
Abstract
Thymic B cells are a unique population of B lymphocytes that reside at the cortico-medullary junction of the thymus, an organ that is specialized for the development and selection of T cells. These B cells are distinct from peripheral B cells both in terms of their origin and phenotype. Multiple lines of evidence suggest that they develop within the thymus from B lineage-committed progenitors and are not recirculating peripheral B cells. Furthermore, thymic B cells have a highly activated phenotype. Because of their location in the thymic medulla, they have been thought to play a role in T cell negative selection. Thymic B cells are capable of inducing negative selection in a number of model antigen systems, including viral super antigen, peptides from immunoglobulin, and cognate self antigen presented by B cell receptor-mediated uptake. These findings establish thymic B cells as a novel and important population to study; however, much work remains to be done to understand how all of these unique aspects of thymic B cell biology inform their function.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thymic B cells were first identified in human thymic sections in 1987 and a year later, a similar population was reported in mice [1, 2]. From these initial descriptions, it was clear that these cells possessed a distinctive phenotype relative to other B cell subsets. First, thymic B cells were detectable very early in ontogeny, even during fetal life. Second, they showed signs of activation, and particularly, the presence of mitotic figures and significant Ki-67 staining. Third, thymic B cells resided exclusively in the thymic medulla, specifically at the cortico-medullary junction.
Much of the thymic B cell research over the last few decades has expanded on these initial findings. The three major questions in the field have been: (1) Given the early appearance of thymic B cells in ontogeny, what are their developmental origin? (2) Despite their surface phenotype, thymic B cells seem refractory to conventional stimulation in vitro, what are the factors governing B cell activation in the thymus? (3) Given the cortico-medullary localization of thymic B cells, do they play a role in T cell negative selection?
Development of thymic B cells
The earliest suggestion that thymic B cells may develop independently of the bone marrow comes from the finding that mature thymic B cells are detectable at embryonic day 18 in mice, prior to the onset of B cell development in the bone marrow [1, 3]. The finding that peripheral B cells inefficiently migrate into the thymus when splenocytes are injected intravenously or during parabiosis, further suggested that this population did not recirculate to the thymus from the periphery [4–6]. Finally, the identification of a B cell progenitor-like population within the thymus gave some support to the idea that there may be an intrathymic pathway for B cell development. Most of our understanding of thymic B cell development comes from analysis of these fairly differentiated B lineage-committed precursors. As we trace this developmental pathway back towards its earliest progenitors in the thymus, it is difficult to make definitive statements about the lineage of these cells. The potential developmental pathways of thymic B cells are summarized in Fig. 1. In this review, we will discuss the better-defined later stages of thymic B cell development before moving on to the earlier, less well-defined stages.

Two models for timing and location of progenitor commitment to the thymic B cell lineage. This figure shows two possible pathways for how cells might undergo B lineage restriction within the thymus. (1) Circulating progenitors (such as the CLP-2 and CTP described in the text) enter the thymus already partially restricted to the T or B cell lineage, and (2) Lineage decision occurs intrathymically from multipotent progenitors
B lineage committed progenitors in the thymus can be identified by their B220low CD43+ surface Ig− phenotype [4, 7, 8]. When these cells are purified and injected intrathymically, they readily develop into B220highCD43−IgM+ cells with similar levels of CD5 expression as mature thymic B cells [4, 7]. The progenitors can also be expanded intrathymically by the addition FLT3L and IL-7 in thymic organ cultures [8]. In vitro, these thymic progenitors differentiate into B cells in the presence of IL7, but only when cultured on thymic stromal cells [9]. Culture on bone marrow stromal cells, or even thymus-derived stromal cell lines did not support B cell development from these progenitors. Characterization of the thymic stromal cells that supported B cell development in these experiments found that they were largely cortical in nature, suggesting that these progenitors, like T lineage progenitors, may initially develop in the cortex before migrating into the medulla [9]. Supporting this interpretation is the fact that while IgM+ cells are located in the medulla, B220 staining appears to extend into the cortex [4].
Two main findings define our understanding of how these progenitors are regulated by the thymic environment. First, early disruption of T cell development leads to an outgrowth of thymic B cell progenitors, and mature thymic B cells. This outgrowth is observed most readily in TCRβ−/− but can also be observed in CD3−/− mice [4, 10]. These data would suggest that a shared niche is occupied by thymocytes and B cells, and when early T cell development is disrupted, B cells expand to fill that niche. However, the expansion of mature thymic B cells in these cases is always modest relative to the loss of total thymic cellularity, indicating the niche for thymic B cells is much smaller than the niche for thymocytes as a whole, and suggesting that while there may be competition for thymic niches between thymocytes and thymic B cells (and potentially their progenitors), other factors likely contribute to the different levels of T and B cells development observed in the thymus.
The second finding is that the numbers of thymic B cell progenitors and mature thymic B cells expand when Notch signaling is disrupted [11, 12]. This relates closely to the first point, since Notch signaling is critical for proper T lineage commitment and Notch deficiency clearly disrupts T cell development. These data are often interpreted as evidence that thymic B cell development is the fate of cells that do not encounter Notch signaling in the thymus; however while there is expansion of B cells in the Notch1 deficient animals, these B cells are phenotypically distinct from the B cells seen in WT mice. Most notably in Notch−/− chimera mice, very few thymic B cells express CD5 and many express high levels of AA4, a marker of BM bone marrow development that is surprisingly absent from WT thymic B cells, even at the progenitor stage [5, 12]. This phenotype is further clarified in mice where Cpa3-Cre causes deletion of Notch1 during the DN1-2 stage of early T cell development. It was hypothesized that deletion of Notch in these early progenitors would divert them towards a B cell fate, and this deletion does lead to an increase in the same type of B cells observed in the Notch1 total knockout (predominantly CD19lowB220lowAA4+ progenitors) [13]. However, when Notch1 deletion is examined in the cells that phenotypically resemble WT mature thymic B cells at the single cell level (CD19hiB220hi cells), most of these cells still carry a functional copy of Notch, despite robust deletion in other thymic subpopulations. Instead, most of the Notch-deleted cells go on to become dendritic cells. These results suggest that when T cell differentiation is blocked, thymic T cell progenitors do not simply adopt a thymic B cell fate, and that the thymic T and B cell lineages may diverge quite early, possibly prior to the DN1 thymocyte stage.
It has been shown in mice when Stat5 is constitutively active, there is a very large expansion of thymic B cells [14]. Additionally, it has been shown through targeted mutations of the individual zinc fingers of Ikaros that mice with a variant of Ikaros lacking the 1st finger are almost entirely deficient in thymic B cell development, while most other peripheral B cell compartments appear largely intact, although it should be noted this is only one of the many observed immune defects in these mice [15]. Whether these transcription factors exert their activities through interactions with the Notch pathway, or in the case of Stat5 potentially through the IL7 or FLT3 pathways, remains unclear.
These results clearly raise questions about the nature of the thymic progenitors upstream of these B220+CD43low cells. The literature surrounding the lineage potential of various thymic progenitor populations is extensive, contentious, and very well-reviewed elsewhere [16, 17]. The major consensus seems to be that the classical early thymic progenitor or ETP (CD3−CD8−CD44+CD25− Kithi) lacks B cell potential. This ETP population (divided into DN1a and DN1b) is generally accepted to be the population that gives rise to the bulk of mature thymocytes, due to its T lineage restriction and large capacity for expansion [18]. DN1c and DN1d cells, which are distinguished from the ETP by their differential expression of CD117 and CD24, are present in the thymus at similar frequencies as DN1a and DN1b cells, possess B cell potential, but seem to lack the same proliferative capacity. DN1a/DN1b do not pass through DN1c and DN1d on their way to the DN2 stage of T cell development, and is unclear whether DN1c and DN1d cells are developmentally downstream of the ETP at all, with some groups suggesting they may derive from distinct progenitors [18]. However, despite their B lineage potential, it seems that the DN1c population largely gives rise to dendritic cells intrathymically [18].
At a potentially earlier stage of development, work from Benz and Bleul demonstrated that a very early progenitor in the thymus, distinguished by its expression of the thymus homing chemokine receptor CCR9 and Flt3 retains B cell potential, but that this potential is lost as these cells downregulate CCR9 and Flt3 and acquire more of a traditional ETP phenotype, suggesting that B lineage diversion could occur very early following progenitor importation into the thymus as shown in path 1 of Fig. 1 [19, 20]. It is known that mice that are doubly deficient in CCR9 and CCR7 have dramatic reductions in ETP numbers, but the status of the thymic B cell development in these mice was not reported [21].
In addition to ETPs, there are a number of different cell populations that are capable of homing to the thymus and giving rise to thymocytes, and these cells exist along a broad spectrum of lineage commitment. While some populations are fully T lineage restricted in the blood before they even enter thymus, such as the Circulating Thymic Progenitor (CTP), there are also cells such as CLP-2 (based on their similarity to the Common Lymphoid Progenitor) that are B220+ and retain B cell potential, but still develop into T cells in the thymus [22, 23].
It is still unclear whether any of these pathways directly contribute to the development of the thymic B cell lineage. Most of what we know about their B cell potential is derived from culture on OP-9 cells. However, when the development of these populations is tracked intrathymically, the amount of B cell development is rarely reported. Interestingly the CCR9hi population described by Benz, is capable of differentiating into B cells even in the presence of Notch ligands (1:20 mixed stromal cultures of OP9-DL4 and OP9, however, not on OP9-DL4 exclusively), suggesting that they might be able to give rise to B cells even in the presence of Notch signals in the thymus [19].
It has been reported that in fetal thymic organ culture systems, a single thymic precursor can generate as many as 105 thymocytes in 12 days [24]. So while the relative B cell potential of the various thymic progenitors is low, the size of the thymic B cell pool is also fairly small. Therefore, it is possible that the number of progenitors needed to sustain this population may be quite low.
Thymic B cell activation
From the human histological studies, thymic B cells appear to be highly activated and many have visible mitotic spindles. Further characterization in mice confirms that these cells express a number of activation and costimulatory markers, such as CD5, CD69, CD80, CD86, CD40, and high levels of MHC class II [5, 25, 26]. Despite this activated phenotype, they display a reduced ability to proliferate and produce antibodies in response to classic B cell mitogens such as anti-IgM plus IL-4, and LPS relative to splenic B cells. Class II-restricted T cell blasts are the most efficient stimulators of thymic B cells, and similarly, anti-CD40 and IL-10 are potent stimulators when used in combination, suggesting potential T-B interactions in the thymus [25, 27]. However, it is critical to note that all of these stimuli elicit a blunted response when compared to splenic B cells.
In particular, the CD40-CD40L interaction appears to be important for thymic B cell biology, as thymic B cells in mice lacking CD40 express lower levels of CD69 and CD86 [26]. Additionally, it has been shown that CD40L−/− mice have a tenfold reduction in the total number of thymic B cells, suggesting this pathway may be important for the survival and maintenance of thymic B cells [28]. More recently, it was demonstrated by Fujihara et al. [29] that CD40 is important for thymic B cell proliferation and function in a cell autonomous manner [29].
CD5 expression is one of the most distinguishing features of the activated phenotype of thymic B cells. This marker is also a hallmark of the B1 lineage of B cells, which develop early in the fetal liver, make up a large proportion of the cells in the peritoneal cavity, and a small but detectable proportion of circulating B lymphocytes [30]. This finding led to speculation that thymic B cells were phenotypically and developmentally related to the B-1 lineage [2]. While there may be some phenotypic similarities, it is clear that these two populations are developmentally distinct, as the CD5+ B-1a B cells develop almost exclusively from fetal liver stem cells, while thymic B cells develop equally well from fetal liver and bone marrow progenitors [5, 6, 31]. Even though CD5 is normally only expressed on B-1a B cells in the periphery, it can be induced on B-2 B cells following IgM crosslinking or stimulation through CD40 [32]. Given the aforementioned importance of CD40-CD40L interactions for thymic B cells, this could potentially explain the CD5 expression as well; however, it remains to be seen to what extent thymic B cell activation is dependent on BCR signaling, interactions with T cells, or other signals from the thymic microenvironment.
Thymic B cells as antigen-presenting cells (APCs)
The thymic medulla contains a dense network of professional APCs [33]. Two photon microscopy studies show that upon migration into the medulla, immature thymocytes are highly motile, constantly scanning the medulla for cognate antigen over a period of 3–5 days [34]. Any T cell that encounters cognate antigen during this period initiates some form of tolerogenic gene expression program, with the most common being negative selection, a programmed cell death pathway that deletes the autoreactive cells from the repertoire.
The thymic medulla contains many MHC class II expressing cell types including classical and plasmacytoid dendritic cells, macrophages, B cells, and medullary thymic epithelial cells [2, 33, 35]. While the development of TCR transgenic mice has allowed us to assess the capacity of each of these medullary APCs to participate in negative selection to model antigens, it is still unclear what the relative contribution of each of these medullary APCs is to shaping the normal T cell repertoire.
Several studies have explored the capacity of thymic B cells to present antigens and delete developing thymocytes. Early reports compared the capacity of B cells and DCs to mediate deletion in response to viral superantigens following intrathymic injection into neonatal mice. In this system, intrathymically injected thymic DCs could only induce clonal anergy, whereas thymic B cells were capable of deleting superantigen-reactive thymocytes [36, 37]. The fact that superantigens are preferentially expressed by B cells is a confounding factor in comparing B cells and DCs in this assay [38, 39]. However, when splenic B cells were used, they were completely unable to induce any tolerance, suggesting that this effect is not solely due to superantigen expression in B cells [36, 37].
To further distinguish the capacity of thymic B cells to present antigens vs. being a source of antigens, MHC I-E+ thymic B cells were injected into I-E−/− thymi. I-E is required to present certain superantigens, so in this system only B cells can present antigens. In this context, it was found that thymic B cells are sufficient to mediate deletion in the absence of other presenting thymic APCs [36]. In these studies, the CD5+ thymic B cell pool contains most of this activity, as a CD5-depleted thymic B cell fraction was less efficient at mediating deletion, and interestingly in vitro activation of splenic B cells with anti-IgM and IL-4 enhanced their effectiveness in the assay.
Another study using a similar superantigen system took advantage of the fact that B6 mice are naturally deficient in I-E, to selectively express the restricting I-E allele in different thymic APCs [40]. Transgenic I-E expression leads to clonal deletion of superantigen-reactive T cells when I-E is expressed specifically on B cells or dendritic cells through the use of the human CD19 promoter or the CD11c promoter, respectively, further confirming that endogenous thymic B cells can mediate deletion [40].
While superantigen models are useful for determining whether thymic B cells express the correct costimulatory molecules and are correctly positioned to participate in negative selection, the major caveat is that superantigen-reactive T cells exist at supraphysiological frequencies and receive a signal that is uncharacteristically strong compared to most TCR-peptide/MHC interactions. The use of TCR transgenic mice controls for one of these caveats by allowing the study of deletion in response to peptide presented on MHC class II molecules.
B cell-specific expression of a peptide from myelin oligodendrocyte glycoprotein dramatically enhances negative selection of a MOG-reactive transgenic T cells, and similar results were found using a system based on the influenza hemagglutinin protein as a neo-self antigen; however in both of these systems, it is hard to distinguish B cell-specific antigen presentation from the more general effect of increasing antigen expression in the thymus [41, 42]. TCR transgenics that are specific for peptides derived from immunoglobulin undergo enhanced negative selection when B cells bearing those immunoglobulin determinants are present in the thymus [43, 44]. In these immunoglobulin systems, it is shown that B cells are the dominant cell type presenting the immunoglobulin peptide, with very little contribution from other thymic APCs [44, 45]. Our own work has demonstrated that autoreactive B cells that are specific for the self antigen glucose-6-phosphate isomerase (GPI) process and present that antigen on MHC class II, enhancing the deletion of autoreactive T cells and suggesting that BCR-mediated endocytosis of self antigen may be a unique pathway by which thymic B cells acquire self antigen for presentation [5].
The field has largely focused on the role of thymic B cells in shaping CD4 T cell selection, and relatively little is known about the capacity of thymic B cells to influence CD8 T cell selection. In a superantigen model, it was found that thymic B cells were less effective at deleting CD8+ thymocytes when compared to their ability to delete CD4+ thymocytes [40]. In a model using the MHC class I restricted OT-1 TCR transgenic, it was found that retroviral expression of the target antigen ovalbumin in B cells did not result in increased negative selection of CD8 thymocytes; however, this system depended on the generation of bone marrow chimeras, which in our experience leads to suboptimal thymic B cell reconstitution, so additional confirmation may be necessary [46].
Remaining questions
The unique properties of thymic B cells discussed in this review raise many more biological questions that need to be addressed. In terms of development, what is the relative contribution of fetal liver vs. bone marrow and does it change with age? Can thymic B cells self renew or is some kind of progenitor always needed, and how is the homeostasis of thymic B cells maintained in the adult thymus? What molecules regulate the homing of B cell progenitors into the thymus and their migration through the cortex towards the thymic medulla? Once thymic B cells are in the medulla, are they highly motile-like thymocytes or static-like dendritic cells? How is the thymic B cell repertoire selected? Are they subject to the same kinds of tolerance mechanisms as B cell development in the bone marrow?
Furthermore, our understanding of the activated phenotype of thymic B cells is still very superficial. It will be important to determine to what extent thymic B cell activation is dependent on BCR signaling, interactions with T cells, or other signals from the thymic microenvironment. How does their activated phenotype affect their function?
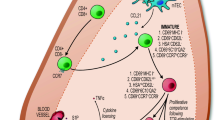
Finally, to understand their role in negative selection, it will become increasingly important to understand the nature of antigens that are being presented by thymic B cells (Fig. 2). The MHC class II presentation pathway in B cells is strongly linked to the B cell receptor. Thymic B cells present cognate self antigens, as well as peptides derived from the B cell receptor, much more efficiently than thymic dendritic cells or other thymic APCs. While it has not been formally demonstrated, it is also possible that B cell-specific proteins could be efficiently presented by thymic B cells (e.g., CD19 or B cell-specific transcription factors). Supporting this possibility is the finding that B cell-specific overexpression of model antigens leads to enhanced negative selection. Ultimately, how does presentation of these unique antigens shape the T cell repertoire?

Sources of antigens for thymic B cell presentation. Thymic B cells present cognate self antigens (blue), peptides derived from the B cell receptor (red), and possibly B cell-specific proteins (purple) to autoreactive thymocytes. Arrows indicate cognate interactions between TCR and peptide-MHC
References
Isaacson PG, Norton AJ, Addis BJ (1987) The human thymus contains a novel population of B lymphocytes. Lancet 2:1488–1491
Miyama-Inaba M, Kuma S, Inaba K, Ogata H, Iwai H, Yasumizu R, Muramatsu S, Steinman RM, Ikehara S (1988) Unusual phenotype of B cells in the thymus of normal mice. J Exp Med 168:811–816
Nango K, Inaba M, Inaba K, Adachi Y, Than S, Ishida T, Kumamoto T, Uyama M, Ikehara S (1991) Ontogeny of thymic B cells in normal mice. Cell Immunol 133:109–115
Akashi K, Richie LI, Miyamoto T, Carr WH, Weissman IL (2000) B lymphopoiesis in the thymus. J Immunol 164:5221–5226
Perera J, Meng L, Meng F, Huang H (2013) Autoreactive thymic B cells are efficient antigen-presenting cells of cognate self-antigens for T cell negative selection. Proc Natl Acad Sci USA 110:17011–17016. doi:10.1073/pnas.1313001110
Than S, Inaba M, Inaba K, Fukuba Y, Adachi Y, Ikehara S (1992) Origin of thymic and peritoneal Ly-1 B cells. Eur J Immunol 22:1299–1303. doi:10.1002/eji.1830220527
Mori S, Inaba M, Sugihara A, Taketani S, Doi H, Fukuba Y, Yamamoto Y, Adachi Y, Inaba K, Fukuhara S, Ikehara S (1997) Presence of B cell progenitors in the thymus. J Immunol 158:4193–4199. doi:10.3109/10837450.2013.829099
McKenna HJ, Morrissey PJ (1998) Flt3 ligand plus IL-7 supports the expansion of murine thymic B cell progenitors that can mature intrathymically. J Immunol 160:4801–4809
Sugihara A, Inaba M, Mori SI, Taketani S, Adachi Y, Hisha H, Inaba K, Toki J, Horio T, Gershwin ME, Ikehara S (2000) Differentiation from thymic B cell progenitors to mature B cells in vitro. Immunobiology 201:515–526. doi:10.1016/S0171-2985(00)80071-6
Ceredig R (2002) The ontogeny of B cells in the thymus of normal, CD3 epsilon knockout (KO), RAG-2 KO and IL-7 transgenic mice. Int Immunol 14:87–99
Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, Aguet M (1999) Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 10:547–558
Wilson A, MacDonald HR, Radtke F (2001) Notch 1-deficient common lymphoid precursors adopt a B cell fate in the thymus. J Exp Med 194:1003–1012
Feyerabend TB, Terszowski G, Tietz A, Blum C, Luche H, Gossler A, Gale NW, Radtke F, Fehling HJ, Rodewald H-R (2009) Deletion of Notch1 converts pro-T cells to dendritic cells and promotes thymic B cells by cell-extrinsic and cell-intrinsic mechanisms. Immunity 30:67–79. doi:10.1016/j.immuni.2008.10.016
Goetz CA, Harmon IR, O’Neil JJ, Burchill MA, Johanns TM, Farrar MA (2005) Restricted STAT5 activation dictates appropriate thymic B versus T cell lineage commitment. J Immunol 174:7753–7763
Schjerven H, McLaughlin J, Arenzana TL, Frietze S, Cheng D, Wadsworth SE, Lawson GW, Bensinger SJ, Farnham PJ, Witte ON, Smale ST (2013) Selective regulation of lymphopoiesis and leukemogenesis by individual zinc fingers of Ikaros. Nat Immunol 14:1073–1083. doi:10.1038/ni.2707
Bhandoola A, von Boehmer H, Petrie HT, Zúñiga-Pflücker JC (2007) Commitment and developmental potential of extrathymic and intrathymic t cell precursors: plenty to choose from. Immunity 26:678–689. doi:10.1016/j.immuni.2007.05.009
Zediak VP, Maillard I, Bhandoola A (2005) Closer to the source: notch and the nature of thymus-settling cells. Immunity 23:245–248. doi:10.1016/j.immuni.2005.08.008
Porritt HE, Rumfelt LL, Tabrizifard S, Schmitt TM, Zúñiga-Pflücker JC, Petrie HT (2004) Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity 20:735–745. doi:10.1016/j.immuni.2004.05.004
Benz C, Bleul CC (2005) A multipotent precursor in the thymus maps to the branching point of the T versus B lineage decision. J Exp Med 202:21–31. doi:10.1084/jem.20050146
Sambandam A, Maillard I, Zediak VP, Xu L, Gerstein RM, Aster JC, Pear WS, Bhandoola A (2005) Notch signaling controls the generation and differentiation of early T lineage progenitors. Nat Immunol 6:663–670. doi:10.1038/ni1216
Zlotoff DA, Sambandam A, Logan TD, Bell JJ, Schwarz BA, Bhandoola A (2009) CCR7 and CCR9 together recruit hematopoietic progenitors to the adult thymus. Blood 115:1897–1905. doi:10.1182/blood-2009-08-237784
Krueger A, von Boehmer H (2007) Identification of a T lineage-committed progenitor in adult blood. Immunity 26:105–116. doi:10.1016/j.immuni.2006.12.004
Martin CH, Aifantis I, Scimone ML, von Andrian UH, Reizis B, von Boehmer H, Gounari F (2003) Efficient thymic immigration of B220+ lymphoid-restricted bone marrow cells with T precursor potential. Nat Immunol 4:866–873. doi:10.1038/ni965
Kingston R, Jenkinson EJ, Owen JJ (1985) A single stem cell can recolonize an embryonic thymus, producing phenotypically distinct T-cell populations. Nature 317:811–813
Inaba M, Inaba K, Adachi Y, Nango K, Ogata H, Muramatsu S, Ikehara S (1990) Functional analyses of thymic CD5+ B cells. Responsiveness to major histocompatibility complex class II-restricted T blasts but not to lipopolysaccharide or anti-IgM plus interleukin 4. J Exp Med 171:321–326
Ferrero I, Anjuère F, Martín P, Martínez del Hoyo G, Fraga ML, Wright N, Varona R, Márquez G, Ardavín C (1999) Functional and phenotypic analysis of thymic B cells: role in the induction of T cell negative selection. Eur J Immunol 29:1598–1609. doi:10.1002/(SICI)1521-4141(199905)29:05<1598:AID-IMMU1598>3.0.CO;2-O
Inaba M, Inaba K, Fukuba Y, Mori S, Haruna H, Doi H, Adachi Y, Iwai H, Hosaka N, Hisha H (1995) Activation of thymic B cells by signals of CD40 molecules plus interleukin-10. Eur J Immunol 25:1244–1248. doi:10.1002/eji.1830250517
Akirav EM, Xu Y, Ruddle NH (2011) Resident B cells regulate thymic expression of myelin oligodendrocyte glycoprotein. J Neuroimmunol 235:33–39. doi:10.1016/j.jneuroim.2011.03.013
Fujihara C, Williams JA, Watanabe M, Jeon H, Sharrow SO, Hodes RJ (2014) T Cell-B cell thymic cross-talk: maintenance and function of thymic B Cells requires cognate CD40-CD40 ligand interaction. J Immunol 193:5534–5544. doi:10.4049/jimmunol.1401655
Kantor AB, Herzenberg LA (1993) Origin of murine B cell lineages. Annu Rev Immunol 11:501–538. doi:10.1146/annurev.iy.11.040193.002441
Kantor AB, Stall AM, Adams S, Herzenberg LA (1992) Differential development of progenitor activity for three B-cell lineages. Proc Natl Acad Sci USA 89:3320–3324
Wortis HH, Teutsch M, Higer M, Zheng J, Parker DC (1995) B-cell activation by crosslinking of surface IgM or ligation of CD40 involves alternative signal pathways and results in different B-cell phenotypes. Proc Natl Acad Sci USA 92:3348–3352
van Ewijk W (1984) Immunohistology of lymphoid and non-lymphoid cells in the thymus in relation to T lymphocyte differentiation. Am J Anat 170:311–330. doi:10.1002/aja.1001700307
Le Borgne M, Ladi E, Dzhagalov I, Herzmark P, Liao YF, Chakraborty AK, Robey EA (2009) The impact of negative selection on thymocyte migration in the medulla. Nat Immunol 10:823–830. doi:10.1038/ni.1761
Wu L, Shortman K (2005) Heterogeneity of thymic dendritic cells. Semin Immunol 17:304–312. doi:10.1016/j.smim.2005.05.001
Inaba M, Inaba K, Hosono M, Kumamoto T, Ishida T, Muramatsu S, Masuda T, Ikehara S (1991) Distinct mechanisms of neonatal tolerance induced by dendritic cells and thymic B cells. J Exp Med 173:549–559
Mazda O, Watanabe Y, Gyotoku J, Katsura Y (1991) Requirement of dendritic cells and B cells in the clonal deletion of Mls-reactive T cells in the thymus. J Exp Med 173:539–547
Molina IJ, Cannon NA, Hyman R, Huber BT (1989) Macrophages and T cells do not express Mlsa determinants. J Immunol 143:39–44
Ahmed A, Scher I, Smith AH, Sell KW (1977) Studies on non-H-2 linked lymphocyte activating determinants. I. Description of the cell type bearing the Mls product. J Immunogenet 4:201–213
Kleindienst P, Chretien I, Winkler T, Brocker T (2000) Functional comparison of thymic B cells and dendritic cells in vivo. Blood 95:2610–2616
Frommer F, Waisman A (2010) B cells participate in thymic negative selection of murine auto-reactive CD4+ T cells. PLoS One 5:e15372. doi:10.1371/journal.pone.0015372
Morlacchi S, Soldani C, Viola A, Sarukhan A (2011) Self-antigen presentation by mouse B cells results in regulatory T-cell induction rather than anergy or clonal deletion. Blood 118:984–991. doi:10.1182/blood-2011-02-336115
Detanico T, Heiser RA, Aviszus K, Bonorino C, Wysocki LJ (2011) Self-tolerance checkpoints in CD4 T cells specific for a peptide derived from the B cell antigen receptor. J Immunol 187:82–91. doi:10.4049/jimmunol.1002287
Rudensky AY, Mazel SM, Yurin VL (1990) Presentation of endogenous immunoglobulin determinant to immunoglobulin-recognizing T cell clones by the thymic cells. Eur J Immunol 20:2235–2239. doi:10.1002/eji.1830201012
Zöller M (1990) Intrathymic presentation of nominal antigen by B cells. Int Immunol 2:427–434
Werner-Klein M, Dresch C, Marconi P, Brocker T (2007) Transcriptional targeting of B cells for induction of peripheral CD8 T cell tolerance. J Immunol 178:7738–7746
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Perera, J., Huang, H. The development and function of thymic B cells. Cell. Mol. Life Sci. 72, 2657–2663 (2015). https://doi.org/10.1007/s00018-015-1895-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1895-1