognosis of human pancreatic cancer. J Gastroenterol Hepatol 21:850–858. doi:10.1111/j.1440-1746.2006.04074.x
Ma J, Jiang Y, Jiang Y et al (2008) Expression of nerve growth factor and tyrosine kinase receptor A and correlation with perineural invasion in pancreatic cancer. J Gastroenterol Hepatol 23:1852–1859. doi:10.1111/j.1440-1746.2008.05579.x
Kolokythas A, Cox DP, Dekker N, Schmidt BL (2010) Nerve growth factor and tyrosine kinase A receptor in oral squamous cell carcinoma: is there an association with perineural invasion? J Oral Maxillofac Surg 68:1290–1295. doi:10.1016/j.joms.2010.01.006
Kobayashi K, Ando M, Saito Y et al (2015) Nerve growth factor signals as possible pathogenic biomarkers for perineural invasion in adenoid cystic carcinoma. Otolaryngol Head Neck Surg 153:218–224. doi:10.1177/0194599815584762
Davidson B, Reich R, Lazarovici P et al (2003) Expression and activation of the nerve growth factor receptor TrkA in serous ovarian carcinoma. Clin Cancer Res 9:2248–2259
Davidson B, Konstantinovsky S, Nielsen S et al (2004) Altered expression of metastasis-associated and regulatory molecules in effusions from breast cancer patients: a novel model for tumor progression. Clin Cancer Res 10:7335–7346. doi:10.1158/1078-0432.CCR-04-0183
Zhang Y, Dang C, Ma Q, Shimahara Y (2005) Expression of nerve growth factor receptors and their prognostic value in human pancreatic cancer. Oncol Rep 14:161–171
Adriaenssens E, Vanhecke E, Saule P et al (2008) Nerve growth factor is a potential therapeutic target in breast cancer. Cancer Res 68:346–351. doi:10.1158/0008-5472.CAN-07-1183
Lagadec C, Meignan S, Adriaenssens E et al (2009) TrkA overexpression enhances growth and metastasis of breast cancer cells. Oncogene 28:1960–1970. doi:10.1038/onc.2009.61
Lagadec C, Romon R, Tastet C et al (2010) Ku86 is important for TrkA overexpression-induced breast cancer cell invasion. Proteomics Clin Appl 4:580–590. doi:10.1002/prca.200900148
Romon R, Adriaenssens E, Lagadec C et al (2010) Nerve growth factor promotes breast cancer angiogenesis by activating multiple pathways. Mol Cancer 9:157. doi:10.1186/1476-4598-9-157
Nico B, Mangieri D, Benagiano V et al (2008) Nerve growth factor as an angiogenic factor. Microvasc Res 75:135–141. doi:10.1016/j.mvr.2007.07.004
Nakagawara A, Arima M, Azar CG et al (1992) Inverse relationship between trk expression and N-myc amplification in human neuroblastomas. Cancer Res 52:1364–1368
Nakagawara A, Arima-Nakagawara M, Scavarda NJ et al (1993) Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med 328:847–854. doi:10.1056/NEJM199303253281205
Suzuki T, Bogenmann E, Shimada H et al (1993) Lack of high-affinity nerve growth factor receptors in aggressive neuroblastomas. J Natl Cancer Inst 85:377–384
Ho R, Minturn JE, Simpson AM et al (2011) The effect of P75 on Trk receptors in neuroblastomas. Cancer Lett 305:76–85. doi:10.1016/j.canlet.2011.02.029
Tacconelli A, Farina AR, Cappabianca L et al (2004) TrkA alternative splicing: a regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 6:347–360. doi:10.1016/j.ccr.2004.09.011
Schramm A, Schowe B, Fielitz K et al (2012) Exon-level expression analyses identify MYCN and NTRK1 as major determinants of alternative exon usage and robustly predict primary neuroblastoma outcome. Br J Cancer 107:1409–1417. doi:10.1038/bjc.2012.391
Barker PA, Lomen-Hoerth C, Gensch EM et al (1993) Tissue-specific alternative splicing generates two isoforms of the trkA receptor. J Biol Chem 268:15150–15157
Farina AR, Tacconelli A, Cappabianca L et al (2009) The alternative TrkAIII splice variant targets the centrosome and promotes genetic instability. Mol Cell Biol 29:4812–4830. doi:10.1128/MCB.00352-09
Sasahira T, Ueda N, Yamamoto K et al (2013) Trks are novel oncogenes involved in the induction of neovascularization, tumor progression, and nodal metastasis in oral squamous cell carcinoma. Clin Exp Metastasis 30:165–176. doi:10.1007/s10585-012-9525-x
Sinkevicius KW, Kriegel C, Bellaria KJ et al (2014) Neurotrophin receptor TrkB promotes lung adenocarcinoma metastasis. Proc Natl Acad Sci USA 111:10299–10304. doi:10.1073/pnas.1404399111
Vanhecke E, Adriaenssens E, Verbeke S et al (2011) Brain-derived neurotrophic factor and neurotrophin-4/5 are expressed in breast cancer and can be targeted to inhibit tumor cell survival. Clin Cancer Res 17:1741–1752. doi:10.1158/1078-0432.CCR-10-1890
Okugawa Y, Tanaka K, Inoue Y et al (2013) Brain-derived neurotrophic factor/tropomyosin-related kinase B pathway in gastric cancer. Br J Cancer 108:121–130. doi:10.1038/bjc.2012.499
Tanaka K, Okugawa Y, Toiyama Y et al (2014) Brain-derived neurotrophic factor (BDNF)-induced tropomyosin-related kinase B (Trk B) signaling is a potential therapeutic target for peritoneal carcinomatosis arising from colorectal cancer. PLoS One 9:e96410. doi:10.1371/journal.pone.0096410
Segal RA, Goumnerova LC, Kwon YK et al (1994) Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proc Natl Acad Sci USA 91:12867–12871
Eggert A, Grotzer MA, Ikegaki N et al (2001) Expression of the neurotrophin receptor TrkB is associated with unfavorable outcome in Wilms’ tumor. J Clin Oncol 19:689–696
Sclabas GM, Fujioka S, Schmidt C et al (2005) Overexpression of tropomysin-related kinase B in metastatic human pancreatic cancer cells. Clin Cancer Res 11:440–449
Kawamura K, Kawamura N, Okamoto N, Manabe M (2013) Suppression of choriocarcinoma invasion and metastasis following blockade of BDNF/TrkB signaling. Cancer Med 2:849–861. doi:10.1002/cam4.158
Eggert A, Grotzer MA, Ikegaki N et al (2002) Expression of the neurotrophin receptor TrkA down-regulates expression and function of angiogenic stimulators in SH-SY5Y neuroblastoma cells. Cancer Res 62:1802–1808
Lucarelli E, Kaplan D, Thiele CJ (1997) Activation of trk-A but not trk-B signal transduction pathway inhibits growth of neuroblastoma cells. Eur J Cancer 33:2068–2070
Kawamura K, Kawamura N, Okamoto N et al (2013) Suppression of choriocarcinoma invasion and metastasis following blockade of BDNF/TrkB signaling. Cancer Med 2(6):849–861. doi:10.1002/cam4.158
Kermani P, Rafii D, Jin DK et al (2005) Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest. 115(3):653–663
Jin H, Pan Y, Zhao L et al (2007) p75 neurotrophin receptor suppresses the proliferation of human gastric cancer cells. Neoplasia 9(6):471–478
Tabassum A, Khwaja F, Djakiew D (2003) The p75(NTR) tumor suppressor induces caspase-mediated apoptosis in bladder tumor cells. Int J Cancer 105(1):47–52
Khwaja F, Tabassum A, Allen J, Djakiew D (2006) The p75(NTR) tumor suppressor induces cell cycle arrest facilitating caspase mediated apoptosis in prostate tumor cells. Biochem Biophys Res Commun 341(4):1184–1192
Verbeke S, Meignan S, Lagadec C et al (2010) Overexpression of p75(NTR) increases survival of breast cancer cells through p21(waf1). Cell Signal 22:1864–1873. doi:10.1016/j.cellsig.2010.07.014
Johnston AL, Lun X, Rahn JJ et al (2007) The p75 neurotrophin receptor is a central regulator of glioma invasion. PLoS Biol 5(8):e212
Truzzi F, Marconi A, Lotti R et al (2008) Neurotrophins and their receptors stimulate melanoma cell proliferation and migration. J Invest Dermatol 128(8):2031–2040. doi:10.1038/jid.2008.21
Denkins Y, Reiland J, Roy M et al (2004) Brain metastases in melanoma: roles of neurotrophins. Neuro Oncol 6(2):154–165
Marchetti D, McQuillan DJ, Spohn WC et al (1996) Neurotrophin stimulation of human melanoma cell invasion: selected enhancement of heparanase activity and heparanase degradation of specific heparan sulfate subpopulations. Cancer Res 56(12):2856–2863
Civenni G, Walter A, Kobert N et al (2011) Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res 71(8):3098–3109. doi:10.1158/0008-5472.CAN-10-3997
Kiyosue T, Kawano S, Matsubara R et al (2013) Immunohistochemical location of the p75 neurotrophin receptor (p75NTR) in oral leukoplakia and oral squamous cell carcinoma. Int J Clin Oncol 18(1):154–163. doi:10.1007/s10147-011-0358-4
Lewis Kelso R, Colome-Grimmer MI, Uchida T et al (2006) p75(NGFR) immunostaining for the detection of perineural invasion by cutaneous squamous cell carcinoma. Dermatol Surg 32(2):177–183
Tomellini E, Lagadec C, Polakowska R, Le Bourhis X (2014) Role of p75 neurotrophin receptor in stem cell biology: more than just a marker. Cell Mol Life Sci 71(13):2467–2481. doi:10.1007/s00018-014-1564-9
Xiong J, Zhou LI, Lim Y et al (2015) Mature brain-derived neurotrophic factor and its receptor TrkB are upregulated in human glioma tissues. Oncol Lett 10(1):223–227
Xiong J, Zhou L, Lim Y et al (2013) Mature BDNF promotes the growth of glioma cells in vitro. Oncol Rep 30(6):2719–2724. doi:10.3892/or.2013.2746
Lawn S, Krishna N, Pisklakova A et al (2015) Neurotrophin signaling via TrkB and TrkC receptors promotes the growth of brain tumor-initiating cells. J Biol Chem 290(6):3814–3824. doi:10.1074/jbc.M114.599373
Wang L, Rahn JJ, Lun X et al (2008) Gamma-secretase represents a therapeutic target for the treatment of invasive glioma mediated by the p75 neurotrophin receptor. PLoS Biol 6(11):e289. doi:10.1371/journal.pbio.0060289
Ahn BY, Saldanha-Gama RF, Rahn JJ et al (2015) Glioma invasion mediated by the p75 neurotrophin receptor (p75NTR/CD271) requires regulated interaction with PDLIM1. Oncogene. doi:10.1038/onc.2015.199 (Epub ahead of print)
Forsyth PA, Krishna N, Lawn S et al (2014) p75 neurotrophin receptor cleavage by α- and γ-secretases is required for neurotrophin-mediated proliferation of brain tumor-initiating cells. J Biol Chem 289(12):8067–8085. doi:10.1074/jbc.M113.513762
Scala S, Wosikowski K, Giannakakou P et al (1996) Brain-derived neurotrophic factor protects neuroblastoma cells from vinblastine toxicity. Cancer Res 56(16):3737–3742
Jaboin J, Kim CJ, Kaplan DR, Thiele CJ (2002) Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3′-kinase pathway. Cancer Res 62(22):6756–6763
Matsumoto K, Wada RK, Yamashiro JM et al (1995) Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res 55(8):1798–1806
Li Z, Jaboin J, Dennis PA, Thiele CJ (2005) Genetic and pharmacologic identification of Akt as a mediator of brain-derived neurotrophic factor/TrkB rescue of neuroblastoma cells from chemotherapy-induced cell death. Cancer Res 65(6):2070–2075
Li Z, Tan F, Thiele CJ (2007) Inactivation of glycogen synthase kinase-3beta contributes to brain-derived neutrophic factor/TrkB-induced resistance to chemotherapy in neuroblastoma cells. Mol Cancer Ther 6(12 Pt 1):3113–3121
Yilmaz T, Jiffar T, de la Garza G et al (2010) Theraputic targeting of Trk supresses tumor proliferation and enhances cisplatin activity in HNSCC. Cancer Biol Ther 10(6):644–653. doi:10.4161/cbt.10.6.12782
Kupferman ME, Jiffar T, El-Naggar A et al (2010) TrkB induces EMT and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene 29(14):2047–2059. doi:10.1038/onc.2009.486
Redmer T, Welte Y, Behrens D et al (2014) The nerve growth factor receptor CD271 is crucial to maintain tumorigenicity and stem-like properties of melanoma cells. PLoS One 9(5):e92596. doi:10.1371/journal.pone.0092596
Ricci A, De Vitis C, Noto A et al (2013) TrkB is responsible for EMT transition in malignant pleural effusions derived cultures from adenocarcinoma of the lung. Cell Cycle 12(11):1696–1703. doi:10.4161/cc.24759
Wang W, Zhao H, Zhang S et al (2009) Patterns of expression and function of the p75(NGFR) protein in pancreatic cancer cells and tumours. Eur J Surg Oncol 35(8):826–832. doi:10.1016/j.ejso.2008.10.013
Davidson B, Reich R, Lazarovici P et al (2004) Altered expression and activation of the nerve growth factor receptors TrkA and p75 provide the first evidence of tumor progression to effusion in breast carcinoma. Breast Cancer Res Treat 83(2):119–128
Descamps S, Toillon RA, Adriaenssens E et al (2001) Nerve growth factor stimulates proliferation and survival of human breast cancer cells through two distinct signaling pathways. J Biol Chem 276(21):17864–17870
Tagliabue E, Castiglioni F, Ghirelli C et al (2000) Nerve growth factor cooperates with p185(HER2) in activating growth of human breast carcinoma cells. J Biol Chem 275(8):5388–5394
Verbeke S, Tomellini E, Dhamani F et al (2013) Extracellular cleavage of the p75 neurotrophin receptor is implicated in its pro-survival effect in breast cancer cells. FEBS Lett 587(16):2591–2596. doi:10.1016/j.febslet.2013.06.039
Yin B, Ma ZY, Zhou ZW et al (2015) The TrkB+ cancer stem cells contribute to post-chemotherapy recurrence of triple-negative breast cancers in an orthotopic mouse model. Oncogene 34(6):761–770. doi:10.1038/onc.2014.8
Louie E, Chen XF, Coomes A et al (2013) Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene 32(35):4064–4077. doi:10.1038/onc.2012.417
Fujikawa H, Tanaka K, Toiyama Y et al (2012) High TrkB expression levels are associated with poor prognosis and EMT induction in colorectal cancer cells. J Gastroenterol 47(7):775–784. doi:10.1007/s00535-012-0532-0
Sasahira T, Ueda N, Kurihara M et al (2013) Tropomyosin receptor kinases B and C are tumor progressive and metastatic marker in colorectal carcinoma. Hum Pathol 44(6):1098–1106. doi:10.1016/j.humpath.2012.09.016
Ødegaard E, Staff AC, Abeler VM et al (2007) The activated nerve growth factor receptor p-TrkA is selectively expressed in advanced-stage ovarian carcinoma. Hum Pathol 38(1):140–146
Matusica D, Skeldal S, Sykes AM et al (2013) An intracellular domain fragment of the p75 neurotrophin receptor (p75(NTR)) enhances tropomyosin receptor kinase A (TrkA) receptor function. J Biol Chem 288(16):11144–11154. doi:10.1074/jbc.M112.436469
Kommaddi RP, Thomas R, Ceni C et al (2011) Trk-dependent ADAM17 activation facilitates neurotrophin survival signaling. FASEB J 25(6):2061–2070. doi:10.1096/fj.10-173740
Ceni C, Kommaddi RP, Thomas R et al (2010) The p75NTR intracellular domain generated by neurotrophin-induced receptor cleavage potentiates Trk signaling. J Cell Sci 123(Pt 13):2299–2307. doi:10.1242/jcs.062612
Gallia GL, Tyler BM, Hann CL et al (2009) Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol Cancer Ther 8(2):386–393. doi:10.1158/1535-7163.MCT-08-0680
Eyler CE, Foo WC, LaFiura KM et al (2008) Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 26(12):3027–3036. doi:10.1634/stemcells.2007-1073
Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361(1473):1545–1564
Bonni A, Brunet A, West AE et al (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286(5443):1358–1362
Xing J, Kornhauser JM, Xia Z et al (1998) Nerve growth factor activates extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways to stimulate CREB serine 133 phosphorylation. Mol Cell Biol 18(4):1946–1955
Atwal JK, Singh KK, Tessier-Lavigne M et al (2003) Semaphorin 3F antagonizes neurotrophin-induced phosphatidylinositol 3-kinase and mitogen-activated protein kinase kinase signaling: a mechanism for growth cone collapse. J Neurosci 23(20):7602–7609
Ruggeri P, Farina AR, Di Ianni N et al (2014) The TrkAIII oncoprotein inhibits mitochondrial free radical ROS-induced death of SH-SY5Y neuroblastoma cells by augmenting SOD2 expression and activity at the mitochondria, within the context of a tumour stem cell-like phenotype. PLoS One 9(4):e94568. doi:10.1371/journal.pone.0094568 (eCollection 2014)
Murillo-Sauca O, Chung MK, Shin JH et al (2014) CD271 is a functional and targetable marker of tumor-initiating cells in head and neck squamous cell carcinoma. Oncotarget 5(16):6854–6866
Tomellini E, Touil Y, Lagadec C et al (2015) Nerve growth factor and proNGF simultaneously promote symmetric self-renewal, quiescence, and epithelial to mesenchymal transition to enlarge the breast cancer stem cell compartment. Stem Cells 33(2):342–353. doi:10.1002/stem.1849
Morrison SJ, Kimble J (2006) Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441(7097):1068–1074
Nykjaer A, Lee R, Teng KK et al (2004) Sortilin is essential for proNGF-induced neuronal cell death. Nature 427(6977):843–848
Vaegter CB, Jansen P, Fjorback AW et al (2011) Sortilin associates with Trk receptors to enhance anterograde transport and neurotrophin signaling. Nat Neurosci 14(1):54–61. doi:10.1038/nn.2689
Demont Y, Corbet C, Page A et al (2012) Pro-nerve growth factor induces autocrine stimulation of breast cancer cell invasion through tropomyosin-related kinase A (TrkA) and sortilin protein. J Biol Chem 287(3):1923–1931. doi:10.1074/jbc.M110.211714
Yan Y, Zuo X, Wei D (2015) Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med 4(9):1033–1043. doi:10.5966/sctm.2015-0048
Chanmee T, Ontong P, Kimata K, Itano N (2015) Key roles of hyaluronan and its CD44 receptor in the stemness and survival of cancer stem cells. Front Oncol 5:180. doi:10.3389/fonc.2015.00180.eCollection
Zöller M (2011) CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer 11(4):254–267. doi:10.1038/nrc3023
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676
Park IH, Zhao R, West JA et al (2008) Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451(7175):141–146
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15(3):178–196. doi:10.1038/nrm3758
Chaffer CL, Marjanovic ND, Lee T et al (2013) Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154(1):61–74. doi:10.1016/j.cell.2013.06.005
Herreros-Villanueva M, Zhang JS, Koenig A et al (2013) SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2:e61. doi:10.1038/oncsis.2013.23
Guo W, Keckesova Z, Donaher JL et al (2012) Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148(5):1015–1028. doi:10.1016/j.cell.2012.02.008
Ye X, Tam WL, Shibue T et al (2015) Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 525(7568):256–260. doi:10.1038/nature14897
Götz R, Sendtner M (2014) Cooperation of tyrosine kinase receptor TrkB and epidermal growth factor receptor signaling enhances migration and dispersal of lung tumor cells. PLoS One 9(6):e100944. doi:10.1371/journal.pone.0100944.eCollection
Bao W, Wang HH, Tian FJ et al (2013) A TrkB-STAT3-miR-204-5p regulatory circuitry controls proliferation and invasion of endometrial carcinoma cells. Mol Cancer 12:155. doi:10.1186/1476-4598-12-155
Bao W, Qiu H, Yang T et al (2013) Upregulation of TrkB promotes epithelial-mesenchymal transition and anoikis resistance in endometrial carcinoma. PLoS One 8(7):e70616. doi:10.1371/journal.pone.0070616
Howe EN, Cochrane DR, Richer JK (2011) Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res 13(2):R45. doi:10.1186/bcr2867
Smit MA, Geiger TR, Song JY et al (2009) A twist-snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol Cell Biol 29(13):3722–3737. doi:10.1128/MCB.01164-08
Smit MA, Peeper DS (2011) Zeb1 is required for TrkB-induced epithelial-mesenchymal transition, anoikis resistance and metastasis. Oncogene 30(35):3735–3744. doi:10.1038/onc.2011.96
Ricci A, Mariotta S, Pompili E et al (2010) Neurotrophin system activation in pleural effusions. Growth Factors 28(4):221–231. doi:10.3109/08977191003677402
Basak SK, Veena MS, Oh S et al (2009) The malignant pleural effusion as a model to investigate intratumoral heterogeneity in lung cancer. PLoS One 4(6):e5884. doi:10.1371/journal.pone.0005884
Mancini R, Giarnieri E, De Vitis C et al (2011) Spheres derived from lung adenocarcinoma pleural effusions: molecular characterization and tumor engraftment. PLoS One 6(7):e21320. doi:10.1371/journal.pone.0021320
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the University of Lille, the Institut National du Cancer (ARC_INCa_LNCC_8068) and the SIRIC ONCOLille.
Rights and permissions
About this article

Cite this article
Chopin, V., Lagadec, C., Toillon, RA. et al. Neurotrophin signaling in cancer stem cells. Cell. Mol. Life Sci. 73, 1859–1870 (2016). https://doi.org/10.1007/s00018-016-2156-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2156-7