
Abstract
Patients suffering from tauopathies including frontotemporal dementia (FTD) and Alzheimer’s disease (AD) present with intra-neuronal aggregation of microtubule-associated protein Tau. During the disease process, Tau undergoes excessive phosphorylation, dissociates from microtubules and aggregates into insoluble neurofibrillary tangles (NFTs), accumulating in the soma. While many aspects of the disease pathology have been replicated in transgenic mouse models, a region-specific non-transgenic expression model is missing. Complementing existing models, we here report a novel region-specific approach to modelling Tau pathology. Local co-administration of the pore-former polymeric 1,3-alkylpyridinium salts (Poly-APS) extracted from marine sponges, and synthetic full-length 4R recombinant human Tau (hTau) was performed in vitro and in vivo. At low doses, Poly-APS was non-toxic and cultured cells exposed to Poly-APS (0.5 µg/ml) and hTau (1 µg/ml; ~22 µM) had normal input resistance, resting-state membrane potentials and Ca2+ transients induced either by glutamate or KCl, as did cells exposed to a low concentration of the phosphatase inhibitor Okadaic acid (OA; 1 nM, 24 h). Combined hTau loading and phosphatase inhibition resulted in a collapse of the membrane potential, suppressed excitation and diminished glutamate and KCl-stimulated Ca2+ transients. Stereotaxic infusions of Poly-APS (0.005 µg/ml) and hTau (1 µg/ml) bilaterally into the dorsal hippocampus at multiple sites resulted in hTau loading of neurons in rats. A separate cohort received an additional 7-day minipump infusion of OA (1.2 nM) intrahippocampally. When tested 2 weeks after surgery, rats treated with Poly-APS+hTau+OA presented with subtle learning deficits, but were also impaired in cognitive flexibility and recall. Hippocampal plasticity recorded from slices ex vivo was diminished in Poly-APS+hTau+OA subjects, but not in other treatment groups. Histological sections confirmed the intracellular accumulation of hTau in CA1 pyramidal cells and along their processes; phosphorylated Tau was present only within somata. This study demonstrates that cognitive, physiological and pathological symptoms reminiscent of tauopathies can be induced following non-mutant hTau delivery into CA1 in rats, but functional consequences hinge on increased Tau phosphorylation. Collectively, these data validate a novel model of locally infused recombinant hTau protein as an inducer of Tau pathology in the hippocampus of normal rats; future studies will provide insights into the pathological spread and maturation of Tau pathology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tau, a microtubule-associated protein which stabilises axonal microtubules and regulates axonal transport [1, 2], aggregates into insoluble neurofibrillary tangles (NFTs) as the core pathology in what is commonly referred to as tauopathies and includes frontal temporal dementia (FTD) and Alzheimer’s disease (AD). Whilst mutations within the Tau gene are causative to FTD [3], in AD, Tau pathology occurs in the absence of identified Tau mutations, most likely downstream from the accumulation of a variety of β-amyloid species [4]. At the core of NFTs, Tau is abnormally and excessively phosphorylated [5, 6] and this correlates with disease-related cognitive impairments [7, 8].
Tau is phosphorylated at multiple serine/threonine (ser/thr) [9, 10] and tyrosine residues [11–13] by several protein kinases including glycogen synthase kinase 3 β (GSK3β) [10–14] and CDK5 [15]. Overphosphorylated Tau loses its restricted expression within axons, accumulates in somatodendritic compartments [16, 17] and correlates with a loss of microtubule binding and a shift in Tau solubility to a state of insolubility [10–18]. Thus, excessive Tau phosphorylation alone may be necessary and sufficient for pathological impairment of neural function. Although some studies in Tau mutant mice indeed correlate the onset of behavioural deficits [19, 20] as well as disruptions in critical cellular signalling pathways [21] with soluble phospho-Tau levels in the absence of an elevation of insoluble Tau levels, such a demonstration without the confounding influences of FTD mutations and/or gross Tau overexpression remains elusive. Equally, the demonstration of improved memory upon the suppression of FTD Tau expression, despite continued formation/persistence of NFTs [22, 23] further supports the notion that soluble Tau phosphorylation is strongly connected with neuronal dysfunction.
Kinase-mediated Tau phosphorylation is opposed by phosphatases, of which protein phosphatase 2A (PP2A) principally regulates Tau phosphorylation in vivo [14, 24, 25], though in vitro Tau is a suitable substrate for PP1, PP2A, PP2B and PP5 [26–30]. In AD brains, there is strong evidence for impaired phosphatase activity, primarily due to a reduction in PP2A expression and activity, but also as a consequence of up-regulation of the endogenous inhibitor of PP2A (I PP2A1 ), alongside a modest decrease in PP5 activity [30–33]. Intriguingly, several mutations in exons 9, 10, 12 and 13 of the Tau gene confer reduced affinity for PP2A [34], providing a possible common pathway of excessive Tau phosphorylation via diminished dephosphorylation between sporadic and familial tauopathies. Indeed, the recent use of statins as potential therapeutic strategy has demonstrated protective effects in both FTD Tau variants and Okadaic acid (OA)-mediated Tau toxicity via a shared mechanism augmenting Tau dephosphorylation [35].
In AD, Tau pathology advances in a stereotypical fashion, led by the spread of phospho-Tau-positive pre-tangle neurons and later the appearance of NFTs. Initially, pathology is seen within the transentorhinal cortex before spreading to the hippocampus and then to neocortical regions [16–36]. We have recently reported a novel mouse expressing truncated but non-mutated Tau 296–390, which genuinely replicates pathology of Braak staging and presents with a predominantly cognitive phenotype [37]. Although it remains unclear how this expression pattern is realised, pathology seems to invade other brain regions following their connectivity pathways in a prion-like fashion. Release from pre- and uptake into post-synaptic structures is a widely proposed feature for mimicking AD in experimental models and has been achieved in vivo by the systemic intraperitoneal or the local inoculation of aggregated beta amyloid (Aβ) seeds or synthetic pre-aggregated Aβ into brains of transgenic APP mice, but seeding was ineffective in non-transgenic mice [38, 39]. Similarly, Tau extracts recovered from P301L mutant mice or preformed synthetic Tau fibrils initiated widespread advanced Tau pathology when injected into young transgenic Tau-overexpressing animals, which alone have only modest changes in phospho-Tau levels and no shift in Tau solubility [40–42]. Yet, trans-synaptic pathological spread of Tau has been demonstrated in wild-type rats locally infected via lentivirus delivering wild-type hTau [43].
The increased use of rats has resulted in several benefits to AD research [44] due to higher similarity with human genetics and cell physiology compared to mice [45, 46]. Especially for the investigation of Tau, rats express not only the 4R Tau isoforms of mice [47], but also the 3R isoforms in adulthood [48] akin to the human proteome [49]. Such human homology may be of critical importance when modelling certain aspects of tauopathies given the putative differences in pathological processing of 3R or 4R isoforms of Tau [49].
Consequently, we aimed at the induction of Tau pathology in intact normal rats by the local administration of synthetic full-length hTau with or without the promotion of increased Tau phosphorylation via OA-mediated phosphatase inhibition. Reasoning that the first clinical signs are dependent on Tau spread from entorhinal cortex to hippocampus, with Tau pathology occurring as early as stage 2 in CA1 [50]. We therefore targeted CA1 for infusion to establish proof of principle for Tau-mediated neuronal deficits and cognitive impairment. The selection of CA1 was further supported by its well-characterised role in learning and memory with underlying changes in synaptic plasticity readily detectable by electrophysiological means.
As a delivery tool, we co-administered polymeric 1,3-alkylpyridinium salts (Poly-APS) to facilitate access of the bulky Tau protein (1.69A: [51]) across the neuronal cell membrane. Poly-APS isolated from a range of haplosclerid marine sponges [52] forms transient pores within cell membranes [53, 54], including rat hippocampal neurons [55] and calculated by the Renkin equation to be 2.9 nm (29 Angstrom) in diameter [56]. Chemically, Poly-APS consists of a unique repeating structure incorporating a positively charged pyridinium group linked from the pyridinium N to the 3-position on the next pyridinium via an alkyl chain, typically 5–12 carbons in length. The connecting chain is mainly saturated alkyl, but has also been shown to contain additional methyl branches or double bonds [57, 58]. In many cases, the connecting chain is a consistent length (i.e., the monomers are identical) but in at least one case it has been shown that the monomer units contain saturated alkyl chains of differing lengths from C5 to C11 [59]. Overall polymer length can vary from ca 20 monomer units (MW ca 5 kDa) to much larger, with reports of 100 monomer units (ca 20 kDa) [60] to potentially >100 kDa [61]. In most cases, mixtures with different degrees of polymerisation are found, giving cocktails of Poly-APS toxins with a large range of molecular weights. Poly-APS has previously been used for the intracellular delivery of enhanced green fluorescent protein (EGFP) cDNA [54]; in this study, the transient pores enabled the entry of recombinant hTau from the extracellular space, thus modestly elevating total Tau levels within the cell. Here, we report the initial confirmation and technical validation of Poly-APS-mediated hTau loading in cultured hippocampal neurons and in rats. In vivo, persistent Tau pathology and behavioural deficits were observed in a phosphorylation-dependent manner providing the basis for the future characterisation of Tau-mediated pathological spread and maturation.
Methods
Drug preparation
Poly-APS was purified from the marine sponge Haliclona sarai (previously Reneira sarai) as previously described [52]. This preparation contained a mixture of mainly two polymers with molecular weights of 5.5 and ~19 kDa [number of units in each polymer; n = 29 and n = 99, with a connecting alkyl chain of (CH2)8]. Test solutions containing Poly-APS were prepared from a stock solution of 5 mg/ml Poly-APS in distilled water. Tau (Tau441 or hTau; recombinant, Sigma-Aldrich, Poole UK) was reconstituted in distilled water to a concentration of 1 mg/ml (21.8 mM) and stored at −80 °C until the day of use. Okadaic acid (OA; Sigma-Aldrich, Gillingham, UK) was dissolved in 1 % dimethylsulfoxide (DMSO, Sigma-Aldrich) to a stock concentration of 100 µM and stored at −20 °C. Glutamate (Glu; Tocris, Bristol, UK) was dissolved in equimolar concentrations of NaOH to a stock concentration of 100 mM and stored at −20 °C.
In vitro experiments
Cell culture and culture treatment
For functional experiments (electrophysiology and Ca2+ imaging), primary mixed hippocampal cultures were prepared from 1- to 3-day-old Sprague–Dawley rats [62, 63]. Cells were plated on poly-1-lysine-coated coverslips and bathed in minimal essential medium (Invitrogen Paisley, UK) containing 0.002 mM l-glutamine (Sigma-Aldrich) and 10 % foetal bovine serum (Invitrogen). Cultures were maintained for up to 1 week at 37 °C in humidified air with 5 % CO2.
For immunocytochemistry, primary hippocampal cultures were prepared from embryonic (day 18) Wistar rat brains. Cells were plated on coverslips coated with poly-l-lysine (30 μg/ml) and laminin (2 μg/ml) at a density of 75,000/well. Hippocampal cultures were grown in Neurobasal A medium (NB) supplemented with B27 and L-alanyl-l-glutamine (Invitrogen). The cultures were maintained for up to 13 days at 37 °C in humidified air with 5 % CO2.
Poly-APS-mediated intracellular macromolecule delivery was achieved using a similar incubation protocol as used to transfect HEK-293 cells with cDNA [54]. Cultures were incubated for 3 h in NaCl-based recording solution (mM: NaCl, 130; KCl, 3.0; CaCl2, 2.0; MgCl2, 0.6; NaHCO3 1.0, HEPES 10.0, glucose 5.0) containing 5 µg/ml bovine serum albumin (BSA), aiding the recovery of cells from Poly-APS treatment [54]. In initial studies, the 5 µg/ml BSA containing NaCl-based solution was supplemented with 1 mM Lucifer yellow (LY; Sigma-Aldrich). Incubations were conducted either in the presence or absence of 0.5 µg/ml Poly-APS for 3 h at 4 °C to minimise the contribution of endocytosis to LY uptake [64]. For all functional experiments, hTau loading was achieved by application of 1 ml, 0.5 µg/ml Poly-APS, 0.05 mg/ml BSA containing HEPES buffered saline (HBS) with the addition of hTau 441 at 1 µg/ml. Incubations were conducted for 3 h at room temperature before wash and return to sterile culture medium. Two further groups, Poly-APS and Poly-APS+hTau-treated neurons, were returned to culture medium containing 1.2 nM OA for 24 h followed by recording. A small number of culture dishes were stained for pan Tau and Tau441 expression; these dishes were incubated with 0.005 µg/ml Poly-APS solution containing Tau441 at a concentration of 2 µg/ml. Cultures were washed in pre-warmed HBS after completion of the 3 h Poly-APS incubation and fixed in ice-cold methanol, supplemented with 1 mM EDTA at −20 °C for 5 min, followed by a 5-min incubation in 4 % formaldehyde/4 % sucrose in 0.1 mM phosphate buffered saline (PBS) at room temperature. For details of immunostaining see below.
In vitro cell culture electrophysiology
Whole-cell recordings [65] of hippocampal neurons were conducted at room temperature (~21 °C) to determine membrane potential, input resistance (evaluated from 100 to 300 ms electrotonic potentials evoked by −50 to −200 pA current commands) and the properties of action potentials as described previously [53–59]. Whole-cell configurations were achieved using low resistance (4–10 MΩ) borosilicate glass pipettes and the voltage clamp amplifier (Axoclamp-2A, Molecular Devices, Wokingham, UK) operated at a sampling rate of 15–20 kHz. Both pH and osmolarity of extracellular and patch solution (mM: KCl, 140; EGTA, 5; CaCl2, 0.1; MgCl2, 2.0; HEPES, 10.0; ATP, 2.0) were kept neutral and at 315 mOsmol/l. The electrophysiological data were stored on digital audio tape and subsequently analysed using CED voltage clamp software (Version 6, Cambridge Electronic Design, Cambridge, UK). For monitoring changes in membrane potential or holding current, continuous records were obtained on a chart recorder (Gould 2200 s pen recorder). For experimental groups and total cell numbers, see Table 1.
All data are given as mean ± SEM, and statistical significance (alpha set to 5 %) was determined using the Student’s two-tailed t test, paired or independent where appropriate, and P values are reported in the text.
Ca2+ imaging
Fura-2 Ca2+ imaging was performed as previously described [62, 63]. After loading with Poly-APS and/or hTau/OA, cultures were incubated in HBS containing 5 µM Fura-2-AM for 1 h at room temperature and then imaged via fluorescence time-lapse microscopy (Olympus BX5W1, 40×, Olympus, Southend-on-Sea, UK). Fura-2 was stimulated with alternating wavelengths of light (340/380 nm) from a Lambda DG-4 illumination system (Sutter Instruments, Novata, USA), and emission at 510 nm captured by a digital CCD camera (Orca-ER; Hamamatsu, Japan) connected to a computer running Openlab (V.4.02, Improvision, Coventry, UK). Ratiometric measurements from regions of interest (ROI) positioned over the cell bodies of neurons were recorded every 5 s. Cell depolarisation was induced with a 1-min 50 µM Glu or a 2-min 50 mM KCl (VWR, Lutterworth, UK) containing HBS application through a gravity-fed perfusion system at a rate of ~5 ml/min. For experimental groups, see Table 1. All solutions contained the sodium channel blocker Tetrodotoxin (TTX, 0.5 μM, Abcam Biochemicals, Cambridge, UK) to prevent spontaneous discharges. Ca2+ transients were measured as peak change in Fura-2 fluorescence (Fpeak − Fbaseline/Fbaseline = %ΔF/F). ROIs which produced <10 %ΔF/F were deemed to be non-responders and were excluded from further analysis. The overall responder rate was used as a metric for the viability of the cultures following Poly-APS treatment [63]. Data are shown as mean ± SEM, statistical significance was determined by a non-parametric Kruskal–Wallis test and post hoc analysis via a Dunn’s multiple comparison test. Alpha was set to 5 %.
In vivo/ex vivo experiments
Animals and design
Three-month-old male Lister-hooded rats (Harlan, UK) weighting 250–300 g at the start of the study were used. Animals were group housed in pairs or trios prior to surgery and singly caged afterwards. They were maintained on a 12-h day–night cycle (lights on at 7 am) and ad libitum accesses to food and water; a summary of groups and numbers is given in Table 2. Experiments were carried out in accordance with the European Communities Council Directive (63/2010/EC) and a project license with local ethical approval under the UK Scientific Procedures Act (1986).
In practice, all rats underwent stereotaxic surgery for administration of Poly-APS/hTau/OA (see Fig. 1 and details below). Independent of treatment, behavioural testing commenced 2 weeks post-surgery and lasted for 11 days spread out over 2 weeks. This was then followed 2–3 days later by tissue harvest for histochemistry or during a further week for ex vivo electrophysiology.

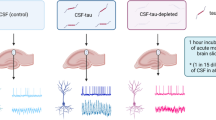
Surgical and behavioural protocol for in vivo loading of hippocampal neurons with human Tau through Poly-APS. a Coronal sections taken from rat brain atlas [125] and defined infusion sites (S1–S3). All infusions were bilateral. From Bregma in mm: Site 1 (S1); anterior–posterior (A-P) −0.33, lateral (L) ± 0.16, depth (D) −0.28, Site 2 (S2); A-P −0.45, L ± 0.30, D −0.30, Site 3 (S3); A-P −0.56, L ± 0.40, D −0.32. b Sequence of drug infusion: 2 µl of 0.005 µg/ml Poly-APS and 2 µl of 1 µg/ml hTau were infused at a rate of 0.4 µl/min with an interval of 5 min into all sites. In groups receiving 1.2 nM okadaic acid (OA), this was infused via S2 over a 7-day period via osmotic minipumps. A 7-day washout period following the removal of the pump was given before behavioural assessment. c Water maze spatial reference memory training/testing protocol: all groups were trained for reference memory for 4 trials (t) per day (d), and recall was then tested in 2 probe trials 24 h and 96 h post-completion of acquisition, 4 reversal trials (Rt) per day were given followed by reversal probe trial (RProbe) (see text for details)
Surgery
Surgery was performed in line with previous work [66]. In total, 52 rats (44 for behavioural work and 8 for immunohistochemistry) were anaesthetised with an intraperitoneal (i.p.) injection of tribromoethanol (1 ml/100 g) with regular top-ups as required and placed in a large parallel rail stereotaxic frame (Stoelting, Wood Dale, IL, USA). Three infusion sites along the dorsoventral axis of the hippocampus were identified in each hemisphere (Fig. 1a, from Bregma: (S1) anterior–posterior (AP) −0.33 mm, lateral (L) ± 0.16 mm, depth (D) −0.28 mm; (S2) AP −0.45 mm, L ± 0.30 mm, D −0.30 mm; (S3) AP −0.56 mm, L ± 0.40 mm, D −0.32 mm). Animals were acutely infused with 2 µl of solution using a Hamilton syringe and PHD 2000 infusion pump (Harvard Apparatus, Cambridge, UK) at a flow rate of 0.4 µl/min bilaterally. For optimal vector spread, a 5-min interval was allowed before a second infusion of 2 µl (Fig. 1b). The acute infusion cannula consisted of a double barrel stainless steel cannula (21 gauge) connected via polyethylene tubing with two syringes. Poly-APS (0.005 μg/ml) was administered first and in some groups followed by hTau441 (1 μg/ml) through the second barrel, before moving to the next site. In all OA groups, this was followed by the attachment of a 7-day osmotic micropump (Alzet, Charles River UK, Kent, UK; flow rate 0.5 ± 0.1 µl/h over 7 days, reservoir volume 90 ± 10 µl) containing 1.2 nM OA infused at site 2 (Fig. 1a, b). All drugs were dissolved in sterile artificial cerebrospinal fluid (aCSF; (in mM): 129.5 NaCl, 1.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1.5 KH2PO4, 25 NaHCO3 and 10 Glucose (pH 7.4)). Animals were given at least 1-week recovery/washout after OA infusion prior to commencement of behavioural testing.
Water maze apparatus and testing procedure
Spatial reference memory was tested using an open-field water maze protocol established previously [67]. The water maze (circular pool, 150 cm diameter, 50 cm height) was filled with water (23°C ± 1) and a circular clear Perspex platform was placed in one of the four pool quadrants. The platform was submerged 1 cm below surface of the water. The animal’s swim paths were tracked by an overhead CCTV camera, captured and analysed using PC-based Ethovision 3.1Pro (Noldus, Wageningen, The Netherlands).
Animals were trained for acquisition (Fig. 1c) to a randomly assigned fixed platform (all pool quadrants used as targets, fully counterbalanced by treatment cohort). Acquisition training consisted of four trials (max 90 s) per day (Monday–Thursday), in which the rats were released at one of four cardinal sites (N, E, S, W) in a pseudorandom fashion. Upon finding the platform or if the trial time had elapsed, following a guided swim, animals were left on the platform for 30 s, dried and returned to their holding cage for an inter-trial interval (ITI) of 90 s. Two probe trials were conducted 24 and 96 h (not shown) post-training, in which the platform was removed and animals were released into the maze from the site directly opposite to the platform for a 60-s free swim.
The following day, a 2-day reversal training paradigm was conducted, in which the platform position was shifted to the opposite quadrant; all other parameters remained the same. Similar to the acquisition phase, a probe trial was administered 24 h following completion of the reversal training.
Behavioural parameters extracted from the swim paths included the overall path length of each trial as a spatial proxy, the swim speed as a motor and activity read-out, and thigmotaxis (wall hugging in the outer 10 cm of the pool) as an index of failure to implement a spatial search strategy. These were averaged for each group and are presented as mean per trial and analysed using 2-way repeated measures analysis of variance (ANOVA) with treatment as between subject and trial as within-subject factor and was followed by appropriate planned post hoc Bonferroni corrected t tests. We also analysed the cumulative performance in each group. Maximum performance (floor level) was derived from the Poly-APS group over the last 4 trials (168 cm) and the percentage of animals that reach this performance level in each trial was plotted and fitted to a nonlinear function. Probe trials were analysed for % time in target relative to non-target quadrants. Alpha was set to 5 %.
Ex vivo slice electrophysiology
Following completion of behavioural assessment, tissue from selected groups was harvested for ex vivo slice electrophysiology. Animals were terminally anaesthetised with halothane, decapitated, brains removed, hippocampi dissected out and placed in ice-cold aCSF [68, 69]. Hippocampal slices were prepared (~400 µm thick) using a McIlwain tissue chopper, and transferred to pre-warmed aCSF (37 °C) where they remained for a minimum of 1 h before recording. All solutions were continuously gassed (5 % O2: 95 % CO2) and heated (37 °C).
Extracellular field recordings of population spikes were obtained using a monopolar stimulating electrode (WPI, 0.5 MΩ) positioned in the Schaffer-collateral fibre pathway and a borosilicate glass aCSF-filled AgCl recording electrode (3–4 MΩ) was inserted into the CA1 pyramidal cell body layer. Input/output curves of basal synaptic transmission were established by application of stimulations (20 μs durations) increasing in 5 V increments from 30 V until the evoked responses were saturated (85 V).
To measure synaptic plasticity, slices were stimulated at 50 % maximal stimulation every 30 s to determine stable baseline responses (±10 % variation) for >10 min. Long-term potentiation (LTP) was induced by a theta (5 Hz) tetanus (150 bursts of four stimuli (100 Hz), 200 μs inter-burst interval for 30 s). Post-tetanus responses were recorded every 30 s for the proceeding 60 min. Two responses recorded at 30-s intervals were averaged to provide a mean response per min and calculated relative to baseline (in percentage). The ex vivo recorded amplitude of the system-relevant output summed as population spike was used as a proxy for synaptic plasticity of hippocampal synapses [68–70].
Data are expressed as group mean ± SEM and were analysed using factorial two-way ANOVA with drug treatment as between subject and step-wise increment of input or time as repeated within-subject measure. The level of tetanus-induced LTP was determined at 60 min post-tetanus with one-way ANOVA comparison of treatments. As for behaviour, alpha was set to 5 %.
Tissue fixation
For histological brain sections, animals were deeply anaesthetised with Nembutal until loss of reflex function followed by intra-cardial perfusion with cold (4 °C) 0.9 % NaCl for 3–5 min, paraformaldehyde (4 % in 0.1 M phosphate buffer, pH 7.4) for 5–7 min, and finally with PBS containing 5 % glycerol with 2 % DMSO for 3–4 min. Brains were removed and placed for 1 h in fixative and then immersed for cryoprotection in 10 % followed by 20 % glycerol and 2 % DMSO in phosphate buffer. Brains were positioned in a brain matrix (BAS Inc., Lafayette, USA) and evenly sliced into uniform blocks. Coronal sections were cut throughout the dorsal hippocampal region at 40-μm thickness with a cryostat (Leica Biosystems, Germany) and series of sections were collected of each rat and processed.
Immunochemistry
For immunochemical determination, the following antibodies and dilutions were applied: for total Tau, monoclonal anti-Tau, clone Tau46 (1:1000, Sigma); for human-specific Tau, monoclonal anti-Tau, clone TAU-2 (1:1000; Sigma); for Tau phosphorylation, anti-p-Tau(Ser396) PS396 (1:200, Santa Cruz Biotechnology Inc). From 5 series of the sections collected, one set was processed for Tau46 immunoreactivity, the second for p-Tau(Ser-396) immunoreactivity, the third for TAU-2 immunoreactivity, the fourth was Nissl stained to verify injection sites and the fifth corresponding series of sections was stored at −20 °C in a cryoprotective solution as a reserve.
Cultures and ex vivo free-floating sections were incubated in PBS, pH 7.4 with the primary antibody overnight at 4 °C and subsequently with goat anti-rabbit biotin-conjugated IgG (diluted 1:1000, Chemicon, Int., Inc.) or goat anti-mouse biotin-conjugated IgG (diluted 1:1000, Sigma) followed by peroxidase-conjugated streptavidin (1:1000, The Binding Site Ltdmt.) for 1 h each. Sections for p-Tau(Ser-396) were incubated in PBS containing 1 mM sodium orthovanadate (Na3VO4) with appropriate antibodies (as above). Sections were washed 3 times for 5 min, incubated for 30 min with peroxidase conjugated with streptavidin, washed, and amplified using tyramide signal reagent (PerkinElmer LAS, Inc.). After another wash and peroxidase incubation, slices reacted with 0.05 % diaminobenzidine (DAB) in the presence of 0.01 % hydrogen peroxide. Finally, sections were mounted on glass microscope slides, air-dried, dehydrated in alcohol-xylene serial solutions and cover-slipped.
Controls for the immunohistochemical procedure were obtained running some slides through the entire procedure with the omission of primary or secondary antibodies. No staining was observed in these control slides. Images were captured using bright field microscopy (Nikon Eclipse NiE equipped with Nikon DS.-Ri1 camera and NIS Elements software, Surrey, UK).
Results
Poly-APS enables intracellular delivery of Tau protein
The transient pore-forming properties of Poly-APS, when applied to cultured rat hippocampal neurons, have been previously characterised [55]. Using a similar loading protocol to that employed for the transfection of HEK 293 cells with cDNA [54], the ability of Poly-APS to deliver membrane-impermeable macromolecules into the intracellular compartment was established. We first explored the permeability of hippocampal cultures for Lucifer Yellow (1 mM LY at 4 °C). The presence of Poly-APS during the incubation substantially enhanced LY uptake and it appeared ubiquitous throughout the cytoplasm and processes of neurons (Fig. 2a). Cultures treated without the pore-former showed no LY loading. In line with these observations, delivery of the human 2N4R Tau protein under similar conditions confirmed the penetration of Tau into pyramidal cells as visualised by a total Tau (clone Tau-46) and human Tau-specific (clone TAU-2) immunostaining (Fig. 2b). Immunoreactivity toward clone Tau-46 appeared strongly enhanced in cultures treated with Poly-APS and hTau, thus confirming the ability of Poly-APS to facilitate intracellular loading with hTau. Similarly, Poly-APS and hTau treatment resulted in robust Tau-2 staining in all compartments of pyramidal cells, when there was virtually no human-specific Tau labelling in controls.

Poly-APS-mediated intracellular macromolecule delivery in hippocampal cultures. a Cultures incubated with 1 mM Lucifer yellow (LY) in the presence of 0.5 µg/ml Poly-APS for 3 h resulted in strong expression of intracellular LY in the soma and processes of neurons (right column). Only a superficial staining with LY is seen in absence of Poly-APS (left column). b, i Incubation of cells with Poly-APS and 1 µg/ml of human Tau (hTau) for 3 h drastically increased immunoreactivity towards the Tau-46 pan Tau antibody, in contrast to untreated cultures (controls). b, ii Human Tau immunoreactivity was established with the TAU-2 antibody only on Poly-APS-treated cultures. Bar = 50 µm
Phosphorylation of Poly-APS-delivered hTau alters electrophysiological properties and Ca2+ signalling in cultured hippocampal neurons
Having established the successful intracellular delivery of hTau into cultured rat hippocampal neurons, the modulation of basic membrane properties and excitability changes caused by exposure of cells to Poly-APS, hTau and OA was assessed. Treatment of neonatal rat hippocampal cultures with Poly-APS, or Poly-APS in combination with hTau or OA had no effect on resting-membrane potentials, input resistance to depolarising stimuli or action potential firing properties (Fig. 3a, b). However, combining the intracellular delivery of hTau via Poly-APS with a 24-h OA incubation dramatically reduced the excitability of hippocampal neurons and prevented action potential firing even when cells were held by constant current injection at −70 mV before application of depolarising current stimulus (Fig. 3a, b). Treatment with Poly-APS+hTau+OA collapsed resting-membrane potentials from −63 ± 5 mV (control) to −22 ± 5 mV (Fig. 3b; n = 11 for Poly-APS+hTau+OA and n = 12 for controls, p < 0.001). Similarly, the input resistance was lowered from 1192 ± 183 (n = 12) to 550 ± 105 MΩ (n = 11; p < 0.01) in Poly-APS+hTau+OA-treated neurons. Despite the reduction in input resistance and a collapse of resting-membrane potential, Poly-APS+hTau+OA-treated neurons were still viable (see responder rates, Fig. 3c, d). Using fura-2 imaging, Ca2+ homeostasis and Ca2+ signalling was assessed. Whilst none of the treatments affected resting Ca2+ levels (p > 0.05, data not shown), Ca2+ transients evoked by a 1-min application of 50 mM KCl or evoked by a 2-min application of glutamate (Glu) were reduced following Poly-APS+hTau+OA treatment (Fig. 3c, d). Overall comparison of KCl-induced peak Ca2+ influx (%ΔF/F) revealed a significant effect of treatment (p < 0.001, n = 18–39, see Table 1), and the post hoc multiple comparison highlighted a significant reduction in neurons exposed to Poly-APS+hTau+OA compared to Poly-APS only (Fig. 3d; p < 0.01, n = 26 for Poly-APS+hTau+OA and n = 34 for Poly-APS). No other comparison proved significant (p’s > 0.05). Similarly, responses towards 50 µM Glu were also affected by treatment (Fig. 3d; p < 0.0001, n = 20–39) and Glu-induced Ca2+ responses were significantly lower in Poly-APS+hTau+OA-treated cultures in comparison to all other groups (Fig. 3d, p’s < 0.05). Neither Poly-APS+OA nor Poly-APS+hTau altered the glutamate response (p > 0.05).

Physiological voltage clamp and calcium responses of hippocampal cultures treated with Poly-APS, Tau (hTau), and/or OA. a Membrane potential in response to depolarising and hyperpolarising stimulation under current clamp. Poly-APS+hTau+OA-treated neurons failed to discharge action potentials in response to 450 pA current injection, from a holding potential of −70 mV. In comparison, injection of a 50 pA current into Poly-APS neurons resulted in a chain of action potentials. Likewise, the altered input resistance is highlighted by the disproportionate hyperpolarisation of membrane potentials established in response to −450 pA current step command. b Comparison of input resistance (mean + SEM) and resting-membrane potential (mean − SEM) following different Poly-APS treatments. Only neurons treated with Poly-APS+hTau+OA demonstrated depressed input resistance and reduced membrane potential, compared to all other conditions. Cultures treated with Poly-AP +hTau+OA also showed a reduced Ca2+ influx in response to a 1-min application of KCl (c; 50 mM) or a 2 min application of glutamate (d; Glu; 50 µM) as measured by %ΔF/F (Mean + SEM) via Fura-2 Ca2+ imaging. Insets display example traces from each treatment. Scale in seconds (s) and ratiometric value (340/380). Percentage of neurons responding is shown above the respective bars. *p < 0.05, **p < 0.01 and ***p < 0.001
Poly-APS enables intracellular delivery of hTau into rat hippocampal CA1 in vivo
Since passage of hTau through the cell membrane was enabled by Poly-APS in vitro, we next explored the efficacy of Poly-APS to deliver hTau intracellularly in vivo. Three-month-old hooded Lister rats underwent intra-hippocampal surgery, in which Poly-APS/hTau was administered at three sites (S1-3; see methods, Fig. 1a), with a subset of animals receiving an additional 7-day bilateral infusion of OA into the CA1 (S2 administration site, Fig. 1a) via indwelling cannulae to prompt Tau over phosphorylation. Following completion of OA infusion and a 7-day wash out period, rats were sacrificed and Tau expression and phosphorylation examined via immunohistochemistry. Relative to untreated controls, intensity of the pan Tau Tau-46 antibody immunostaining appeared elevated in Poly-APS+hTau groups confirming the intracellular accumulation of Tau in rat CA1 neurons (Fig. 4a). While controls did not return immunoreactivity for the human-specific TAU-2 antibody (Fig. 4b), there was a clear hTau labelling after Poly-APS+hTau treatment with a similar outcome independent of OA administration. Tau phosphorylation was also assessed using the p-Tau(S396) antibody. Phospho-Tau in Poly-APS-treated subjects was observed in cellular compartments different from controls (Fig. 4c). Somewhat fainter in controls and stronger in Poly-APS+hTau-treated rats, p-Tau(S396) was distributed in both cell bodies and neuronal fibres, showing a characteristic pattern of the morphological organisation of the pyramidal layer in CA1 area. The additional administration of OA caused a redistribution of p-Tau from neuronal processes to the soma. This is reminiscent of the age-dependent redistribution of Tau in rats [71] observed in the soma of hippocampal, cortical and basal forebrain neurons and could here be a result of excessive phosphorylation of loaded hTau. Critically, no overt lesions or tumour growth was observed in OA-treated animals as has been reported following the infusion of OA at much higher concentrations (mM) compared to nM used here; [72, 73].

Qualitative analysis of hTau loading and Tau phosphorylation following in vivo loading. Digital images of hippocampal CA1 region from brains of representative examples of untreated controls (left column), animals treated with Poly-APS+hTau (middle column), or treated with Poly-APS+hTau+OA (right column; for details, see "Methods"). a Tau-46 antibody labelling revealed an increase in Tau immunoreactivity in sections taken from both Poly-APS+hTau and Poly-APS+hTau+OA-treated animals. b Selective staining in Poly-APS+hTau groups with human-specific Tau-2 antibody. c Phosphorylated Tau [p-Tau(Ser396)] was confirmed in Poly-APS+hTau cohorts. However, a translocation of Tau from the axonal processes into the soma is apparent in sections taken from animals also treated with OA. Images taken at 10× and 40× (Bar = 50 µm)
Spatial learning deficits in Poly-APS+hTau animals is phosphorylation dependent
The differences in compartmentalisation of hTau in CA1 neurons may translate to functional differences. We reasoned that in agreement with human patients, phosphorylation of Tau may lead to stronger behavioural/cognitive decay than Tau exposure alone. This was confirmed in a spatial reference memory test conducted in the open-field water maze. In the five treatment conditions, three global observations in terms of path length were made: (1) all cohorts showed spatial learning and achieved floor level after 4 days of training (Fig. 5a, c; F(15,624) = 62.85, p < 0.0001 for factor trial); (2) control groups (Poly-APS, OA or hTau) were comparable to each other (Fig. 5a; no treatment effect); (3) there was a subtle but reliable difference in the experimental groups (Fig. 5c; effect of treatment: F(2,384) = 9.1; p < 0.0001; interaction between treatment and trial: F(30,384) = 1.5; p = 0.047). Planned comparison between treatments finally confirmed that Poly-APS+hTau+OA was significantly different from all other Poly-APS groups (F’s > 12; p’s < 0.01) or any control group (F’s > 8, p’s < 0.01). Since both treatment and trial interacted, we confirmed that variability occurred in a trial-dependent manner and indeed experimental groups differed for the average of trials 1 and 2 of each day (F’s > 6; p < 0.05), but not for the average of trials 3 and 4 (F’s < 1).

Subtle long-term memory deficits in rats with overphosphorylated hTau loading in hippocampus. Mean ± SE. a Path length to acquire a spatial reference memory task in the water maze of control groups consisting of S1–S3 injection of Poly-APS, or injection of hTau, or a minipump administration of OA into S2. There was no difference between cohorts. b Wall hugging behaviour as percent time in outer perimeter (10 %) of pool plotted over trials. All control groups readily abandoned thigmotaxis in favour of more spatial strategies. c Path length to acquire a spatial reference memory task in experimental groups consisting of Poly-APS as representative control, Poly-APS+hTau, and Poly-APS+hTau+OA. Only the latter group was impaired, mainly during trial 1 of each day. Note that there was no performance deficit on day 4 (trials 13-16). d Wall hugging behaviour was higher in Poly-APS+hTau+OA-treated subjects during trials 1–8, but normal in the Poly-APS + hTau cohort. e Swim speed during acquisition learning was not different between treatment groups, but peaked on the first daily trials. f Cumulative performance expressed as percent animals of each treatment (ordinate) reaching floor level in the respective trial (abscissa). All cohorts presented the same time course of learning except the Poly-APS+hTau+OA group which was retarded and displayed a rightward shift in the learning curve. g, h Probe trial data expressed as percent time spent in target (Tg) or other (O) quadrants (average for three quadrants) 24 h post-learning. All treatment groups presented a spatial bias for target which was significantly above chance ($ = p < 0.05, t test against chance) and different from other quadrants (asterisks, p < 0.05, paired t test). i Example probe trial swim paths for Poly-APS control and Poly-APS+hTau+OA experimental group; the target quadrant is shaded. OA okadaic acid
Drug exposure had no effect on swim speed (Fig. 5e), but some alterations in thigmotaxis became apparent in the OA and Poly-APS+hTau+OA groups. Okadaic acid increased thigmotaxis relative to other control groups (Fig. 5b, F’s > 7.1; p’s < 0.01), while Poly-APS+hTau+OA increased thigmotaxis relative to all groups (Fig. 5d, F > 7.9; p’s < 0.01).
A more in-depth analysis of swim path length across all training trials was conducted by calculation of the cumulative performance to achieve floor level (Fig. 5f). As is summarised in Fig. 5f, the number of achievers progressively increased in the different treatment conditions until reaching maximum level of 100 %. Only the Poly-APS+hTau group levelled out at 90 % due to one single rat not reaching criterion throughout. Of note is the rightward shift of the sigmoidal curve of the Poly-APS+hTau+OA group indicating an overall learning deficit in the early phase of training (trials 1–7); this was followed by a steeper part of the sigmoid so that 100 % of animals in this group did reach maximal performance at trial 10. Statistical comparison of the nonlinear fit was based on the null hypothesis that all data sets return the same mean. This hypothesis was rejected [F(4,70) = 36; p < 0.0001) due to a reliably different fit for the Poly-APS+hTau+OA group relative to all other treatments (F’s > 42; P’s < 0.0001), thus strongly supporting the notion that the rightward shift in performance was dependent on the phosphorylation of the exogenously delivered Tau molecules.
In the probe trial 24 h post-acquisition (platform removed), control (Fig. 5g, i) and experimental groups (Fig. 5h, i) presented with a significant spatial bias for the target platform (p’s < 0.05 compared to chance; unpaired t test, two tailed) and was significantly above the average time spent in the other three quadrants (p’s < 0.05; paired t test, two tailed). There was no effect of treatment on the % time spent in the target quadrant (F < 1) and no differences were found following a second probe trial conducted 96 h post-acquisition (data not shown).
Subtle impairment in behavioural flexibility in hTau group is phosphorylation dependent
Subsequent reversal training, in which the submerged platform was shifted to the opposite quadrant, yielded results similar to the original acquisition (Fig. 6). Globally, all cohorts acquired the task (main effects of trial in all comparisons: p’s < 0.001). The control cohorts (Fig. 6a) acquired the novel location within 2 days and achieved floor level performance (<124 cm) already in trial 6 (see Fig. 6f, F’s < 1 for treatment and interaction with trial). In the experimental groups, however (Fig. 6c), there was a slowing of reversal learning in the Poly-APS+hTau+OA-treated animals for the path length across the trials and the treatment factor interacted with trial [F(14,192) = 2.6; p = 0.002]. Importantly, the difference was solely due to an impairment in the Poly-APS+hTau+OA cohort compared to Poly-APS + hTau and Poly-APS alone groups (F’s > 2.8; p’s < 0.05 for effects including treatment as factor) mainly due to poor performance in trial two (asterisk in Fig. 6c; p < 0.001). Thigmotaxis and swim speed were unaffected by treatment, but significantly varied over the course of reversal learning (Fig. 6b, d–f’s > 4.4; p’s < 0.001). As for the analysis of animals in each treatment condition and their achievement of floor level performance, we observed a much higher variability than for acquisition learning (compare Figs. 5f, 6f). Unexpected was a lag in performance in the Poly-APS alone group (small rightward shift), but we observed a more dramatic change in the time course in the Poly-APS+hTau+OA group (Fig. 6f). Again, the null hypothesis of similar means in reversal learning was rejected [(F 3,30) = 18; p < 0.0001] and the Poly-APS+hTau+OA reliably differed from all other cohorts (F’s > 3.6; p’s < 0.05). A subsequent probe test conducted 24 h after the last reversal trial returned a strong spatial preference for the reversal target in all control and experimental groups apart from Poly-APS+hTau+OA animals (Fig. 6g, h, comparative swim paths shown in 6I). These triple infused rats spent equal amounts of time in each pool quadrant (lack of asterisks in Fig. 6h) providing strong evidence for impairment in spatial flexibility and memory.

Impaired behavioural flexibility in rats with overphosphorylated hTau loading in hippocampus. Mean ± SE. a Reversal path length in control groups (Poly-APS, hTau, OA). There was no difference between cohorts but note the heightened path length at first trial of second day (trial 5) in all groups. b Wall hugging behaviour was low in control cohorts during reversal learning. c Reversal path length in experimental groups revealed impairment in Poly-APS+hTau+OA group with noticeable difference in trial 2 (asterisk p < 0.05; Bonferroni corrected post hoc test against either Poly-APS alone or Poly-APS + hTau). d Heightened wall hugging behaviour in Poly-APS+hTau+OA-treated subjects. e Swim speed during reversal learning was not different between treatment groups. f Cumulative performance expressed as percent animals of each treatment (ordinate) reaching reversal floor level in the respective trial (abscissa). Cohorts followed different learning curves. A shallow learning curve was revealed for Poly-APS+hTau+OA-treated subjects. g, h Probe trial data expressed as percent time spent in target (Tg) or other (O) quadrants (average for three quadrants) conducted 24 h after reversal learning. All treatment groups presented with a spatial bias for the reversal target except the Poly-APS + hTau + OA group. No spatial bias was indexed for this group ($ = p < 0.05, t test against chance; asterisks, p < 0.05, paired t test for within-group comparison of quadrant time). i Example probe trial swim paths for Poly-APS control and Poly-APS+hTau+OA experimental group; target quadrant is shaded. Note the focal swim of the Poly-APS animal in comparison to the random search of the Poly-APS+Tau+OA animal
Tau phosphorylation impairs synaptic plasticity ex vivo
Hippocampal slices were prepared from selected cohorts at the end of the behavioural testing (Fig. 7). To remain within a comparable time window, we focussed on the experimental cohorts as no cognitive differences were observed in control groups. Ex vivo recorded population spikes in CA1 were probed for alterations in basal synaptic transmission (input–output relationship) and synaptic plasticity (LTP). In the experimental groups, a step-wise increase in intensity of Schaffer collateral stimulation led to an increase in spike amplitudes, which attained a plateau at 75–85 volts (Fig. 7a). There was a small leftward shift in the input–output relationship for the Poly-APS+hTau+OA-treated tissue, but there was no overall difference between groups (F’s < 1.2; p’s > 0.3 with factor treatment). Following the establishment of a stable baseline, LTP was induced by a theta burst stimulation protocol. Both Poly-APS and Poly-APS + hTau slices displayed robust long-lasting LTP up to 60 min post-tetanus (Fig. 7b). Although there was a post-tetanic elevation in the population spike amplitudes for Poly-APS+hTau+OA exposed slices, this was significantly below the level of the other experimental groups (F’s > 1.6; p’s < 0.004 for interactions relative to Poly-APS and Poly-APS+hTau) confirming that behavioural deficit is paralleled by an impairment in synaptic plasticity.

Altered synaptic plasticity as a result of loaded hTau phosphorylation in rat hippocampal slices ex vivo. Mean ± SE. a Despite somewhat higher population spike amplitudes in Poly-APS+hTau+OA group, this was not reliable for the input–output relationship of Schaffer collateral → CA1 synapses. b Time course of population spike amplitudes (% of baseline) in hippocampal slice ex vivo. Black bar indicates application of theta burst stimulation to induce LTP (for details see methods). Slices prepared from Poly-APS+hTau+OA show significantly reduced level of LTP compared to controls. Insets display representative recordings from Poly-APS control and Poly-APS+hTau+OA groups at baseline (i), immediately post-tetanus (ii) and at 60 min (iii)
Discussion
Poly-APS delivers macromolecules into neurons in vivo
In this report, we extend our previous investigations into the use of Poly-APS as a pore-forming agent into cultured rat hippocampal neurons [55] and provide evidence for its utility for intracellular loading of neurons with hTau protein. This was confirmed in both neonatal cultures and for the first time in hippocampal cells in vivo in rats. Poly-APS exerted notable potential as a delivery agent, despite the induction of transient Ca2+ permeable inward currents in conjunction with a suppression of membrane excitation and action potential generation. A recovery period of 24 h was sufficient to completely restore membrane properties [55]. Here, such recovery was also observed in cultures with Poly-APS and co-treatment including hTau or OA. Equally safe was the dose of 0.005 μg/ml Poly-APS used to achieve transfection in vivo. We also explored the toxicity profile of a dose 100-fold higher (5 μg/ml), which also did not result in lasting behavioural effects in our rats (data not shown) but opted for a lower concentration in connection with hTau and OA. This intriguing mechanism of in vivo intracellular macromolecule delivery is clearly an attractive alternative over currently explored methods including (1) viral delivery of human Tau genes, which requires long expression times before Tau shows built-up [74] and expression is isotype and receptor dependent, or (2) the seeding of Tau protein extracted and re-inoculated [42, 75] for which the mechanisms and thus the prerequisites mediating the initiation of intracellular uptake are currently elusive. The Poly-APS pore-forming method thus has the potential for ubiquitous use in any animal of choice and the compound itself appears when opting for low doses to enable full recovery of the transfected target cells. Moreover, it may be applied in conjunction with any macromolecule/drug, given the fact that pores of 29 Angstrom enable sizable complex molecules to pass through the pores. This is a clear advantage over viral approaches which require the expression of genetic material over longer periods of time and each macromolecule of interest would need the design and cloning of a novel viral construct.
Limitations to the Poly-APS approach may arise from its chemical properties such that selective macromolecules may directly interact with the lining or the surface charge of the pore and thereby lower its kinetics or block it altogether [76]. Its non-toxic dose range may be limited further owing to its mix in polymer length determining some of its biological properties [77–79] (including anticholinesterase activity at higher concentrations [80] and complex in vivo toxicity >2.7 mg/kg lethal in rats, [81]). Based on the localised detection of hTau within the CA1 stratum pyramidale and the preferential entry of both Luficier yellow and hTau into neurons in mixed cultures, it would appear that Tau loading more readily occurs in neurons and not glial cells. We currently have no explanation for this preference and this requires further investigatory work. However, Tau pathology in human AD is very infrequent in glia [82, 83]. Moreover, the current hypothesis of the seeding and regional expression assumes that Tau pathology occurs along the fibre paths of neurons, not glia [43].
Nevertheless, the overall intriguing outcome of this work is the observation that a transient inhibition of phosphatases via OA was sufficient to render the exogenously applied hTau neurotoxic with consequential long-lasting impairments in cell physiology, cognition and synaptic plasticity. At the same time, delivery of exogenous hTau per se using Poly-APS, at least at intracellular concentrations achieved here and within the short time frame under scrutiny, was not detrimental to basic neuronal properties, synaptic plasticity or learning and memory.
Exogenous hTau and phosphatase activity interact to induce physiological changes in neurons in vitro
Of all protocols, physiological impairments such as suppression of neuronal excitability, reduced input resistance and membrane potential were found in cultured hippocampal neurons only when intracellular hTau loading was combined with the application of OA. OA is a commonly used non-specific inhibitor of Ser/Thr phosphatases active in the nM range (PP2A<PP5<PP1) [84–86], which alone can lead to cytoskeletal disruption, changes in synaptic signalling, excessive Tau phosphorylation and toxicity when applied in vitro [62, 87–89]. Here, OA was applied in a concentration (1.2 nM) low enough to avoid such toxicity and this corroborates our previous work, in which KCl-mediated Ca2+ signalling was unaffected in cultures treated with OA alone [62]. The intact electrophysiology and Ca2+ signalling in Poly-APS+hTau-treated cultures indicates that intracellular delivery of hTau alone (at least at low concentrations) is benign to neuronal function, a finding which is in agreement with the preserved KCl-induced Ca2+ signalling reported following wild-type hTau expression in vitro [90]. Only in the combined group of Poly-APS+hTau+OA was neuronal excitation persistently compromised (Fig. 3), suggesting an interaction between loaded hTau and phosphatase inhibition.
In contrast to wild-type hTau, mutant variants of hTau can induce suppression of neuronal excitability in the absence of phosphatase inhibition (P301L/R406W; [91]); this may be explained by their reduced affinity for PP2A [34]. Collectively, our in vitro data provide compelling evidence that hTau can be delivered into cells via pore-former Poly-APS, and Tau proteins overphosphorylated by phosphatase inhibition lead to mechanistic changes such that the principal characteristics of neurons are compromised.
Phosphorylation of hTau in hippocampus leads to cognitive deficits in rats
The in vivo impairment of cognition required the interaction of hTau with OA, and neither molecule was sufficient on its own. In this context, the redistribution of phosphorylated Tau (p-Tau(S396)) into the soma in the Poly-APS+hTau+OA group, but not in the Poly-APS+hTau only group, is of particular importance. This somatodendritic localisation of phospho-Tau is reminiscent of the age-dependent redistribution of murine Tau in 28-month-old laboratory rats [71] and is in line with the toxic potential of mis-compartmentalisation of Tau reported by others (for review see [92]). Such old animals also present with cognitive decline and diminished plasticity. In our model, when OA is absent, hTau may not be readily phosphorylated in the relative short time between hTau loading and tissue harvest. Typically, Tau phosphorylation in transgenic Tau models of non-mutant Tau appears to have a slow build up taking months rather than weeks [93–95] and this time course may have been accelerated by OA. Thus, our work supports these observations in that wild-type hTau must first become abnormally phosphorylated to render the species pathological.
Perhaps the closest comparison can be made with models in which Tau loading was achieved by adeno-associated or lentiviral gene transfer in rats [96–99]. Again, a slow onset of Tau pathology including increased phosphorylation and conformational changes were detected after at least 7 weeks with a more full blown histopathology revealed 8 months after infusion; any cognitive consequences in these models remain unclear. A similar approach may be taken in our Poly-APS-mediated hTau model in future. Previous studies employing PP2A inhibitors, which report behavioural deficits in the water maze following acute intra-hippocampal [100–102] or chronic [103] administration, typically employ high doses of drug (ranging from 100 nm to ~320 µM) and assess learning and/or memory during or closely after administration (24 h post-injection). As a consequence of such an experimental design, determination of the putative mechanism underlying the memory disruption in prior studies is difficult and may include alterations such as indirect up-regulation of kinase activity [104], receptor modulation [105], deficient Ca2+ signalling, inhibition of LTP [62] or indeed hippocampal lesioning as reported 24 h post-OA injection at high mM range [73, 74].
Both a low dose of OA and time course of administration were selected specifically because they proved non-toxic [106]. Functional consequences in neuronal plasticity and learning and memory were only apparent when hippocampal neurons were pre-loaded with hTau. Acute toxicity of intracellular hTau and/or OA infusion are unlikely to explain the functional change given that respective control groups behaved normally, yet cellular alterations were long lasting and observed more than 3–4 weeks after the termination of the OA containing minipumps.
Phosphorylation of Tau is of physiological relevance as a biomarker for Alzheimer’s disease given that (1) total Tau levels are increased in hippocampus of AD patients, (2) phosphorylated Tau is increased in hippocampus of AD sufferers [107–110]; and (3) there is an inverse relationship between p-Tau in cerebrospinal fluid and mini-mental score in AD patients at Braak stage ≥3 [111]. This is congruent with the ‘hippocampal’ stage of the disease when first cognitive defects become apparent [36, 112].
Towards this end, it appears that our infusion model qualitatively reiterates: (1) the human AD pathology of excessively phosphorylated Tau. A more quantitative analysis would be warranted, which should also include a more extended time course with the aim to explore possible longitudinal spreading of the pathology along anatomical pathways in line with the recent Tau seeding studies. While this was not the overall aim in this proof-of-principle study, it would be interesting to also evaluate whether loading of hTau will, over extended time periods, lead to tangle-like aggregates in hippocampal cells. (2) Aspects of cognitive impairment seen in early AD. There were subtle deficits in spatial learning and flexibility in the Poly-APS+hTau+OA group; all other treatment groups were unaffected. Early diagnostic stages of AD present with difficulties in forming novel event memories within familiar spatio-temporal contexts [113, 114]. (3) Physiological changes likely to underlie the cognitive decline. In line with both cognitive and histopathological endpoints, synaptic plasticity was only compromised in tissue harvested from animals receiving Poly-APS + hTau + OA. The concept of impaired synaptic plasticity is widely held responsible for early impairments in learning and memory. LTP is one mechanism associated with memory [115, 116] and many AD and FTD models report deficits in learning and memory alongside those in LTP [69, 117–119].
In terms of cognitive deficit, it is noteworthy that animals in the Poly-APS+hTau+OA group failed predominantly in the first daily trials, but there was significant improvement in within-session learning. This is interesting as it seems to suggest an overall deficient consolidation process which would enable the storage of the acquired information long term (Riedel et al. 1999). However, most memory studies recently conducted using genetic models of Alzheimer mice (including our own: [37, 120, 121] have not distinguished between trials and this information may have gone amiss. Given these mouse models do not express their pathology in a region-specific manner, their deficits may have a more global origin. By contrast, we here purposefully selected CA1 for the transfection of hTau and our previous work has highlighted the role of the hippocampus and particularly CA1 in cohesion and long-term consolidation [66] by selective distinction between first and last daily trials [122, 123]. Previous work was concerned with the GABAergic input to the principle cells and led to a similar consolidation deficit as the Poly-APS+hTau+OA applied here.
Further application of Poly-APS-mediated Tau
Future work needs to establish in more detail (1) the time course of tauopathy over longer periods; (2) quantitatively what levels of Tau are achieved and how far the injection spreads within CA1; (3) what Tau species (oligomeric or fibrils or others) are generated within the cell and how they are modulated by phosphorylation; (4) optimisation of intracellular delivery may further be achieved by synthetic Poly-APS with preselected polymeric length (as recently described [124]). In addition, Poly-APS-mediated intracellular macromolecule delivery should be exploited in greater detail as it provides high flexibility for (1) the region of administration, (2) the Tau species or gene construct of interest that is delivered (for example mutant or wild type; full length or truncated), (3) enables the co-administration of other disease-related toxins (for example synuclein, amyloid, etc.), (4) avoids complications arising from hyperexpression of multiple gene copies, and (5) may be adopted to any experimental species of choice.
References
Pryer NK, Walker RA, Skeen VP, Bourns BD, Soboeiro MF, Salmon ED (1992) Brain microtubule-associated proteins modulate microtubule dynamic instability in vitro Real-time observations using video microscopy. J cell Sci 103(4):965–976
Niewiadomska G, Baksalerska-pazera M, Lenarcik I (1996) Riedel G (2006) Compartmental protein expression of Tau, GSK-3beta and TrkA in cholinergic neurons of aged rats. J Neural Transm Vienna Austria 113(11):1733–1746
Spillantini MG, Goedert M (2000) Tau mutations in familial frontotemporal dementia. Brain J Neurol 123(5):857–859
Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci USA 99(9):6364–6369
Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A (1988) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein Tau. Proc Natl Acad Sci USA 85(11):4051–4055
Crowther RA (1991) Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci USA 88(6):2288–2292
Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60(9):1495–1500
Guillozet AL, Weintraub S, Mash DC, Mesulam MM (2003) Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol 60(5):729–736
Morishima-kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, Titani K, Ihara Y (1995) Hyperphosphorylation of Tau in PHF. Neurobiol Aging 16(3):365–371 (discussion 371–80)
Wang JZ, Grundke-Iqbal I, Iqbal K (2007) Kinases and phosphatases and Tau sites involved in Alzheimer neurofibrillary degeneration. The European journal of neuroscience 25(1):59–68
Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, Bird IN, Varndell IM, Sheppard PW, Everall I, Anderton BH (2002) Rapid tyrosine phosphorylation of neuronal proteins including Tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases. J Neurosci Off J Soc Neurosci 22(1):10–20
Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, van Hoesen G, Ksiezak-Reding H (2004) Phosphorylation of Tau by fyn: implications for Alzheimer’s disease. J Neurosci Off J Soc Neurosci 24(9):2304–2312
Derkinderen P, Scales TM, Hanger DP, Leung KY, Byers HL, Ward MA, Lenz C, Price C, Bird IN, Perera T, Kellie S, Williamson R, Noble W, van Etten RA, Leroy K, Brion JP, Reynolds CH, Anderton BH (2005) Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament Tau and in fetal Tau with c-Abl as the candidate tyrosine kinase. J Neurosci Off J Soc Neurosci 25(28):6584–6593
Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G (2014) Tau protein modifications and interactions: their role in function and dysfunction. Int J Mol Sci 15(3):4671–4713
Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40(3):471–483
Braak H, Braak E (1996) Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl 165:3–12
Lauckner J, Frey P, Geula C (2003) Comparative distribution of Tau phosphorylated at Ser262 in pre-tangles and tangles. Neurobiol Aging 24(6):767–776
Biernat J, Mandelkow EM (1999) The development of cell processes induced by Tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol Biol Cell 10(3):727–740
Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, Andorfer C, Rosenberry TL, Lewis J, Hutton M, Janus C (2007) Accumulation of pathological Tau species and memory loss in a conditional model of tauopathy. J Neurosci Off J Soc Neurosci 27(14):3650–3662
Flunkert S, Hierzer M, Loffler T, Rabl R, Neddens J, Duller S, Schofield EL, Ward MA, Posch M, Jungwirth H, Windisch M, Hutter-Paier B (2013) Elevated levels of soluble total and hyperphosphorylated Tau result in early behavioral deficits and distinct changes in brain pathology in a new Tau transgenic mouse model. Neuro-degenerative diseases 11(4):194–205
Fox LM, William CM, Adamowicz DH, Pitstick R, Carlson GA, Spires-Jones TL, Hyman BT (2011) Soluble Tau species, not neurofibrillary aggregates, disrupt neural system integration in a Tau transgenic model. J Neuropathol Exp Neurol 70(7):588–595
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, Deture M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Sci (New York, NY) 309(5733):476–481
Sydow A, Mandelkow EM (2010) ‘Prion-like’ propagation of mouse and human Tau aggregates in an inducible mouse model of tauopathy. Neuro-degenerative Dis 7(1–3):28–31
Planel E, Miyasaka T, Launey T, Chui DH, Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y, Takashima A (2004) Alterations in glucose metabolism induce hypothermia leading to Tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J Neurosci Off J Soc Neurosci 24(10):2401–2411
Zhu D, Kosik KS, Meigs TE, Yanamadala V, Denker BM (2004) Galpha12 directly interacts with PP2A: evidence FOR Galpha12-stimulated PP2A phosphatase activity and dephosphorylation of microtubule-associated protein Tau. J Biol Chem 279(53):54983–54986
Sontag E, Nunbhakdi-Craig V, Bloom GS, Mumby MC (1995) A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J Cell Biol 128(6):1131–1144
Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC (1996) Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron 17(6):1201–1207
Liao H, Li Y, Brautigan DL, Gundersen GG (1998) Protein phosphatase 1 is targeted to microtubules by the microtubule-associated protein Tau. J Biol Chem 273(34):21901–21908
Gong CX, Liu F, Wu G, Rossie S, Wegiel J, Li L, Grundke-Iqbal I, Iqbal K (2004) Dephosphorylation of microtubule-associated protein Tau by protein phosphatase 5. J Neurochem 88(2):298–310
Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2005) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of Tau phosphorylation. Eur J Neurosci 22(8):1942–1950
Sontag E, Hladik C, Montgomery L, Luangpirom A, Mudrak I, Ogris E, White CL III (2004) Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol Exp Neurol 63(10):1080–1091
Sontag E, Luangpirom A, Hladik C, Mudrak I, Ogris E, Speciale S, White CL III (2004) Altered expression levels of the protein phosphatase 2A ABalphaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol 63(4):287–301
Tanimukai H, Grundke-Iqbal I, Iqbal K (2005) Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am J Pathol 166(6):1761–1771
Goedert M, Satumtira S, Jakes R, Smith MJ, Kamibayashi C, White CL III, Sontag E (2000) Reduced binding of protein phosphatase 2A to Tau protein with frontotemporal dementia and parkinsonism linked to chromosome 17 mutations. J Neurochem 75(5):2155–2162
Li R, Xu DE, Ma T (2015) Lovastatin suppresses the aberrant tau phosphorylation from FTDP-17 mutation and okadaic acid-induction in rat primary neurons. Neuroscience 294:14–20
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259
Melis V, Zabke C, Stamer K, Magbagbeolu M, Schwab K, Marschall P, Veh RW, Bachmann S, Deiana S, Moreau P, Davidson K, Harrington KA, Rickard JE, Horsley D, Garman R, Mazurkiewicz M, Niewiadomska G, Wischik CM, Harrington CR, Riedel G, Theuring F (2014) Different pathways of molecular pathophysiology underlie cognitive and motor tauopathy phenotypes in transgenic models for Alzheimer’s disease and frontotemporal lobar degeneration. Cell Mol Life Sci CMLS. doi:10.1007/s00018-014-1804-z
Jucker M, Walker LC (2011) Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol 70(4):532–540
Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, Dearmond SJ, Prusiner SB, Giles K (2012) Purified and synthetic Alzheimer’s amyloid beta (Abeta) prions. Proc Natl Acad Sci USA 109(27):11025–11030
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11(7):909–913
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53(3):337–351
Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, Duytschaever H, Ver Donck L, Torremans A, Sluydts E, Van Acker N, Kemp JA, Mercken M, Brunden KR, Trojanowski JQ, Dewachter I, Lee VM, Moechars D (2015) Intracerebral injection of preformed synthetic Tau fibrils initiates widespread tauopathy and neuronal loss in the brains of Tau transgenic mice. Neurobiol Dis 73:83–95
Dujardin S, Lecolle K, Caillierez R, Begard S, Zommer N, Lachaud C, Carrier S, Dufour N, Auregan G, Winderickx J, Hantraye P, Deglon N, Colin M, Buee L (2014) Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol Commun 2:14
Do Carmo S, Cuello AC (2013) Modeling Alzheimer’s disease in transgenic rats. Mol Neurodegener 8:37
Lin JH (1995) Species similarities and differences in pharmacokinetics. Drug metabolism and disposition: the biological fate of chemicals 23(10):1008–1021
Jacob HJ, Kwitek AE (2002) Rat genetics: attaching physiology and pharmacology to the genome. Nat Rev Genet 3(1):33–42
McMillan P, Korvatska E, Poorkaj P, Evstafjeva Z, Robinson L, Greenup L, Leverenz J, Schellenberg GD, D’Souza I (2008) Tau isoform regulation is region- and cell-specific in mouse brain. J Comp Neurol 511(6):788–803
Hanes J, Zilka N, Bartkova M, Caletkova M, Dobrota D, Novak M (2009) Rat Tau proteome consists of six Tau isoforms: implication for animal models of human tauopathies. J Neurochem 108(5):1167–1176
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33(1):95–130
Braak E, Braak H (1997) Alzheimer’s disease: transiently developing dendritic changes in pyramidal cells of sector CA1 of the Ammon’s horn. Acta Neuropathol 93(4):323–325
Cehlar O, Skrabana R, Kovac A, Kovacech B, Novak M (2012) Crystallization and preliminary X-ray diffraction analysis of Tau protein microtubule-binding motifs in complex with Tau5 and DC25 antibody Fab fragments. Acta Crystallogr Sect F Struct Biol Cryst Commun 68(10):1181–1185
Sepcic K, Batista U, Vacelet J, Macek P, Turk T (1997) Biological activities of aqueous extracts from marine sponges and cytotoxic effects of 3-alkylpyridinium polymers from Reniera sarai Comparative biochemistry and physiologyPart C. Pharmacol Toxicol Endocrinol 117(1):47–53
McClelland D, Evans RM, Abidin I, Sharma S, Choudhry FZ, Jaspars M, Sepcic K, Scott RH (2003) Irreversible and reversible pore formation by polymeric alkylpyridinium salts (poly-APS) from the sponge Reniera sarai. Br J Pharmacol 139(8):1399–1408
Tucker SJ, McClelland D, Jaspars M, Sepcic K, Macewan DJ, Scott RH (2003) The influence of alkyl pyridinium sponge toxins on membrane properties, cytotoxicity, transfection and protein expression in mammalian cells. Biochim Biophys Acta 1614(2):171–181
Koss DJ, Hindley KP, David KC, Mancini I, Guella G, Sepcic K, Turk T, Rebolj K, Riedel G, Platt B, Scott RH (2007) A comparative study of the actions of alkylpyridinium salts from a marine sponge and related synthetic compounds in rat cultured hippocampal neurones. BMC Pharmacol 7:1
Malovrh P, Sepcic K, Turk T, Macek P (1999) Characterization of hemolytic activity of 3-alkylpyridinium polymers from the marine sponge Reniera sarai Comparative biochemistry and physiologyPart C. Pharmacol Toxicol Endocrinol 124(2):221–226
Schmitz FJ, Hollenbeak KH, Campbell DC (1978) Marine natural products: halitoxin, toxic complex of several marine sponges of the genus Haliclona. J Org Chem 43:3916–3922
Berlinck RG, Ogawa CA, Almeida AM, Sanchez MA, Malpezzi EL, Costa LV, Hajdu E, De Freitas JC (1996) Chemical and pharmacological characterization of halitoxin from Amphimedon viridis (Porifera) from the southeastern Brazilian coast Comparative biochemistry and physiology Part C. Pharmacol Toxicol Endocrinol 115(2):155–163
Scott RH, Whyment AD, Foster A, Gordon KH, Milne BF, Jaspars M (2000) Analysis of the structure and electrophysiological actions of halitoxins: 1,3 alkyl-pyridinium salts from Callyspongia ridleyi. J Membr Biol 176(2):119–131
Sepcic K, Guella G, Mancini I, Pietra F, Serra MD, Menestrina G, Tubbs K, Macek P, Turk T (1997) Characterization of anticholinesterase-active 3-alkylpyridinium polymers from the marine sponge Reniera sarai in aqueous solutions. J Nat Prod 60(10):991–996
Davies-Coleman MT, Faulkner DJ, Dubowchik GM, Roth GP, Polson C, Fairchild C (1993) A New EGF-Active Polymeric Pyridinium Alkaloid from the Sponge Callyspongia fibrosa. J Org Chem 58:5925–5930
Koss DJ, Hindley KP, Riedel G, Platt B (2007) Modulation of hippocampal calcium signalling and plasticity by serine/threonine protein phosphatases. J Neurochem 102(4):1009–1023
Koss DJ, Riedel G, Platt B (2009) Intracellular Ca2 + stores modulate SOCCs and NMDA receptors via tyrosine kinases in rat hippocampal neurons. Cell Calcium 46(1):39–48
Scharschmidt BF, Lake JR, Renner EL, Licko V, van Dyke RW (1986) Fluid phase endocytosis by cultured rat hepatocytes and perfused rat liver: implications for plasma membrane turnover and vesicular trafficking of fluid phase markers. Proc Natl Acad Sci USA 83(24):9488–9492
Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch 391(2):85–100
Riedel G, Micheau J, Lam AG, Roloff EL, Martin SJ, Bridge H, de Hoz L, Poeschel B, McCulloch J, Morris RG (1999) Reversible neural inactivation reveals hippocampal participation in several memory processes. Nat Neurosci 2(10):898–905
Robinson L, McKillop-Smith S, Ross NL, Pertwee RG, Hampson RE, Platt B, Riedel G (2008) Hippocampal endocannabinoids inhibit spatial learning and limit spatial memory in rats. Psychopharmacology 198(4):551–563
Christie LA, Riedel G, Algaidi SA, Whalley LJ, Platt B (2005) Enhanced hippocampal long-term potentiation in rats after chronic exposure to homocysteine. Neurosci Lett 373(2):119–124
Koss DJ, Drever BD, Stoppelkamp S, Riedel G, Platt B (2013) Age-dependent changes in hippocampal synaptic transmission and plasticity in the PLB1(Triple) Alzheimer mouse. Cellular and molecular life sciences : CMLS 70(14):1273–1279
Algaidi SA, Christie LA, Jenkinson AM, Whalley L, Riedel G, Platt B (2006) Long-term homocysteine exposure induces alterations in spatial learning, hippocampal signalling and synaptic plasticity. Exp Neurol 197(1):8–21
Niewiadomska G, Baksalerska-Pazera M, Riedel G (2006) Cytoskeletal transport in the aging brain: focus on the cholinergic system. Rev Neurosci 17(6):581–618
Mudher AK, Perry VH (1998) Using okadaic acid as a tool for the in vivo induction of hyperphosphorylated Tau. Neuroscience 85(4):1329–1332
van Dam AM, Bol JG, Binnekade R, van Muiswinkel FL (1998) Acute or chronic administration of okadaic acid to rats induces brain damage rather than Alzheimer-like neuropathology. Neuroscience 85(4):1333–1335
Dayton RD, Wang DB, Cain CD, Schrott LM, Ramirez JJ, King MA, Klein RL (2012) Frontotemporal lobar degeneration-related proteins induce only subtle memory-related deficits when bilaterally overexpressed in the dorsal hippocampus. Exp Neurol 233(2):807–814
Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM (2013) Anti-Tau antibodies that block Tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80(2):402–414
McLaggan D, Adjimatera N, Sepcic K, Jaspars M, Macewan DJ, Blagbrough IS, Scott RH (2006) Pore forming polyalkylpyridinium salts from marine sponges versus synthetic lipofection systems: distinct tools for intracellular delivery of cDNA and siRNA. BMC Biotechnol 6:6
Paleari L, Trombino S, Falugi C, Gallus L, Carlone S, Angelini C, Sepcic K, Turk T, Faimali M, Noonan DM, Albini A (2006) Marine sponge-derived polymeric alkylpyridinium salts as a novel tumor chemotherapeutic targeting the cholinergic system in lung tumors. Int J Oncol 29(6):1381–1388
Berne S, Pohleven F, Turk T, Sepcic K (2008) Induction of fruiting in oyster mushroom (Pleurotus ostreatus) by polymeric 3-alkylpyridinium salts. Mycol Res 112(Pt 9):1085–1087
Elersek T, Kosi G, Turk T, Pohleven F, Sepcic K (2008) Influence of polymeric 3-alkylpyridinium salts from the marine sponge Reniera sarai on the growth of algae and wood decay fungi. Biofouling 24(2):137–143
Grandic M, Araoz R, Molgo J, Turk T, Sepcic K, Benoit E, Frangez R (2013) Toxicity of the synthetic polymeric 3-alkylpyridinium salt (APS3) is due to specific block of nicotinic acetylcholine receptors. Toxicology 303:25–33
Turk T, Frangez R, Sepcic K (2007) Mechanisms of toxicity of 3-alkylpyridinium polymers from marine sponge Reniera sarai. Marine drugs 5(4):157–167
Yamada T, McGeer PL (1990) Oligodendroglial microtubular masses: an abnormality observed in some human neurodegenerative diseases. Neurosci Lett 120(2):163–166
Nishimura M, Tomimoto H, Suenaga T, Namba Y, Ikeda K, Akiguchi I, Kimura J (1995) Immunocytochemical characterization of glial fibrillary tangles in Alzheimer’s disease brain. Am J Pathol 146(5):1052–1058
Bialojan C, Takai A (1988) Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. The Biochemical journal 256(1):283–290
Cohen PT (1997) Novel protein serine/threonine phosphatases: variety is the spice of life. Trends Biochem Sci 22(7):245–251
Borthwick EB, Zeke T, Prescott AR, Cohen PT (2001) Nuclear localization of protein phosphatase 5 is dependent on the carboxy-terminal region. FEBS Lett 491(3):279–284
Malchiodi-Albedi F, Petrucci TC, Picconi B, Iosi F, Falchi M (1997) Protein phosphatase inhibitors induce modification of synapse structure and Tau hyperphosphorylation in cultured rat hippocampal neurons. J Neurosci Res 48(5):425–438
Kim D, Su J, Cotman CW (1999) Sequence of neurodegeneration and accumulation of phosphorylated Tau in cultured neurons after okadaic acid treatment. Brain Res 839(2):253–262
Tanaka T, Zhong J, Iqbal K, Trenkner E, Grundke-Iqbal I (1998) The regulation of phosphorylation of Tau in SY5Y neuroblastoma cells: the role of protein phosphatases. FEBS Lett 426(2):248–254
Furukawa K, Wang Y, Yao PJ, Fu W, Mattson MP, Itoyama Y, Onodera H, D’Souza I, Poorkaj PH, Bird TD, Schellenberg GD (2003) Alteration in calcium channel properties is responsible for the neurotoxic action of a familial frontotemporal dementia Tau mutation. J Neurochem 87(2):427–436
Stoppelkamp S, Bell HS, Palacios-Filardo J, Shewan DA, Riedel G, Platt B (2011) In vitro modelling of Alzheimer’s disease: degeneration and cell death induced by viral delivery of amyloid and Tau. Exp Neurol 229(2):226–237
Zempel H, Mandelkow E (2014) Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci 37(12):721–732
Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Burki K, Davies P (2000) Characterization of pathology in transgenic mice over-expressing human genomic and cDNA Tau transgenes. Neurobiology of disease 7(2):87–98
Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P (2003) Hyperphosphorylation and aggregation of Tau in mice expressing normal human Tau isoforms. J Neurochem 86(3):582–590
Polydoro M, Acker CM, Duff K, Castillo PE, Davies P (2009) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of Tau pathology. The Journal of neuroscience : the official journal of the Society for Neuroscience 29(34):10741–10749
Khandelwal PJ, Dumanis SB, Herman AM, Rebeck GW, Moussa CE (2012) Wild type and P301L mutant Tau promote neuro-inflammation and alpha-Synuclein accumulation in lentiviral gene delivery models. Molecular and cellular neurosciences 49(1):44–53
Dayton RD, Wang DB, Cain CD, Schrott LM, Ramirez JJ, King MA, Klein RL (2012) Frontotemporal lobar degeneration-related proteins induce only subtle memory-related deficits when bilaterally overexpressed in the dorsal hippocampus. Exp Neurol 233(2):807–814
Caillierez R, Begard S, Lecolle K, Deramecourt V, Zommer N, Dujardin S, Loyens A, Dufour N, Auregan G, Winderickx J, Hantraye P, Deglon N, Buee L, Colin M (2013) Lentiviral delivery of the human wild-type Tau protein mediates a slow and progressive neurodegenerative Tau pathology in the rat brain. Molecular therapy : the journal of the American Society of Gene Therapy 21(7):1358–1368
Dujardin S, Lecolle K, Caillierez R, Begard S, Zommer N, Lachaud C, Carrier S, Dufour N, Auregan G, Winderickx J, Hantraye P, Deglon N, Colin M, Buee L (2014) Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism relevance to sporadic tauopathies. Acta Neuropathol Commun 2:14
He J, Yamada K, Zou LB, Nabeshima T (2001) Spatial memory deficit and neurodegeneration induced by the direct injection of okadaic acid into the hippocampus in rats. J Neural Transm Vienna Austria 108(12):1435–1443
Sun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, Wang JZ (2003) Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced Tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience 118(4):1175–1182
He J, Yang Y, Xu H, Zhang X, Li XM (2005) Olanzapine attenuates the okadaic acid-induced spatial memory impairment and hippocampal cell death in rats. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 30(8):1511–1520
Arendt T, Holzer M, Fruth R, Bruckner MK, Gartner U (1995) Paired helical filament-like phosphorylation of Tau, deposition of beta/A4-amyloid and memory impairment in rat induced by chronic inhibition of phosphatase 1 and 2A. Neuroscience 69(3):691–698
Millward TA, Zolnierowicz S, Hemmings BA (1999) Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci 24(5):186–191
Herzig S, Neumann J (2000) Effects of serine/threonine protein phosphatases on ion channels in excitable membranes. Physiol Rev 80(1):173–210
Zhang Z, Simpkins JW (2010) An okadaic acid-induced model of tauopathy and cognitive deficiency. Brain Res 1359:233–246
Khatoon S, Grundke-Iqbal I, Iqbal K (1992) Brain levels of microtubule-associated protein Tau are elevated in Alzheimer’s disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem 59(2):750–753
Khatoon S, Grundke-Iqbal I, Iqbal K (1994) Levels of normal and abnormally phosphorylated Tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett 351(1):80–84
Holzer M, Holzapfel HP, Zedlick D, Bruckner MK, Arendt T (1994) Abnormally phosphorylated Tau protein in Alzheimer’s disease: heterogeneity of individual regional distribution and relationship to clinical severity. Neuroscience 63(2):499–516
Thal DR, Holzer M, Rub U, Waldmann G, Gunzel S, Zedlick D, Schober R (2000) Alzheimer-related Tau-pathology in the perforant path target zone and in the hippocampal stratum oriens and radiatum correlates with onset and degree of dementia. Exp Neurol 163(1):98–110
Wang X, Zhu M, Hjorth E, Cortes-Toro V, Eyjolfsdottir H, Graff C, Nennesmo I, Palmblad J, Eriksdotter M, Sambamurti K, Fitzgerald JM, Serhan CN, Granholm AC, Schultzberg M (2015) Resolution of inflammation is altered in Alzheimer’s disease. Alzheimer’s Dement J Alzheimer’s Assoc 11(1):40–50
Bancher C, Braak H, Fischer P, Jellinger KA (1993) Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients. Neurosci Lett 162(1–2):179–182
Freedman M, Oscar-Berman M (1989) Spatial and visual learning deficits in Alzheimer’s and Parkinson’s disease. Brain Cogn 11(1):114–126
Gold CA, Budson AE (2008) Memory loss in Alzheimer’s disease: implications for development of therapeutics. Expert Rev Neurother 8(12):1879–1891
Whitlock JR, Heynen AJ, Shuler MG, Bear MF (2006) Learning induces long-term potentiation in the hippocampus. Science (New York, NY) 313(5790):1093–1097
Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R (2014) Engineering a memory with LTD and LTP. Nature 511(7509):348–352
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, Laferla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39(3):409–421
Rosenmann H, Grigoriadis N, Eldar-Levy H, Avital A, Rozenstein L, Touloumi O, Behar L, Ben-Hur T, Avraham Y, Berry E, Segal M, Ginzburg I, Abramsky O (2008) A novel transgenic mouse expressing double mutant Tau driven by its natural promoter exhibits tauopathy characteristics. Exp Neurol 212(1):71–84
Lo AC, Iscru E, Blum D, Tesseur I, Callaerts-Vegh Z, Buee L, de Strooper B, Balschun D, D’Hooge R (2013) Amyloid and Tau neuropathology differentially affect prefrontal synaptic plasticity and cognitive performance in mouse models of Alzheimer’s disease. J Alzheimer’s Dis JAD 37(1):109–125
Platt B, Drever B, Koss D, Stoppelkamp S, Jyoti A, Plano A, Utan A, Merrick G, Ryan D, Melis V, Wan H, Mingarelli M, Porcu E, Scrocchi L, Welch A, Riedel G (2011) Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PLoS One 6(11):e27068
Ryan D, Koss D, Porcu E, Woodcock H, Robinson L, Platt B, Riedel G (2013) Spatial learning impairments in PLB1Triple knock-in Alzheimer mice are task-specific and age-dependent. Cell Mol life Sci CMLS 70(14):2603–2619
Robinson L, Goonawardena AV, Pertwee RG, Hampson RE, Riedel G (2007) The synthetic cannabinoid HU210 induces spatial memory deficits and suppresses hippocampal firing rate in rats. Br J Pharmacol 151(5):688–700
Robinson L, Goonawardena AV, Pertwee R, Hampson RE, Platt B, Riedel G (2010) WIN55,212-2 induced deficits in spatial learning are mediated by cholinergic hypofunction. Behav Brain Res 208(2):584–592
Houssen WE, Lu Z, Edrada-Ebel R, Chatzi C, Tucker SJ, Sepcic K, Turk T, Zovko A, Shen S, Mancini I, Scott RH, Jaspars M (2010) Chemical synthesis and biological activities of 3-alkyl pyridinium polymeric analogues of marine toxins. J Chem Biol 3(3):113–125
Paxinos G, Watson C (2013) The Rat Brain in stereotaxic coordinates, 7th edn. Academic Press, London
Acknowledgments
This work was supported by the Alzheimer Research Trust (now ARUK) to GR, RHS and BP, as well as grants from the National Science Centre to AG and GN, and by statutory funds from the Nencki Institute of Experimental Biology (Warsaw, Poland) and School of Medical Sciences of the University of Aberdeen (UK). We gratefully acknowledge the help of students involved in this project: Kanola David, Jonathan Jones, Risto Kylanpaa, David McClelland, Rhian Evans, Iona Beange and Sandra Brooks.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Koss, D.J., Robinson, L., Mietelska-Porowska, A. et al. Polymeric alkylpyridinium salts permit intracellular delivery of human Tau in rat hippocampal neurons: requirement of Tau phosphorylation for functional deficits. Cell. Mol. Life Sci. 72, 4613–4632 (2015). https://doi.org/10.1007/s00018-015-1949-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1949-4