
Abstract
Spinal muscular atrophy (SMA) is a devastating motoneuron (MN) disorder caused by homozygous loss of SMN1. Rarely, SMN1-deleted individuals are fully asymptomatic despite carrying identical SMN2 copies as their SMA III-affected siblings suggesting protection by genetic modifiers other than SMN2. High plastin 3 (PLS3) expression has previously been found in lymphoblastoid cells but not in fibroblasts of asymptomatic compared to symptomatic siblings. To find out whether PLS3 is also upregulated in MNs of asymptomatic individuals and thus a convincing SMA protective modifier, we generated induced pluripotent stem cells (iPSCs) from fibroblasts of three asymptomatic and three SMA III-affected siblings from two families and compared these to iPSCs from a SMA I patient and control individuals. MNs were differentiated from iPSC-derived small molecule neural precursor cells (smNPCs). All four genotype classes showed similar capacity to differentiate into MNs at day 8. However, SMA I-derived MN survival was significantly decreased while SMA III- and asymptomatic-derived MN survival was moderately reduced compared to controls at day 27. SMN expression levels and concomitant gem numbers broadly matched SMN2 copy number distribution; SMA I presented the lowest levels, whereas SMA III and asymptomatic showed similar levels. In contrast, PLS3 was significantly upregulated in mixed MN cultures from asymptomatic individuals pinpointing a tissue-specific regulation. Evidence for strong PLS3 accumulation in shaft and rim of growth cones in MN cultures from asymptomatic individuals implies an important role in neuromuscular synapse formation and maintenance. These findings provide strong evidence that PLS3 is a genuine SMA protective modifier.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proximal spinal muscular atrophy (SMA) is a usually devastating motoneuron (MN) disorder with a broad clinical variability raging from the severe type I (SMA I) with onset in the first 6 months of life and inability to sit or walk to the mild type III (SMA III) with onset after 18 months of life and ability to sit and walk [1]. Autosomal-recessive SMA is one of the most common neurodegenerative disorders with an incidence of 1:6000 to 1:10,000 and a carrier frequency of 1:35 in the Caucasian population [2, 3]. Both alleles of the disease-causing gene survival of motor neuron 1 (SMN1) are mutated in all SMA patients [4]. Severity and onset are mainly determined by the copy numbers of the nearly identical SMN2 gene. Higher SMN2 copy numbers correlate with a milder phenotype [3, 5]. SMN2 is predominantly aberrantly spliced due to a single silent variant affecting an exonic splicing enhancer. Thus, only about 10 % of full-length SMN transcripts and protein are generated [6, 7] from each SMN2 copy as compared to SMN1. The ubiquitously expressed housekeeping protein SMN is diffusely distributed in the cytoplasm and in the nucleus where it forms distinct punctual nuclear substructures called “Gemini of coiled bodies” (abbreviated “gems”) [8]. SMN plays an important role in small nuclear ribonucleoprotein particle (snRNP) biogenesis and splicing [9]. In addition, SMN is crucial for motoneuron development particularly for neuromuscular junction (NMJ) maturation and maintenance [9–11].
In less than 1 % of SMA families, homozygously SMN1-deleted individuals are fully asymptomatic despite carrying the same SMN2 copy number as their SMA III-affected siblings [12, 13]. This observation suggests a protective impact of a genetic modifier other than SMN2. We previously identified high expression levels of plastin 3 (PLS3), an actin-binding and -bundling protein, in lymphoblastoid (LB) cell lines of asymptomatic but not of symptomatic siblings using transcriptome-wide differential expression analysis [14]. Intriguingly, PLS3, an X-linked gene, was described not to be expressed in the hematopoietic system but only in solid tissues [15]. This unusual finding of high PLS3 expression in asymptomatic individuals has further been confirmed in native blood whereas primary fibroblasts showed identical expression levels in both SMA III and their asymptomatic siblings [14]. PLS3 overexpression is able to rescue axonal development deficits in smn zebrafish morphants exhibiting axon truncation and early branching [14, 16, 17]. Moreover, overexpression of transgenic PLS3 in SMA mice has shown to ameliorate all tested NMJ and MN functions connected to F-actin dynamics. Most pronounced was the increased neuronal connectivity and delayed axonal pruning in PLS3 overexpressing SMA mice. Moreover the proprioceptive input on MN somata was restored in PLS3 overexpressing SMA mice in comparison to SMA mice. Thus, high PLS3 levels restore impaired MN and NMJ functions caused by reduced SMN levels [18]. In contrast, overall survival rate is only very moderately prolonged in this severe SMA mouse model owing to impairment of other organs such as heart, lung and intestine [18].
The major questions that we aimed to address in this study are: (1) Is PLS3 indeed elevated in MN cultures of asymptomatic individuals versus SMA III patients and thus a genuine SMA modifier candidate? (2) Does PLS3 influence the expression level of SMN in MNs? (3) Which phenotype do asymptomatic MNs exhibit versus SMA III MNs versus MNs of SMA I and control individuals?
Given the inaccessibility of human spinal MNs as main target tissue of SMA, we used induced pluripotent stem cell (iPSC) technology [19, 20] to shed light on these three questions. Somatic human donor cells were converted into a pluripotent state by ectopic overexpression of four transcription factors (4F) of the pluripotency regulatory circuit described by the team of Yamanaka [21]. Mimicking human embryogenesis and neural development, human iPSCs can be redifferentiated into spinal MNs by patterning with the morphogens retinoic acid (RA) for posteriorization and sonic hedgehog (SHH) for ventralization [22]. Several protocols have been established for the controlled derivation of MN cultures from human pluripotent stem cells [23–29]. Previously, a cellular in vitro SMA model was established from a SMA I patient displaying essential features of in vivo SMA pathology such as reduced SMN expression and gem numbers, and impeded MN survival rate [23].
We established a personalised in vitro SMA III disease model of PLS3 discordant families to elucidate PLS3 mode of action in SMA III. In total, adult fibroblasts of three asymptomatic individuals and three SMA III siblings from two PLS3 discordant families, one SMA I patient and five healthy control individuals were reprogrammed by either retrovirus or Sendai virus (SeV) mediated 4F overexpression and thoroughly validated. Subsequently, a neural progenitor cell population (smNPCs, small molecule neural precursor cells) [30] was generated by a small molecule-based protocol and subsequently differentiated into MNs. In four phenotypic groups (controls, SMA I and PLS3 discordant SMA III and asymptomatic siblings), SMA disease-related features (MN survival, gem numbers, and SMN expression on RNA and protein levels), growth cone architecture and PLS3 expression were analysed during MN development in order to recapitulate SMA pathology and to understand potential protective mechanisms.
Materials and methods
Patients’ material
Fibroblast cell lines were established from skin biopsies of three SMA III patients and three asymptomatic siblings overexpressing PLS3 who belong to two SMA discordant families (Fig. 1a and Online Resource Tab. S1) [12, 14, 31]. In addition, iPCS lines reprogrammed from fibroblasts of an independent SMA I patient [32] and five unrelated control individuals (Online Resource Tab. S1; [33]) were used. Informed written consent was obtained from all patients or care givers according to the Declaration of Helsinki, and the study was approved by the ethics committee of the University Hospital of Cologne.

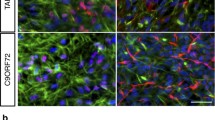
Determination of MN numbers by HB9 and ISL1 immunocytochemical staining in mixed MN cultures. a Pedigrees of SMA discordant families 1 and 2 (grey symbols asymptomatic; black symbols SMA III; white symbols healthy carriers). Subordinated tables provided SMN1/SMN2 copy numbers, PLS3 expression levels in lymphoblastoid cells and current age. Age of onset in HGK13 was 13 months, in HGK27 13 years and in HGK22 18 years. b Semi-quantitative RT-PCR of PLS3 and HPRT (used for equal loading control) from RNA isolated from lymphoblastoid cell lines of family 1 and 2. c Representative staining from motoneuron-differentiated smNPC lines from each of the four phenotypes (control, SMA I, SMA III and asymptomatic) demonstrated strong expression of MN markers HB9 and ISL1 (green) or neuronal marker SMI-32 (green) on d8 irrespective of their genetic background. PLS3 (red) was also expressed in HB9+ MNs. d Survival of MNs at d27 showed clear differences. In SMA I hardly any ISL1+ (green) or HB9+ (green)/ChAT+ (red) MNs were seen in contrast to control, SMA III and asymptomatic lines. SMI-32 expression was detectable in all four cell lines. Insets depict corresponding pictures with stained nuclei. Nuclei were counterstained with DAPI (blue). Scale bars 100 µm, valid for all images. e Grouping HB9+ or ISL1+ MN numbers according to the four phenotypes did not present any statistically significant difference on d8 in SMA I and SMA III. Only asymptomatic revealed an initially lower MN number. f On d27, all four groups lost HB9+ MNs in comparison to d8 (healthy control 29.8 to 24.8 %; SMA I 26.8 to 0.7 %; SMA III 20.3 to 7.4 %; asymptomatic 16.5 to 5.5 %). Determination of ISL1+ neurons revealed a similar tendency. Between d8 and d27, massive death of ISL1+ MNs was only detectable in SMA I phenotype (19.5 to 2.3 %) whereas the discordant family members exhibited a moderate decline in ISL1+ cell numbers (SMA III 21.5 to 13.9 %; asymptomatic: 11.8 to 6.8 %). Healthy controls even gained a small percentage of ISL1+ MNs (21.2 to 24.6 %). For d8, results showed the MN counting relative to the total number of cells from two to three independent differentiation experiments. For d27, two to three independent differentiation experiments were considered. Mean ratios were computed relative to total number of cells in case of HB9 and total number of β III-tubulin+ neurons in case of ISL1, respectively. Error bars represented ± SEM of two to three independent differentiation experiments. Significance was calculated relative to grouped five healthy controls (Kruskal–Wallis test with Dunn’s post hoc test, p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***)
Generation of iPSCs
Retroviral vectors harbouring SOX2, OCT4, KLF4 and c-MYC [21] were used to generate iPSC lines HGK1 (SMA I) [32], and discordant family 1 members SMA III (HGK13) and asymptomatic (HGK16) by iPierian (South San Francisco, CA, USA). Discordant family 2 iPSC lines from two asymptomatic (HGK21 and HGK28) and two SMA III (HGK22 and HGK27) patients were generated with Sendai virus (SeV) vectors supplied by ID Pharma Co. Ltd (formerly DNAVEC Corporation) [34]. In case of family 2, two independent iPSC clones from each of the four siblings were used in all experiments. In brief, primary fibroblasts were centrifuged for 45 min at 32 °C with 1500 g (spinfection) and cultivated in Advanced DMEM containing 5 % foetal calf serum (FCS) and 1 % GlutaMAX™ (all from Life Technologies) (modified method from [35]). Five days post-infection (d5), SeV infected fibroblasts were trypsinized and seeded onto feeder-coated dishes in DMEM/F-12 containing 10 % knockout serum replacement, 1 % nonessential amino acids (NEAA), 1 % GlutaMAX™, 1 % pyruvate, 0.1 mM β-mercaptoethanol and 6–10 ng/mL basic fibroblast growth factor (bFGF) (all from Life Technologies). Medium was changed every other day until clonal iPSC colonies were manually picked.
Cell culture of human iPSCs
Human iPSC lines were subsequently grown on Matrigel™ (MG)-coated cell culture plates in mTeSR™1 medium (STEMCELL Technologies) with daily medium change. iPSCs were split by Alfazyme (PAA) treatment when cultures were 70–75 % confluent. Medium was supplemented with 10 µM ROCK inhibitor Y-27632 (Tocris Bioscience) post plating to promote single cell survival [36].
DNA extraction and whole-genome karyotyping in iPSCs
Genomic DNA (gDNA) of iPSCs was isolated with DNeasy Blood and Tissue Kit (Qiagen) according to manufacturer’s instructions. Whole-genome single nucleotide polymorphism (SNP) genotyping was conducted for HGK13 and HGK16 at the Cologne Centre of Genomics (University of Cologne) using the Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix). All other iPSC lines were processed with an Illumina® Human610-Quad chip or a HumanCytoSNP-12 chip at the Institute of Human Genetics, University of Bonn. Data analysis was performed with Illumina®BeadStudio.
Isolation of RNA and quantitative real-time RT-PCR (qRT-PCR)
Total RNA was extracted with RNeasy Blood and Tissue Kit with parallel DNase I treatment (Qiagen) according to manufacturer’s instructions. Reverse transcription into cDNA was carried out by QuantiTect Reverse Transcription Kit (Qiagen) or iScript™ Kit (Bio-Rad) according to manufacturers’ instructions. All qRT-PCRs were performed with LightCycler 1.5 instrument using Fast Start DNA Master SYBR® Green I Kit (Roche). All primer sequences are listed in Online Resource Tab. S2.
Retroviral transgene silencing by semi-quantitative reverse transcription (RT)-PCR
Total RNA was isolated using TRIzol (Life Technologies) or RNeasy Mini Kit (Qiagen). Reverse transcription was carried out with the iScript Kit (BioRad) plus DNase I (Promega) according to the manufacturer’s manual. qRT-PCR was performed using the iCycler (BioRad) applying standard protocols as outlined previously [32]. All results were normalised to GAPDH. PCR samples were analysed by agarose gel electrophoresis with 1.2 % agarose gels supplemented with 0.1 µL ethidium bromide per mL gel in TAE buffer.
Undirected differentiation of iPSCs into three germ layers
To induce spontaneous differentiation, human iPSC colonies were detached by collagenase (1 mg/mL) treatment and transferred to non-adhesive Petri dishes in knockout-DMEM containing 20 % knockout serum replacement, 1 % NEAA and 1 % GlutaMAX™ to induce embryoid body (EB) formation. After 6 days, EBs were plated onto gelatine-coated dishes in DMEM containing 10 % FCS, 1 % pyruvate and 1 % NEAA for further differentiation into meso- and endodermal or neuroectodermal lineages. Cells were cultured for further 10 days and subsequent immunocytochemical analysis [37].
Teratoma formation
To test ability of teratoma formation, 1 × 106 iPSCs were resuspended as clumps in PBS and injected into the testicular capsule of immune-compromised SCID/Beige mice. After 8–10 weeks, tumours were isolated, fixed using 4 % paraformaldehyde (PFA) and immunohistologically analysed by haematoxylin/eosin staining.
Western blot analysis
Whole cell lysates were sampled with radio-immunoprecipitation assay (RIPA) buffer (Sigma–Aldrich). SDS-PAGE (Tris-HCl) and subsequent wet blotting were performed according to standard protocols. The following primary antibodies were used: mouse anti β-actin (1:60,000, Sigma–Aldrich), mouse anti-SMN (1:100, immunoGlobe) and rabbit anti-PLS3 (1:50; [14]) (Online Resource Tab. S3).
PLS3 siRNA knock-down
Predesigned negative siRNA (Cat: 1022076) and PLS3 siRNA (Cat: SI03174136) were purchased from Qiagen. Control and PLS3 siRNA stocks were prepared according to manufacturer’s instructions. 2 × 105 cells were seed into a six-well plate 1 day prior to transfection. 50 pmol of siRNA and 4µ of DharmaFECT 1 transfection reagent (Cat: T-2001-01, GE Life Science) were diluted in OptiMEM (Cat: 31985-070, Life Science) according to manufacturer’s instruction and used for the knockdown experiment. Glioblastoma cells (U373) were transfected with non-silencing negative control and PLS3 siRNAs. After 6 h of transfection, the medium was changed and cells were incubated at 37 °C for 48 h. The efficiency of the knockdown experiment was verified by Western blotting.
Generation and cultivation of small molecule neural precursor cells (smNPCs)
Generation and cultivation of smNPCs was performed as described [30]. Briefly, iPSC colonies were detached by collagenase treatment and transferred to a non-adherent Petri dish in knockout-DMEM containing 20 % knockout serum replacement, 1 % NEAA, 1 % GlutaMAX™, 1 mM β-mercaptoethanol, 10 µM ROCK inhibitor Y-27632 (Merck), 0.5 µM LDN193189 (Axon MedChem), 10 µM SB431542 (Sigma–Aldrich), 0.5 µM purmorphamine (Pur) (Merck) and 3 µM CHIR99021 (Stemgent) (adapted from [30] ). After 2 days, medium was changed to medium without Y-27632. On d4 of culture, dual SMAD signalling inhibition was stopped by withdrawing LDN193189 and SB431542. Finally, EB-like spheres were collected and mildly chopped by trituration. Spherical fragments were plated on MG-coated dishes in 1:1 DMEM/F-12:Neurobasal® medium containing 0.5 % N2 Supplement, 0.5 % GlutaMAX™, 1 % B27® supplement minus vitamin A (B27-RA) (all Life Technologies) supplemented with 10 µM ROCK inhibitor Y-27632, 150 µM L-ascorbic-acid-2-phosphat (LAAP) (Sigma–Aldrich), 0.5 µM Pur and 3 µM CHIR99021. After five passages, medium for stably growing smNPC lines was switched to standard medium (1:1 Advanced DMEM/F-12:Neurobasal® medium containing 1 % N2 Supplement, 1 % GlutaMAX™, 2 % B27-RA (all Life Technologies), 5 µg/mL bovine serum albumin (BSA) (Carl Roth) supplemented with 150 µM LAAP, 1 µM Pur and 3 µM CHIR99021). Regular splitting was carried out by Accutase (PAA) treatment in a ratio of 1:8–1:12 in previous standard medium which was changed every other day.
Motoneuronal differentiation from smNPCs
Based on the original protocol [30], a modified MN differentiation protocol was developed. In order to pre-pattern smNPCs towards a ventral-caudal fate, 1 µM retinoic acid (RA) (Sigma–Aldrich) and 1 µM smoothened agonist (SAG) (Calbiochem) were added to standard smNPC medium with daily medium change for at least 2 weeks. To differentiate smNPCs into MNs, smNPCs were seeded onto 6-well plates coated with 15 µg/mL poly-l-ornithine/10 µg/mL laminin (PO/Ln) in standard smNPC medium. Once cells reached 70 % confluence, medium was changed to 1:1 Advanced DMEM/F-12:Neurobasal® medium containing 1 % N2 Supplement, 1 % GlutaMAX™, 2 % B27-RA, 5 µg/mL BSA supplemented with 150 µM LAAP, 1 µM Pur, 1 µM RA and 1 µM SAG initiating motoneuronal patterning for the next 7 days. On d7 of differentiation, cells were singularised by alfazyme treatment (5–7 min, 37 °C). Bigger cell clumps were retained by a cell strainer (40 µm). For final maturation during the following weeks, 150,000 single cells per well of a chamber slide (Millipore) coated with PO/Ln and 5 µg/mL fibronectin (Fn) were plated in maturation medium containing Neurobasal® medium, 1 % N2 Supplement, 1 % GlutaMAX™, 1 % NEAA, 2 % B27-RA plus 10 µM ROCK inhibitor Y-27632, 10 ng/mL brain-derived neurotrophic factor (BDNF), 10 ng/mL glial cell line-derived neurotrophic factor (GDNF), 10 ng/mL insulin-like growth factor 1 (IGF-1) (all R&D System), 0.2 mM LAAP and 500 µM dibutyryladenosine cyclic monophosphate (db-cAMP) (Enzo Life Sciences). At least two million cells were seeded per 3.5 cm-dish (PO/Ln/Fn-coated) for later RNA or protein harvest. Medium was changed the next day. To thrust neural progenitor cells into neuronal differentiation, 10 µM Notch-signalling pathway inhibitor DAPT (Sigma–Aldrich) was added from d8 to d12 [38, 39]. Moreover, 0.5 µM Pur and 50 nM RA were supplemented from d8 to d15 to ensure proper maturation of potentially present pMN-patterned progenitor cells [27, 29]. Maturation medium was changed every other day. MNs were maturated until d27 for 3 weeks (i.e., 21 days) in total.
Immunocytochemistry
Cells were fixed with 4 % PFA in 1 × phosphate buffered saline (PBS) for 10 min at room temperature and blocked in 10 % FCS in Hank’s buffered salt solution (HBSS) containing Ca2+/Mg2+ (Life Technologies). Primary antibodies were administered over-night in 5 % FCS in HBSS. After washing three times with HBSS, samples were incubated with the secondary antibodies in 5 % FCS in HBSS at room temperature for 1 h. For visualisation of the actin cytoskeleton in growth cones, samples were incubated with phalloidin conjugated with Alexa555 in 5 % FCS in HBSS at room temperature for 1 h. If necessary, staining solutions were additionally supplemented with 0.1 % Triton X-100 (Sigma–Aldrich). Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) and samples were mounted with Mowiol 4–88 mounting solution (Carl Roth). Pictures were taken with an AxioCamMRn and further processed with Axiovision software (Zeiss). Confocal laser scanning microscopy was performed using Zeiss Meta 510 microscope. Pictures were processed with ZEN software and further analysed with ImageJ and Fiji software. Information about primary and respective secondary antibodies is given in Online Resource Tab. S3.
For quantitative analysis, 7–10 pictures were randomly chosen. In each image section, the total number of vital cells was ascertained by counting DAPI+ nuclei or β III-tubulin+ neurons, respectively. Then, the epitope of interest was quantified to compute the ratio of positive cells relative to the numbers of total cells or total neurons (d8: HB9: 500–2000 cells; ISL1: 600–2400 cells in total; d27: HB9: 400–1900 cells; ISL1: 300–900 neurons in total). In MN cultures (d27) counting was performed in areas where it was feasible.
For gem quantification, gems and total numbers of nuclei were counted to further calculate their ratio. Fibroblasts: 150–300 cells in total; iPSCs: 950–1300 cells in total; smNPCs: 1200–1700 cells in total; MN cultures: 200–450 cells (d8) and 300–550 cells (d27) in total.
Statistical analyses
Counting from immunocytochemical stainings and expression rates from molecular-biological experiments were expressed as mean values ± SEM. All data were analysed by Kruskal–Wallis non-parametric test with Dunn’s post hoc correction for multiple comparisons. Statistical significance of measurements was determined by GraphPad Prism software. Differences were considered significant when p < 0.05. Statistical significance was calculated relative to control line 30 m-r12 (when plotting individual cell lines) or grouped five healthy control lines (when plotting phenotype groups), respectively.
Results
SMA discordant families
All individuals included in this study except for the controls showed homozygous deletions of SMN1 and were expected to develop SMA based on SMN2 copy number and current age [4, 13]. However, three females were fully asymptomatic despite carrying only three SMN2 copies as their affected male siblings (Fig. 1a and Online Resource Tab. S1) [12, 14, 31]. The three asymptomatic siblings showed increased PLS3 levels in lymphoblastoid cell lines (Fig. 1b and [14]) but not in fibroblasts compared to their affected siblings (Fig. 2a and [14]), implying a protective role of PLS3 concerning SMA. Similar results were found in five further SMA discordant families [14].

Determination of number of SMN containing gems in the four phenotype classes throughout reprogramming and MN differentiation. a Immunocytochemical staining of cell lines representing the four phenotype classes in different cell populations during in vitro MN development. Principally, SMN (green) was either diffusely spread in the cytoplasm or visible as distinct dot-like nuclear accumulations, i.e., gems (arrows). Nuclei were counterstained with DAPI (blue). Dashed line enclosed area magnified in insets (scale bar 20 µm, valid for all images). b Overview of gem number quantification in different cell populations among four phenotypic classes. Overt SMN deficiency in SMA I resulted in poor gem formation and highly significant gem number reduction compared to control. However, more SMN2 copies (3–4) in SMA III visibly led to a medium gem number relative to control. Notably, asymptomatic siblings shared almost equal gem numbers with their SMA III-affected brothers. Error bars represented ± SEM of duplicates in fibroblasts (n = 2) and triplicates in iPSCs and smNPCs (n = 3). MN cultures displayed data of two to three independent MN differentiation experiments. Significance was calculated relative to grouped two healthy controls (fibroblasts, iPSCs, smNPCs) and to grouped five healthy controls (MN culture d8 and d27), respectively (Kruskal–Wallis test, p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***)
Generation and characterisation of iPSC lines from individuals of SMA discordant families
Human fibroblast cell lines of SMA discordant family 1 comprising an asymptomatic sibling (ML101) and an SMA III patient (ML102) as well as of an independent SMA I patient (ML17) [32] were reprogrammed by retroviral infection via pMXs-based vectors with the classical four Yamanaka factors (4F: OCT4, SOX2, KLF4, c-MYC) [19, 21] at iPierian Inc. (San Francisco, USA). One clone per line was delivered. Similarly, healthy unrelated control lines 30 m-r12 [33] and 35 m-r1 were generated and fully characterised previously. Additionally, non-integrating Sendai virus (SeV) vector system containing 4F [40] was employed to reprogram human fibroblast cell lines from SMA discordant family 2 (asymptomatic ML11 and ML13 and SMA III patients ML12 and ML14) (Fig. 1a and Online Resource Tab. S1). Two independent clones for each of the four siblings belonging to family 2 were selected, validated and used in all experiments. In addition, unrelated healthy control cell lines 51f-s6, 61f-s2 and 62 m-s4 were generated and validated in the same manner (Online Resource Tab. S1).
Representative iPSC lines clearly demonstrated human ESC-like morphology and characteristics including a small cell body with high nucleus/cytoplasm ratio, flat growth, typical colony shape, activity of alkaline phosphatase (AP) [21, 41] and expression of specific surface antigens (e.g., SSEA-3, SSEA-4, TRA-1-60, TRA-1-81) as pluripotency markers [41] (Online Resource Fig. S1A).
By SNP-array analysis, all new iPSC lines were scrutinised for any potential genomic aberration (Online Resource Fig. S1B and Tab. S1). If the genomic variant occurred in all iPSC clones and the original fibroblast cell line, the variant was regarded as an inherited intrinsic feature of this individual cell line already present in the donor genome. All iPSC lines applied in this study were genomically intact according to SNP-array analysis.
Assessment of SeV nucleocapsid protein NP expression via semi-qRT-PCR [42] proved loss of SeV vector activity in SeV-derived iPSC lines at later passages (>p7) (Online Resource Fig. S1C and Tab. S1). Human iPSC lines generated by retrovirus transduction were successfully examined for proviral transgene silencing (Online Resource Tab. S1).
To test the developmental potential of pluripotent SCs in vitro, iPSC colonies were grown in suspension as embryoid bodies (EBs). Upon withdrawal of pluripotency-maintaining growth factors, iPSCs spontaneously commenced to differentiate into cells of all three germ layers within those EBs [37]. When plated and immunocytochemically analysed after further growth, representative iPSC lines exhibited presence of tissue of endodermal (α-fetoprotein, AFP), mesodermal (smooth muscle actin, SMA) and neuroectodermal origin (β III-tubulin) (Online Resource Fig. S2A and Tab. S1).
We further verified pluripotency in human iPSCs by the teratoma formation assay [41]. All tested human iPSC lines formed tissues of the three germ layer lineages displaying adenoid goblet cell-like cells (endodermal tissue), cartilage (mesodermal tissue) and neural rosettes (ectodermal tissue) (Online Resource Fig. S2B and Tab. S1).
iPSCs give rise to stably expandable self-renewing neural precursor population of smNPCs
The establishment of stably expandable fully patternable neural progenitor cell population comprised the advantage to monitor the entire developmental process of MN generation in vitro: this enables to examine possible PLS3-mediated changes in different cell populations of an in vitro SMA cell culture model from original fibroblasts via iPSCs to NPCs and ultimately to MN cultures. Recently, a novel neural precursor cell type (smNPCs) was introduced with the advantageous feature of robust cost-effective proliferation and solid MN differentiation capability [30]. All 16 iPSC lines were converted into corresponding smNPC lines expressing CNS neural stem cell (NSC) markers like SOX1, SOX2, nestin and PAX6 as well as the early NSC marker FORSE-1 (Online Resource Fig. S3). By default, smNPCs comprise an early pre-rosette NPC population [30]. Hence, expression of classical rosette-markers PLZF and ZO-1 did not reveal the typical petal-shape arrangements with centred ZO-1 accumulation (Online Resource Fig. S3) further confirming smNPC identity [30].
Motoneuronal differentiation of smNPCs revealed a SMA phenotype
All smNPC lines were subjected to a modified differentiation regimen adapted from the original protocol [30]. Upon administration of RA and SAG in the smNPC culture medium, smNPCs were pre-patterned towards MN precursor fate for at least 2 weeks. Then, smNPCs underwent regular patterning for 1 week (d0–d6). On d7 of differentiation, cells were singularised and seeded for final maturation.
Immunocytochemical labelling of patterned smNPCs on d8 confirmed expression of classical markers of pMN-domain precursors such as OLIG2 and NKX6.1 [22] (Online Resource Fig. S4). Motoneuron-differentiated smNPC lines exhibited robust expression of pan-NSC markers SOX2 and nestin [43, 44] while early CNS marker PAX6 [45] was moderately expressed (Online Resource Fig. S4). Sufficient posteriorization was confirmed by broad presence of HOXB4 [29]. In addition, OLIG2+/NKX6.1+ double positive precursors indicated correct ventral pMN-regionalisation [22]. Finally, low expression of NKX2.2, a marker of the most ventral neural tube progenitor domain p3, disclosed an exact hit of the desired pMN domain confirming the shift of originally anterior smNPCs into the ventral-posterior pMN domain.
Classical MN markers HB9 and ISL1 and the neuronal marker SMI-32 [22] showed strong expression in all four phenotype classes (control, SMA I, SMA III, asymptomatic) in smNPC-derived MN cultures on d8 (Fig. 1c). Irrespective of their genetic background, representative cell lines largely exhibited ISL1+/β III-tubulin+ MNs as well as SMI-32+ neurons. Furthermore, HB9+ MNs showed detectable PLS3 expression strongly suggesting that this in vitro disease model is suitable for further studies on PLS3-specific influence in SMA discordant families (Fig. 1c).
Numbers of HB9+ MNs ranged highest in healthy controls (29.8 %) closely followed by SMA I (27.2 %) (Fig. 1d). Discordant family members showed fewer HB9+ MN in SMA III (20.3 %) and asymptomatic lines (16.5 %). Similarly, higher number of ISL1+ MNs were present in controls (21.2 %), SMA I (19.5 %) and SMA III (21.5 %), while asymptomatic individuals showed a lower number of ISL1+ MNs (11.8 %). Significant differences in MN numbers among phenotype classes were found only between control and asymptomatic on d8 (Fig. 1e). MN numbers for each individual cell line and corresponding phenotype groups are given in Online Resource Figs. S5A-S5B. Notably, SMA I line demonstrated high MN numbers on d8 suggesting that MN generation per se is not impaired by SMN reduction.
The detrimental effect of SMN loss in MN maintenance became obvious after 3 weeks of further maturation (Fig. 1d). On d27, MN cultures were again immunocytochemically stained for MN markers HB9, ISL1, neuronal marker SMI-32 and mature MN marker ChAT in addition. SMA I line hardly contained any HB9+ (0.7 %) or ISL1+ (2.3 %) MNs, respectively, however SMI-32+ neurons were detected (Fig. 1d). In contrast, SMA III and asymptomatic as well as control MNs clearly expressed all aforementioned MN markers. Yet, in comparison to d8 SMA III and asymptomatic MN cultures at d27 exhibited a distinct decline of MN number (Fig. 1f). Quantification unravelled a very significant loss of HB9+ and ISL1+ MNs in SMA I compared to control and an intermediate loss in SMA III and asymptomatic on d27 (Fig. 1f). Quantification of MNs in individual cell lines is given in Online Resource Fig. S5C–D.
In summary, we established an in vitro SMA model of mild SMA III and asymptomatic siblings based on iPSC-differentiation reflecting progressive MN loss in regard to disease severity on d27: A strong reduction in MN numbers was observed in SMA I and a moderate decline in SMA III and asymptomatic individuals on d27. However, no significant difference between SMA III and asymptomatic siblings was found suggesting that the protective effect is not related to the capacity to develop and maintain MNs.
Number of gems correlated with SMN2 copy numbers in phenotype classes throughout MN differentiation
As a sign of SMA pathology, loss of gems correlates with low SMN expression in SMA patients [46]. Consequently, gem numbers were determined in all cell populations of every donor fibroblast line, iPSC lines and their derived smNPC lines and differentiated MN cultures on d8 and d27. Immunocytochemical staining of SMN in the four phenotypic classes demonstrated a diffuse SMN dispersion in the cytoplasm. On the other hand, there were gems present as distinct nuclear agglomerations. Healthy control fibroblast line 30 m and the derived iPSC line 30 m-r12 showed the highest gem numbers in all cell populations (Fig. 3a). In contrast, SMA I fibroblast line ML17 and derivative line HGK1 consistently lacked visible gems. SMA III and asymptomatic lines, however, revealed gems in all cell populations (Fig. 3a). When quantified, the overview of phenotypes illustrated a recurring distribution pattern throughout the different cell populations during MN development (Fig. 3b). Compared to control, gem numbers in SMA I were highly diminished whereas SMA III and asymptomatic were intermediately reduced in comparison to controls (Fig. 3b). While adult somatic cells (i.e., fibroblasts) indicated the overall highest gem numbers among the phenotype classes rapidly proliferating cell populations (iPSCs and smNPCs) revealed the fewest gem numbers in general. However, once subjected to MN differentiation, total gem numbers increased within all phenotype groups exhibiting statistically significant reduction in SMA I, SMA III and asymptomatic compared to controls (Fig. 3b). Quantification of gems in each cell line and respective phenotype groups is depicted in Online Resource Fig. S6.

Overview of SMN and PLS3 protein expression during in vitro MN development. a–d Representative immunoblots from fibroblasts (a), iPSCs (b), smNPCs (c) and MN cultures (d) of all four phenotypes (control, SMA I, SMA III and asymptomatic) stained with antibodies against PLS3 and SMN. Actin was used as equal loading control. (e, f) Quantification of SMN and PLS3 protein relative to β-actin levels. In fibroblasts, iPSCs and smNPCs error bars represented ± SEM of duplicates (n = 2). MN culture (d27) displayed data of two to three independent MN differentiation experiments. Significance was calculated relative to grouped two healthy controls (fibroblasts, iPSCs, smNPCs) and to grouped five healthy controls (MN culture d27), respectively (Kruskal–Wallis test, p < 0.05 = *, p < 0.01 = **, p < 0.0001 = ****)
PLS3 was highly upregulated in mixed MN cultures of asymptomatic individuals while SMN showed similar expression levels in discordant siblings
A cornerstone of SMA pathology in SMN1-deleted individuals is low SMN expression which was already demonstrated in iPSC-derived SMA I models [23, 47]. Therefore, SMN and PLS3 mRNA expression were examined by qRT-PCR while SMN and PLS3 protein amounts were assessed by semi-quantitative Western blotting.
SMN mRNA amount was markedly diminished in SMA I compared to control in all cell populations. SMA III and asymptomatic siblings exhibited similar SMN expression in fibroblasts, iPSCs or smNPCs and reached only moderate expression levels in comparison to controls (Online Resource Fig. S7A–E).
PLS3 mRNA expression was more variable. In fibroblasts, the four phenotype classes expressed low and equal PLS3 levels (Online Resource Fig. S8A–E). Contrarily, in strongly proliferating cell populations like iPSCs and smNPCs PLS3 expression was overall higher. Asymptomatic individuals ranged always at the highest PLS3 expression level in comparison to their affected siblings although at a statistical non-significant level (Online Resource Fig. S8A–E). SMA I and SMA III instead exhibited a lower PLS3 expression in mixed MN cultures in comparison to iPSCs and smNPCs. Most importantly, the asymptomatic group significantly upregulated PLS3 expression by 4.8-fold in mixed MN cultures compared to total controls (Online Resource Fig. S8A–E), by 6.2-fold compared to male controls and 3.5-fold compared to female controls separately. Healthy controls 35 m-r1, 30 m-r12 and 62 m-s4 showed low PLS3 expression upon MN differentiation delivering the least PLS3 amount (Online Resource Fig. S11A).
Next, we analysed the SMN and PLS3 protein expression in the four phenotypic groups and respective four cell populations. SMN protein levels resembled SMN mRNA expression in the four cell populations (Fig. 2a–d). In SMA I cells the SMN level was visibly reduced and always the lowest among the four phenotypes (Fig. 2a–e and Online Resource Fig. S9A–D). SMN expression was comparable between SMA III and asymptomatic siblings and only insignificantly reduced as compared to control. In MN cultures, SMA III and asymptomatic siblings exhibited slightly higher but insignificant elevated SMN expression than control levels. This data excludes SMN levels as the reason for SMA protection in asymptomatic siblings. Phenotypic grouping demonstrated a general low SMN expression rate in adult fibroblasts in which SMN amounts in SMA I was visibly lower compared to control, SMA III and asymptomatic. Highly proliferative iPSCs and smNPCs presented a globally higher SMN expression rate (Fig. 2e).
Unlike SMN expression, PLS3 protein showed a more diverse expression pattern among different cell populations as well as among phenotype groups. In fibroblasts, an almost equal PLS3 expression rate was detected in all phenotype groups (Fig. 2a and Online Resource Fig. S10A). Probing protein samples from iPSCs, smNPCs and MN cultures with anti-PLS3 antibody during immunoblotting demonstrated two bands around the assumed size of 70 kDa. (Figure 2b). PLS3 in vitro siRNA-mediated knock-down identified the upper band as the correct specific PLS3 signal (Online Resource Fig. S11C). In iPSCs and smNPCs, PLS3 expression varied among individuals without presenting any significant difference among the four groups. However, the asymptomatic siblings showed a tendency of higher PLS3 expression in iPSCs and smNPCs than their SMAIII-affected siblings in both SMA discordant families (Fig. 2f and Online Resource Fig. S10B–C). Most importantly, in mixed MN cultures asymptomatic siblings showed highly significant upregulation of PLS3 expression as compared to total controls (Fig. 2d, f and Online Resource Fig. S10D). Moreover, PLS3 levels in asymptomatic exceeded those of their SMA III-affected siblings by 1.7-fold. Taking family 2 separately, the increase in PLS3 in asymptomatic individuals was 1.9-fold as compared to SMA III symptomatic and highly consistent between two different clones for each of the four siblings. Instead, asymptomatic line HGK16 from family 1 did not display a significant PLS3 upregulation in our MN culture model, although the level was clearly elevated in both iPSCs and smNPC as compared to the affected sibling HGK13. Since only one clone was available from this line, we could not verify the result in an independent clone.
PLS3 expression in male and female controls was, consistent with the RNA expression, strikingly low and did not differ between genders. However, at single individual level differences were seen (Online Resource Fig. S11A, B).
Comparison of the PLS3 expression levels among the four cell populations revealed a low and equal expression in fibroblasts while an increased expression in iPSCs and smNPCs was detectable with no significant differences between the groups. Instead, mixed MN cultures showed a highly significant PLS3 upregulation in asymptomatic compared to total controls (Fig. 2f).
These results demonstrated that PLS3 is indeed specifically upregulated in mixed MN cultures of asymptomatic as compared to SMA III siblings and controls advocating the role of PLS3 as genuine protective modifier in SMA.
PLS3 is abundant in growth cones of asymptomatic MN cultures
Originally it was reported that endogenous PLS3 accumulates in F-actin rich growth cones of primary murine MNs [14]. It is being discussed that PLS3 exerts a protective effect in stabilising neuromuscular connectivity and improved MN-synapse architecture as demonstrated in a PLS3 overexpressing mouse model [18]. Therefore, triple immunocytochemical staining of axonal marker microtubule-associated protein tau (MAPT), PLS3 and actin was performed in MN cultures on d27. Subsequently, cellular distribution was examined in growth cones by confocal laser scanning microscopy. MAPT+ axonal protrusions ended in cones with very delicate thin branches containing PLS3 and actin (Fig. 4a–c and Online Resource Fig. S12A). In controls, PLS3 and actin appeared evenly distributed and co-localised (Fig. 4a). SMA III lines displayed a more separate PLS3 and actin distribution in the growth cone (Fig. 4b). On the other hand, asymptomatic lines showed strong PLS3 expression in axon rods and growth cone centre while actin accumulated at the rims (Fig. 4c). Growth cone area size was determined by bordering each growth cone individually. Averaged area size was enlarged in SMA III and asymptomatic compared to control (Online Resource Fig. S12B). Merged pictures revealed co-localisation of PLS3 and actin in growth cones of control, SMA III and asymptomatic in iPSCs derived mixed MN cultures (Fig. 4a–c and Online Resource Fig. S12A). Co-localisation in growth cones was affirmed by Z-stack analyses. Signal intensity levels of PLS3 and actin were determined for calculation of Pearson’s coefficient [48]. However, no overt difference was observable between control, SMA III and asymptomatic (Online Resource Fig. S12C, D). Importantly, average fluorescence mean intensity value of PLS3 per growth cone was significantly reduced in SMA III in comparison to asymptomatic and control MN (Fig. 4d).

Confocal microscopy images from growth cones in maturing MN cultures on d27. a–c Mixed MN cultures from control (a), SMA III (b) and asymptomatic (c) on d27 immunolabelled with PLS3 (green), actin (red) and MAPT (magenta). Single labelling is pictured in the left panels and PLS3 and actin staining are overlaid in the right panels. Growth cones and corresponding fluorescence signal intensities were illustrated in 2.5D profiles (lower left insets). a Control growth cone shows PLS3 and actin almost equally distributed and frequently co-localised at the rim. b In SMA III cell line HGK27.13, presence of PLS3 and actin is demonstrated. c In asymptomatic line HGK28.9, intense PLS3 expression was visualised particularly in the rod end and growth cone central domain whereas actin allocated especially at the edges. Scale bars 10 µm. d Average fluorescence mean intensity signal value of PLS3 per growth cone was significantly diminished in SMA III in comparison to both control and asymptomatic. In contrast, fluorescence mean intensity value of PLS3 in asymptomatic reached control level hinting at an overall higher PLS3 amount in asymptomatic growth cones. Growth cones were analysed from four independent MN differentiation experiments (n = 4). In total, n = 28 (controls 30 m-r12, 35 m-r1, 51f-s6), n = 22 (SMA III HGK27.10, HGK27.13) and n = 20 (asymptomatic HGK21.1, HGK21.8, HGK28.9) growth cones were analysed. Significance was calculated relative to grouped three healthy controls (Kruskal–Wallis test, p < 0.05 = *, p < 0.01 = **)
In conclusion, co-localization of PLS3 and actin is similar in asymptomatic, controls and symptomatic growth cones; however, there may be a significant surplus of PLS3 in asymptomatic as compared to symptomatic, which may counteract impaired processes at NMJ level induced by reduced SMN levels.
Discussion
In this study we successfully applied iPSC technology in establishing a patient-derived motoneuronal in vitro cell model with PLS3-discordant mildly affected SMA III and asymptomatic individuals in comparison to healthy controls and a severe SMA I case. Data revealed cellular deficits upon SMN depletion at decisive steps of in vitro MN differentiation. No significant SMN expression difference was found between asymptomatic and symptomatic siblings in MN cultures excluding SMN alone as protective modifier.
Concerning PLS3 expression and its role in MN we conclude: (1) High PLS3 expression is not required per se during maturation stage of MNs as reflected by MN cultures in controls. (2) High PLS3 levels do not protect against MN loss when SMN levels are dramatically reduced as in SMA Type I. (3) In asymptomatic individuals PLS3 is particularly elevated during MN differentiation peaking in MN cultures (d27) and this correlates with the unusual elevated expression in lymphoblastoid cell lines and native blood [14]. (4) PLS3 expression might be similarly regulated in MNs and blood but not in other cell types like fibroblasts. (5) Growth cones of asymptomatic show an overall higher abundance of PLS3 expression as compared to symptomatic MNs; the excess of PLS3 might counteract impaired processes caused by SMN deficiency at MN and particularly at NMJ level as previously demonstrated [18].
SMA III and asymptomatic show a SMA intermediate phenotype in MN cultures differentiated from iPSCs
iPSC lines from adult fibroblasts of three SMA III patients and three asymptomatic siblings were generated and validated by non-integrative SeV technique [40] or retroviral integration of the four Yamanaka factors [19, 21]. Additionally, we compared our data to previously published SMA I and control lines [32, 33]. Furthermore, a stably expandable self-renewing small molecular neural precursor cell (smNPC) population [30] was derived and successfully differentiated into MN cultures yielding valid MN numbers. Unlike previous studies [23, 49] our data relied on two classical MN markers, i.e., ISL1 and HB9 [22]. Three weeks in vitro maturation resulted in significant decline in ISL1+ as well as HB9+ MN numbers in severely affected SMA I. Likewise, severe SMA mouse models have been published that develop MNs until birth, followed by massive MN death in the postnatal period [50–52] as comparably detected in iPSC-derived SMA I in vitro models [23, 49]. SMN1-deleted SMA III and asymptomatic groups exhibited a noticeable yet smaller decrement in ISL1+ and HB9+ MN numbers after maturation mirroring higher SMN2 copy numbers in these groups. In addition, PLS3 overexpression in asymptomatic did not influence overall MN survival rate suggesting a PLS3-mediated protection rather in other parts of the motor circuit than in MN development itself. Loss of HB9+ MNs in control during maturation likely originated from HB9 downregulation in developing spinal cord [53].
Our data in iPSC-derived SMA I MN models confirmed previously published data showing marked reduction of SMN expression and gem counts [23, 47]. SMN expression level in SMA III and asymptomatic showed mildly reduced levels in iPSC and smNPCs and no difference in MNs as compared to controls. Instead SMN gem numbers in both SMA III and asymptomatic displayed intermediate counts as compared to controls in all developmental stages. Most importantly, no statistical differences in SMN expression and gem counts were found between asymptomatic and SMA III siblings, excluding SMN as the SMA protective modifier in asymptomatic siblings. Similar results were obtained in a recent study [54].
Asymptomatic highly overexpress PLS3 in MN cultures and particularly in growth cones as compared to SMA III-affected siblings
Based on transcriptome studies in lymphoblastoid cell lines and native blood, PLS3 overexpression is regarded as causative for the asymptomatic phenotype in some SMA discordant families [14] but not all ([55] and Wirth et al. unpublished data). In fibroblasts instead, PLS3 showed low and highly similar expression levels in all four phenotypic groups, which confirms previous results [14, 55]. In contrast to fibroblasts from which all iPSC lines derived, we found different PLS3 expression levels during MN differentiation stages with asymptomatic presenting around double as much PLS3 as their symptomatic siblings. PLS3 expression was significantly upregulated in asymptomatic compared to both male and female controls in MN cultures. Interestingly, two control male lines showed extremely low PLS3 expression, suggesting a potential gender-specific downregulation mechanism and supporting the observation of a female-specific protective effect in SMA discordant families [14]. PLS3 level in asymptomatic siblings of family 2 was 1.9-fold elevated as compared to their SMA III-affected siblings and highly consistent between two different clones for each of the four siblings. Instead, asymptomatic line HGK16 from family 1 did not display a significant PLS3 upregulation in mixed MN cultures, although the level was clearly elevated in both iPSCs and smNPC as compared to the affected sibling HGK13. Unfortunately, only one clone of this line was available, making a confirmation in an independent clone impossible. Importantly, despite lower MN counts in asymptomatic HGK21 than HGK28 of family 2 the PLS3 expression was similar and reproducible in all four clones, suggesting an overall PLS3 upregulation in neuronal cells types in asymptomatic individuals. This observation can be considered as particular relevant since there is increasing evidence that not only MN but also interneurons and proprioceptive inputs on MNs are crucial [56].
Since smNPCs already reveal PLS3 upregulation in asymptomatic individuals, it might be speculated that neural lineages and especially MN-specific differentiation may trigger PLS3 overexpression in asymptomatic patients by a so far unknown regulatory mechanisms. Concomitantly with our study, Boza-Morán et al. found in one of their discordant families in which PLS3 has previously been excluded as genetic modifier that also in iPSC-derived MN cultures PLS3 is not upregulated in a milder type IV-affected individual as compared to SMA III-affected siblings [54, 55]. Both studies strengthen the observation that PLS3 expression in blood mirrors expression in MNs, and that both tissues may have a similar mechanism for PLS3 expression regulation which remains to be elucidated in future.
PLS3 expression in iPSCs, smNPCs and MN cultures of SMA I was comparably high, but no positive effect on MN survival was observed. This finding is in agreement with (1) recently published data showing that overexpression of a PLS3 transgene in a severely affected SMA mouse model had no impact on MN number or survival [18] and (2) our previous hypothesis that PLS3 confers full protection only when a certain amount of SMN is present [13]. Indeed asymptomatic individuals never carried only two but at least three SMN2 copies ([14] and Online Resource Tab. S1).
Unlike in zebrafish where decreasing Smn protein amounts accompanied declining Pls3 protein amounts [57], data from our in vitro MN system do not suggest a direct correlation between SMN and PLS3 expression in severely affected SMA patients since SMA I exhibited high PLS3 expression rates despite overt SMN deficiency. Whether moderately elevated SMN levels in asymptomatic MN cultures resulted from possible interaction between SMN and PLS3 remains to be elucidated.
Multiple studies reported defects in NMJ development and maintenance, which were therefore regarded as the hallmark of SMA pathology [58–61]. Interestingly, SMN levels had little impact on SMA pathology after NMJ formation and maturation [11]. Since PLS3 overexpression in SMA mice has shown a major influence on all tested F-actin dependent processes at the NMJ level and on the motor activity of these mice [18], our present data strongly support the view that PLS3 overexpression in MNs can indeed be a genuine protective SMA modifier.
We previously demonstrated that elevated PLS3 levels shift the G/F-actin ratio towards more F-actin and change the dynamics and F-actin dependent functions at MN and NMJ level in a SMA mouse model overexpressing PLS3 [14]. Moreover, co-localisation of PLS3 with F-actin was shown in primary murine MNs [14] and in filopodia of PLS3 overexpressing MEFs [18]. We therefore examined PLS3 and F-actin expression and distribution by confocal microscopy analysis in growth cones from human mixed MN cultures. Indeed, growth cones of controls, SMA III and asymptomatic siblings were shaped according to previous descriptions [62] and displayed presence of PLS3 and actin. MAPT+ protrusions denoted axons ending in growth cones in which actin was located primarily in the peripheral zone and filopodia-like protrusions [62]. In control growth cones, an equal PLS3/actin distribution and co-localisation pattern was found, supporting the conclusion that PLS3 mainly functions in F-actin bundles. In asymptomatic strong co-localisation of PLS3 and F-actin was mainly observed at the rim and central domain of the growth cones. However, co-localisation per se was apparently not differing between the three phenotype groups since Pearson’s coefficient of SMA III and asymptomatic reached the same medium coefficient values indicating a moderate PLS3/actin co-localisation in growth cones. However, the overall average PLS3 fluorescence intensity value, which supposedly corresponds with the PLS3 amount in a growth cone was significantly reduced in SMA III while asymptomatic reached control levels. Thus, the absolute PLS3 amount might indeed be increased in asymptomatic growth cones. This fact might lead to retention of actin filaments and improved F-actin/PLS3 bundling in asymptomatic growth cones and thereby extended NMJ stabilisation. This goes in line with our previous finding that all F-actin related processes at NMJ level are impaired in SMA mice and restored by PLS3 overexpression [18]. Still, additional F-actin-independent roles of PLS3 cannot be excluded [17]. Since PLS3 has several binding partners involved in additional processes such as RNA metabolism, translation, etc. (Wirth lab, unpublished data), abundant PLS3 as found in asymptomatic siblings might exert the SMA protective effect by further mechanisms than mere actin-cytoskeleton stabilisation. Moreover, since the neuronal subtype of the growth cones was not determined further experiments are required to substantiate these findings.
We finally conclude that increased PLS3 in asymptomatic SMN1-deleted individuals, who showed also elevated PLS3 levels in blood, can be considered as a genuine SMA protective modifier and is a reliable biomarker. This opens an avenue for further studies on the mode of action of PLS3, which might ultimately result in the development of novel therapies.
Abbreviations
- PLS3:
-
Plastin 3
- SMA:
-
Spinal muscular atrophy
- SMN1:
-
Survival motor neuron 1
- SMN2:
-
Survival motor neuron 2
- iPSC:
-
Induced pluripotent stem cell
- CNS:
-
Central nervous system
- LAAP:
-
l-ascorbic-acid-2-phosphate
- MN:
-
Motoneuron
- NSC:
-
Neural stem cell
- PO:
-
Poly-l-ornithine
- ROCK:
-
Rho-associated coiled-coil containing protein kinase
- SeV:
-
Sendai virus
References
Munsat TL, Davies KE (1992) International SMA consortium meeting. (26–28 June 1992, Bonn, Germany). Neuromuscul Disord 2:423–428
Pearn J (1978) Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J Med Genet 15:409–413
Feldkotter M et al (2002) Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 70:358–368
Lefebvre S et al (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80:155–165
Wirth B et al (2006) Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet 119:422–428
Lorson CL et al (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA 96:6307–6311
Cartegni L, Krainer AR (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 30:377–384
Liu Q, Dreyfuss G (1996) A novel nuclear structure containing the survival of motor neurons protein. EMBO J 15:3555–3565
Li DK et al (2014) SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin Cell Dev Biol 32:22–29
Burghes AH, Beattie CE (2009) Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10:597–609
Kariya S et al (2014) Requirement of enhanced survival motoneuron protein imposed during neuromuscular junction maturation. J Clin Invest 124:785–800
Hahnen E et al (1995) Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: evidence of homozygous deletions of the SMN gene in unaffected individuals. Hum Mol Genet 4:1927–1933
Wirth B et al (2013) How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Curr Opin Genet Dev 23:330–338
Oprea GE et al (2008) Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320:524–527
Lin CS et al (1993) Human plastin genes. Comparative gene structure, chromosome location, and differential expression in normal and neoplastic cells. J Biol Chem 268:2781–2792
McWhorter ML et al (2003) Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol 162:919–931
Lyon AN et al (2014) Calcium binding is essential for plastin 3 function in Smn-deficient motoneurons. Hum Mol Genet 23:1990–2004
Ackermann B et al (2013) Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum Mol Genet 22:1328–1347
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676
Yu J et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920
Takahashi K et al (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Davis-Dusenbery BN et al (2014) How to make spinal motor neurons. Development 141:491–501
Ebert AD et al (2009) Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457:277–280
Amoroso MW et al (2013) Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J Neurosci 33:574–586
Patani R et al (2011) Retinoid-independent motor neurogenesis from human embryonic stem cells reveals a medial columnar ground state. Nat Commun 2:214
Wichterle H et al (2002) Directed differentiation of embryonic stem cells into motor neurons. Cell 110:385–397
Hu BY, Zhang SC (2009) Differentiation of spinal motor neurons from pluripotent human stem cells. Nat Protoc 4:1295–1304
Li XJ et al (2005) Specification of motoneurons from human embryonic stem cells. Nat Biotechnol 23:215–221
Li XJ et al (2008) Directed differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small molecules. Stem Cells 26:886–893
Reinhardt P et al (2013) Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One 8:e59252
Helmken C et al (2003) Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet 114:11–21
Garbes L et al (2013) VPA response in SMA is suppressed by the fatty acid translocase CD36. Hum Mol Genet 22:398–407
Koch P et al (2011) Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 480:543–546
Ban H et al (2011) Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc Natl Acad Sci USA 108:14234–14239
Aasen T et al (2008) Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol 26:1276–1284
Watanabe K et al (2007) A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25:681–686
Itskovitz-Eldor J et al (2000) Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med 6:88–95
Borghese L et al (2010) Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells 28:955–964
Crawford TQ, Roelink H (2007) The notch response inhibitor DAPT enhances neuronal differentiation in embryonic stem cell-derived embryoid bodies independently of sonic hedgehog signaling. Dev Dyn 236:886–892
Fusaki N et al (2009) Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 85:348–362
Smith KP et al (2009) Pluripotency: toward a gold standard for human ES and iPS cells. J Cell Physiol 220:21–29
Nishimura K et al (2011) Development of defective and persistent Sendai virus vector: a unique gene delivery/expression system ideal for cell reprogramming. J Biol Chem 286:4760–4771
Kim JB et al (2009) Direct reprogramming of human neural stem cells by OCT4. Nature 461:649–653
Michalczyk K, Ziman M (2005) Nestin structure and predicted function in cellular cytoskeletal organisation. Histol Histopathol 20:665–671
Alaynick WA et al (2011) SnapShot: spinal cord development. Cell 146(178–178):e171
Coovert DD et al (1997) The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 6:1205–1214
Corti S et al (2012) Genetic correction of human induced pluripotent stem cells from patients with spinal muscular atrophy. Sci Transl Med 4:165ra162
Dunn KW et al (2011) A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300:C723–C742
Sareen D et al (2012) Inhibition of apoptosis blocks human motor neuron cell death in a stem cell model of spinal muscular atrophy. PLoS One 7:e39113
Hsieh-Li HM et al (2000) A mouse model for spinal muscular atrophy. Nat Genet 24:66–70
Monani UR et al (2000) The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet 9:333–339
Riessland M et al (2010) SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet 19:1492–1506
William CM et al (2003) Regulation of motor neuron subtype identity by repressor activity of Mnx class homeodomain proteins. Development 130:1523–1536
Boza-Moran MG et al (2015) Decay in survival motor neuron and plastin 3 levels during differentiation of iPSC-derived human motor neurons. Sci Rep 5:11696
Bernal S et al (2011) Plastin 3 expression in discordant spinal muscular atrophy (SMA) siblings. Neuromuscul Disord 21:413–419
Mentis GZ et al (2011) Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69:453–467
le Hao T et al (2012) Survival motor neuron affects plastin 3 protein levels leading to motor defects. J Neurosci 32:5074–5084
Murray LM et al (2008) Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet 17:949–962
Kariya S et al (2008) Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet 17:2552–2569
Kong L et al (2009) Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci 29:842–851
Ruiz R et al (2010) Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J Neurosci 30:849–857
Lowery LA, Van Vactor D (2009) The trip of the tip: understanding the growth cone machinery. Nat Rev Mol Cell Biol 10:332–343
Acknowledgments
We thank Dr John Dimos (iPierian) for generating the HGK13 and HGK16 iPSC clones. We thank M. Segschneider, C. Thiele, R. Konang and V. Poppe from the Institute of Reconstructive Neurobiology (RNB, Bonn, Germany) for excellent technical support and A. Leinhaas for carrying out the teratoma assays. Dr A. Schauss and I. Hensen of the Imaging Facility of the Cluster of Excellence in Cellular Stress Responses in Aging-associated Diseases (CECAD, Cologne, Germany) supported the confocal microscopy. This work has been supported by the European Community’s Seventh Framework Program FP7/2007-2013 under grant agreement no 2012-305121 (NeurOmics), the 7FP project SCR&Tox (HEALTH-F5-2010-26675), BIO.NRW (Project StemCellFactory, #005-1403-0106), the Center for Molecular Medicine Cologne (grant no C11), the Hertie Foundation and the Deutsche Forschungsgemeinschaft (Wi 945/13-1; Wi 945/16-1) to B.W.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
No competing financial interests exist.
Additional information
L. Heesen and M. Peitz contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
18_2015_2084_MOESM1_ESM.tif
Supplementary material 1: Figure S1: Validation of human iPSCs from SMA III and asymptomatic siblings. (A) Typical flat colony morphology with dense centre in phase contrast was shown for one representative asymptomatic (HGK21.1) and one representative SMA III (HGK22.17) iPSC line. Pluripotency was proven by AP activity (blue) and via immunostaining with pluripotency surface markers SSEA-3, SSEA-4, TRA-1-60 and TRA-1-81 (all red). Nuclei were counterstained with DAPI (blue). Scale bars 200 µm, valid for all images. (B) Exemplary karyograms of human iPSC lines HGK21.1 (asymptomatic) and HGK22.17 (SMA III) with respective original fibroblast lines schematically displayed every chromosome with the corresponding B allele frequency (upper panel) and Log R ration (lower panel). (C) Semi-quantitative analysis of SeV nucleocapsid (NP) mRNA expression in representative iPSC lines in comparison to 18S rRNA expression as loading control. SeV vector activity vanished at later passages (> p7). Only the positive control (pos ctrl) depicted a strong NP expression (TIFF 62136 kb)
18_2015_2084_MOESM2_ESM.tif
Supplementary material 2: Figure S2: Validation of human iPSCs from SMA III and asymptomatic siblings. (A) Spontaneous undirected differentiation of human iPSCs delivered progeny of all three germ layers in representative cell lines showing endodermal cells (α-fetoprotein AFP, green), mesodermal cells (smooth muscle actin SMA, red) and ectodermal cells (β III-tubulin, red) upon immunocytochemical labelling. (B) Teratoma formation assay confirmed pluripotency in iPSCs by generating derivatives of all three germ layers in vivo. Immunohistologic haematoxylin/eosin (HE) staining in representative samples displayed endoderm (glandular epithelium), mesoderm (cartilage) and ectoderm (neural rosettes). Nuclei were counterstained with DAPI (blue) (A). Scale bars 200 µm (A and B), valid for all images (TIFF 42104 kb)
18_2015_2084_MOESM3_ESM.tif
Supplementary material 3: Validation of smNPCs. iPSC-derived smNPC lines clearly disclosed typical NSC markers PAX6 (green), SOX1 (red), SOX2 (green) and nestin (red) in four representative cell lines upon immunolabelling. Expression of anterior marker FORSE-1 (green) was visualised as well. smNPCs expressed characteristic rosette-markers PLZF (green) and ZO-1 (red), however, lacked the typical petal-like arrangement underscoring their early pre-rosette NSC fate. Nuclei were counterstained with DAPI (blue). Scale bar 100 µm, valid for all images (TIFF 30730 kb)
18_2015_2084_MOESM4_ESM.tif
Supplementary material 4 Immunocytochemistry of four smNPC-derived cultures subjected to MN differentiation. Cultures representing the respective phenotype classes (control 30 m-r12, SMA I patient HGK1, SMA III patient HGK27.13 and asymptomatic sister HGK28.9) proved correct pMN-regionalisation on day 8 of the differentiation protocol. Pan-NSC markers SOX2 (green) and nestin (red) were clearly visible while early CNS marker PAX6 (green) was moderately expressed. Cells broadly exhibited pMN-marker OLIG2 (green) and NKX6.1 (red) whose evident co-localisation confirmed true MN progenitor identity. Moreover, expression of HOXB4 (red) verified sufficient posteriorization. Presence of only few NKX2.2+ cells (green) marked correct domain boundaries meaning that cells were not too ventralised into domain p3. Insets showed corresponding pictures with stained nuclei. Nuclei were counterstained with DAPI (blue). Scale bar 100 µm, valid for all images (TIFF 15294 kb)
18_2015_2084_MOESM5_ESM.tif
Supplementary material 5: Quantification of MN marker expression in smNPC-derived MN cultures on day 8 and day 27. For each individual cell line, MN numbers are given for (A) HB9+ MNs on day 8, (B) ISL1+ MNs on day 8, (C) HB9+ MNs on day 27 and (D) ISL1+ MNs on day 27. Error bars represented ± SEM of two (controls 35 m-r1, 61f-s2, 51f-s6, 62 m-s4) to three (all other lines) independent MN differentiation experiments (A and B) and n = MN differentiation experiments as outlined in tables below (C and D). Significance was calculated relative to 30 m-r12 (Kruskal–Wallis test, p < 0.05 = *) (TIFF 2297 kb)
18_2015_2084_MOESM6_ESM.tif
Supplementary material 6: Determination of gem numbers in different cell populations. Quantification of individual cell lines was displayed in left hand panels in (A) fibroblasts, (B) iPSCs, (C) smNPCs, (D) early MN culture (day 8) and (E) late MN culture (day 27). Right hand panels show grouped phenotypes. Error bars represented ± SEM of duplicates in fibroblasts (n = 2) and triplicates in all other cell populations (n = 3). MN cultures displayed data of two to three independent MN differentiation experiments (n = 3). Significance was calculated relative to 30 m-r12 (individual cell lines) and grouped five healthy controls (phenotype groups), respectively (Kruskal-Wallis test, p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***, p < 0.0001 = ****) (TIFF 4443 kb)
18_2015_2084_MOESM7_ESM.tif
Supplementary material 7 Quantitative analyses of SMN mRNA expression levels. (A) SMN mRNA expression occurred in a similar pattern in fibroblasts, iPSCs and smNPCs with controls at the top, SMA III and asymptomatic in the middle and SMA I at the lowest range, matching the SMN1/SMN2 copy numbers. In MN culture, SMA III and asymptomatic exhibited an elevated SMN level in comparison to control. Quantification of SMN mRNA expression in individual lines of (B) fibroblasts, (C) iPSCs, (D) smNPCs and (E) MN cultures (day 27) is shown. Expression was normalised to total RNA levels and significance calculated relative to 30 m-r12 (individual cell lines) and grouped five healthy controls (phenotype groups), respectively (Kruskal–Wallis test, p < 0.05 = *, p < 0.01 = **). Error bars represented ± SEM of duplicates (n = 2). MN culture displayed data of one to three MN differentiation experiments (TIFF 3619 kb)
18_2015_2084_MOESM8_ESM.tif
Supplementary material 8: Quantitative analyses of PLS3 mRNA expression (A) In fibroblasts, PLS3 mRNA expression levels hardly differed among the classes. Expression rates in iPSCs showed a wider spreading with asymptomatic at the top, SMA I and controls following closely and SMA III at lowest level. While asymptomatic prevailed as strongest expressing group underlining putative overexpression, PLS3 expression continuously declined in controls resulting in barely any detectable PLS3 mRNA in MN cultures. Expression in SMA I also dropped to levels equal to SMA III in MN cultures. Importantly, PLS3 levels in asymptomatic were significantly elevated in comparison to controls. Quantification of PLS3 mRNA expression in individual lines of (B) fibroblasts, (C) iPSCs, (D) smNPCs and (E) MN cultures (day 27) is shown. Expression was normalised to total RNA levels and significance computed relative to 30 m-r12 (individual cell lines) and grouped five healthy controls (phenotype groups), respectively (Kruskal–Wallis test, p < 0.05 = *, p < 0.01 = **). Error bars represented ± SEM of duplicates (n = 2). MN culture displayed data of one to three MN differentiation experiments (TIFF 3735 kb)
18_2015_2084_MOESM9_ESM.tif
Supplementary material 9 Quantitative analyses of SMN protein expression. Western blot analysis from healthy controls, SMA I- and SMA III-affected and asymptomatic discordant siblings in (A) fibroblasts, (B) iPSCs, (C) smNPCs, (D) MN cultures (day 27). Expression was normalised to β-actin protein levels and significance calculated relative to 30 m-r12 (individual cell lines, left hand panels) and grouped five healthy controls (phenotype groups, right hand panels), respectively (Kruskal-Wallis test). In fibroblasts, iPSCs and smNPCs error bars represented ± SEM of duplicates (n = 2). In MN culture (day 27) error bars represented ± SEM of two to three independent MN differentiation experiments (TIFF 4061 kb)
18_2015_2084_MOESM10_ESM.tif
Supplementary material 10 Quantitative analyses of PLS3 protein expression. Western blot analysis from healthy controls, SMA I- and SMA III-affected and asymptomatic discordant siblings in (A) fibroblasts, (B) iPSCs, (C) smNPCs and (D) MN culture (day 27). Note that all asymptomatic cell lines except for HGK16 strikingly elevated PLS3 expression in comparison to SMA III and controls. Expression was normalised to β-actin protein levels and significance calculated relative to 30 m-r12 (individual cell lines, left hand panels) and grouped five healthy controls (phenotype groups, right hand panels), respectively (Kruskal–Wallis test, p < 0.01 = **, p < 0.001 = ***, p < 0.0001 = ****). In fibroblasts, iPSCs and smNPCs error bars represented ± SEM of duplicates (n = 2). In MN culture (day 27) error bars represented ± SEM of two to three independent MN differentiation experiments (TIFF 3854 kb)
18_2015_2084_MOESM11_ESM.tif
Supplementary material 11 Protein expression levels in control cell lines. (A) Representative Western blots from male and female controls visualising SMN and PLS3 protein expression in MN culture on day 27. (B) Graphs visualise PLS3 expression on RNA and protein level in MN cultures on day 27 according to gender of controls. Note that no different PLS3 protein expression was detected between male and female controls. (C) PLS3 in vitro siRNA-mediated knock-down shows the upper band as the correct specific PLS3 signal (marked with an arrow). The lower band is unspecific (TIFF 2958 kb)
18_2015_2084_MOESM12_ESM.tif
Supplementary material 12: Analysis of growth cones in mixed MN cultures on day 27. (A) Growth cones of male control 30 m-r12, female control 51f-s6, SMA III HGK27.13 and asymptomatic HGK21.8 visualising PLS3 (green) and actin (red) in growth cones and MAPT (magenta) in axons. Growth cones were magnified (dashed lines) in PLS3/actin merge in insets. Scale bar 10 µm, valid for all images. (B) Average area size was measured by encircling every growth cone individually. In comparison to controls, average area size was increased in SMA III and asymptomatic yet only in asymptomatic the difference was significant. (C) Co-localisation of PLS3 and actin was calculated by Pearson’s coefficient. However, Pearson’s coefficient accounted for a comparable value (control: 0.52; SMA III: 0.52; asymptomatic: 0.47) among all three phenotype groups indicating a medium co-localisation between PLS3 and actin. Growth cones were analysed from four independent MN differentiation experiments (n = 4). In total, n = 28 (controls 30 m-r12, 35 m-r1, 51f-s6), n = 22 (SMA III HGK27.10, HGK27.13) and n = 20 (asymptomatic HGK21.1, HGK21.8, HGK28.9) growth cones were analysed. Significance was calculated relative to grouped three healthy controls (Kruskal-Wallis test, p < 0.05 = *) (TIFF 33,462 kb)
18_2015_2084_MOESM13_ESM.tif
Supplementary material 13: Overview table listed specific features of iPSC lines such as originating parental fibroblast line, SMA phenotype, age, sex, SMN1/SMN2 copy number, elevated (↑) PLS3 expression levels in blood, reprogramming technique and validation criteria. Note that numbers in HGK denote individual line and additional clone number of the same line (TIFF 2953 kb)
18_2015_2084_MOESM15_ESM.tif
Supplementary material 15: List of primary antibodies and respective secondary antibodies (gt = goat, ms = mouse, rb = rabbit) (TIFF 2967 kb)
Rights and permissions
About this article

Cite this article
Heesen, L., Peitz, M., Torres-Benito, L. et al. Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell. Mol. Life Sci. 73, 2089–2104 (2016). https://doi.org/10.1007/s00018-015-2084-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2084-y