
Abstract
The mode of tumor cell death has significant effects on anti-tumor immunity. Although, previously it was thought that cell death is an inert effect, different investigators have clearly shown that dying tumors can attract, activate and mature professional antigen presenting cells and dendritic cells. In addition, others and we have shown that the type of tumor cell death not only controls the presence or absence of specific tumor antigens, but also can result in immunological responses ranging from immunosuppression to anti-tumor immunity. More importantly, it is possible to enhance anti-tumor immunity both in vitro and in vivo by targeting specific molecular mechanisms such as oligopeptidases and the proteasome. These studies not only extend our knowledge on basic immunological questions and the induction of anti-tumor immunity, but also have implications for all types of cancer treatments, in which rapid tumor cell death is induced. This review is a comprehensive summary of cell death and particularly necrosis and the pivotal role it plays in anti-tumor immunity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction to cell death
Cell death plays a pivotal role in different physiological processes and pathological conditions. Several different types of cell death exist, which differ from each other based on death stimuli, signaling pathways involved in the process, morphological and biochemical changes developed in dying/dead cells and the biological outcome [28, 56].
Cell death is an extremely complex and multifactorial process challenging the attempts to understand, characterize and classify it. Based on the localization of death stimuli, cell death might be intrinsic or extrinsic. Intrinsic death is induced by the signals from within the cell whereas extrinsic death is caused by extracellular stimuli. Depending on the character of primary stimuli and death progression one can distinguish programmed, regulated or accidental cell death. Morphologically apoptosis, necrosis, autophagy or cornification is distinguished as typical cell death mode.
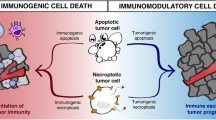
Cell death of tumor cells can induce immunological responses, which can vary from immunosuppression to the induction of a tumor-specific immunity. Apart from the type of the cell death, the location of tumor cell death as well as the local cellular environment of dying tumor cells may affect anti-tumor immunity.
Apoptosis was considered to be a programmed and controlled type of cell death in physiology [50]. It was proposed as a universal clearance mechanism of unwanted, aged, stressed cells from the body, quiescently, without induction of inflammation and injury [50]. As mentioned above apoptosis might be induced by the stimuli coming from outside the cells as well as from intracellular signaling [108]. Extracellular signaling involves activation of death receptors [e.g., FAS (CD95) receptor cascade [5, 14]], or perforins and granzymes released by cytotoxic cells [64]. Intracellular initiators of apoptosis might be DNA damage, release of free radicals and cytochromes from mitochondria, or proteins of Bcl family and p53 (reviewed in [54, 108]). All these intrinsic and extrinsic pathways converge at the intracellular level and lead to caspase activation, which is responsible for programmed degradation of cytoplasmic and nuclear proteins and internucleosomal cleavage of DNA [54, 108]. In the course of apoptosis cell integrity stays relatively intact, organelles are preserved, plasma membrane blebbing occurs and intracellular contents are not released in surrounding tissue [108]. Condensation of cytoplasm leads to cell shrinkage and to generation of ‘apoptotic bodies’, which are carrying ‘eat-me’ signals on the surface and are cleared by major scavengers of body macrophages (reviewed in [87]).
Autophagic cell death is a type of cell death which is accompanied by autophagy, a process usually involved in cell survival but not responsible for execution of death pathway per se [70]. Morphologically autophagy is characterized by cytoplasmic vacuolization and accumulation of autophagic vesicles, autophagosomes. They sequester cytoplasmic content including organelles, fuse with lysosomes and generate autolysosomes in which the content is degraded by lysosomal hydrolases [24]. These processes occur in the absence of chromatin condensation and independent from phagocytosis which is a hallmark of apoptosis. Autophagy has been shown to play significant role in MHC-II or MHC-I-based presentation and involved in the generation of epitopes for cross-priming of tumor cell-associated antigens [46, 62]. Recent studies have shown that autophagy might support the expression of proinflammatory cytokines—type I IFNs, “eat-me” signals or DAMPs such as ATP or HMGB 1 by the cells [68, 100, 101]. They can alarm immune system and trigger innate or adaptive immune responses.
The term necrosis is used for description of an accidental, forced, pathological type of cell death triggered by harsh, external, physical or chemical stimuli including, heat, freeze-thawing, mechanical stress or osmotic shock. Morphologic characteristic of necrosis are cell swelling, loss of nuclear and cytoplasmic integrity, rupture of cellular membranes and release of intracellular contents into extracellular milieu. This leads to recruitment of cells of the immune system and triggers inflammation. Often necrosis is associated with infection and, therefore, is ‘non-sterile’. Although in certain pathological processes it can be induced under aseptic conditions. This includes mostly ischemic necrosis (e.g., myocardial infarction, stroke, tumor necrosis) or necrosis developed during intoxication (e.g., hepatic injury, or aseptic pancreonecrosis) [73, 82, 106]. Accidental necrosis is also characterized with rapid development of death process. It might, therefore, lack morphological and biochemical changes typically associated with initial apoptotic step and which are hallmark of secondary necrosis.
Recent studies have revealed that necrosis can also occur in a tightly controlled, regulated fashion and can be induced by different stimuli including ischemia–reperfusion injury, certain pathogens, signaling through the receptors of TNF superfamily, Toll-like receptors or IFN-γ (reviewed in [103]). Death signaling in typical regulated necrosis termed as necroptosis classically is conducted through receptor-interacting protein kinase 1 (RIPK1–RIPK3) complex [23, 41]. It is accompanied by inhibition of Caspase-8 and generation of necrosome, which is responsible for consequent necrosis induction [23, 41]. Types of regulated necrosis are extending and new definitions or characteristics of them will follow.
Immunogenicity of cell death
Dead or dying cells can play a crucial role in generation of immune responses. They serve as a source of antigen for APCs [2], as well as provide signals in context of which antigen can lead either to immune activation or to tolerance [27, 65].
The terms “immunogenicity of cell death” or “immunogenic cell death” (ICD) is often used by scientists to describe the ability of dead/dying cells (especially of tumor cells) to mount antigen-specific and particularly CD8+ T cell-mediated adaptive immune responses and not simply lead to innate inflammation. CD8+ T cells play significant role in tumor protection and development of this type of immunity is an ultimate goal for successful anticancer therapy. Here we will discuss the ability of necrotic cells to trigger Ag-specific CD8+ T cell immune responses and term ICD will be used for this aim unless otherwise mentioned.
It has been a matter of debate as to which type of cell death influences the activity of the immune system, i.e., which cell death is immunogenic and which is not. This question is of high importance since understanding the immunological consequences of cell death and molecular mechanisms responsible for it can allow manipulating course of death and to trigger desired type of immunity. The “Danger theory”, which was initially proposed by Polly Matzinger postulated that the immune system has an ability to recognize endogenous signals from stressed, injured dying/dead cells and responds to it [67]. Mostly hydrophobic signals, termed damage-associated molecular pattern molecules (DAMPs), are responsible for mounting immune responses according to the danger theory [27, 90]. Since necrosis was known to be a forced accidental type of cell death with loss of cellular integrity, release of intracellular content and associated inflammation, it was considered to be immunogenic as well. This concept was strongly supported by the danger theory. For years, it was generally accepted that DAMPs released during necrosis can lead to local inflammation and generate immune responses. At the same time apoptosis was hypothesized to be lacking DAMPs and, therefore, counted as a non-immunogenic death or even with the potential to tolerize the immune system against self-antigens [67].
However, when the danger theory was tested in practice, particularly in development of cell-based vaccines for anticancer therapy, results were unexpected. Although under certain circumstances immune activation was achieved [88, 112], many attempts to generate successful immune response using necrotic cells failed [6, 18, 29, 31, 40, 45, 72, 89, 112]. In vitro DAMPs released from necrotic cells lead to maturation of antigen presenting cells (APCs) and had innate immune cell activation ability, but in vivo necrotic cell vaccines were showing immunologically inert nature and although local inflammation and infiltration of injection site with immune cells occurred it did not transform into productive adaptive immune response and did not protect animals from tumor development [31, 89]. On the other hand, at least some death stimuli triggering apoptosis were able to mount successful adaptive immunity [2, 72]. Screening of different chemotherapeutic agents or radiation revealed that apoptotic cells generated by these death inducers had a potential of productive immunity [72].
These contradictory observations indicated that the mechanism of immunogenicity of cell death is far more complex and forced scientists to revise the original concept. It was postulated that not every necrosis is immunogenic and not every apoptosis tolerogenic or an immunologically null event. The potential of dead/dying cells to trigger adaptive immune response and to be used as a source of antigen/DAMPs does not directly correlate with the type of cell death and is defined by biochemical and molecular changes occurring in the cell. In recent years, a modernized concept has emerged [7, 42, 55], which defines ICD in general as a result of mutual or consequent processes including ER stress [33], release of “find-me” signals (e.g., ATP [26]), exposure of “eat-me” signals (e.g., calreticulin [32], phosphatidylserine [61]) and DAMPs (HMGB1 [88], F-actin [1]). These molecular changes might occur in the cells undergoing apoptotic or necrotic death. Recent studies have broadly addressed the question of immunogenicity of apoptosis although the immunogenicity of necrosis (particularly of sterile, accidental necrosis, which might be used as a potential tool for generation of efficient and safe tumor cell vaccines) still remains as an open dilemma. Here we will discuss this further.
In vitro accidental necrosis (especially repeated freeze-thawing and/or extended heating of the cells) is the fastest and most efficient way to induce uniform cell death where 100 % of cells will show a hallmark of death (propidium iodide or trypan blue incorporation tested by flow cytometry and light microscopy, unpublished observations). No other cell death inducers including irradiation or chemotherapeutic agents show such efficacy [72]. Irradiated cells, or cells treated by chemotherapeutic agents almost always contain the portion which might be defined morphologically or biochemically as a ‘live cell’ or the cell in an “early apoptotic” or “late apoptotic” stage and such “contamination” has to be taken into account when immunogenicity of distinct cell death is addressed. This issue is becoming very important when considering safety of in vitro-killed tumor cells as cell-based vaccines for treatment of cancer patients.
It is worth to mention that except inappropriate cell death, development of immunosuppression, exhaustion of the effector cells, suboptimal tumor antigen, low immunogenicity of the tumor cells, might be additional factors responsible for a failure of tumor vaccines which can operate either separately or in combination.
Molecular mediators of ICD
Current concept of ICD describes “find-me” signals, “eat-me” signals and danger signals (DAMPs) as an ultimate requirement for immunogenicity of cell death and states that these signals together with ER stress are necessary for ICD induction. According to our current understanding, essential components responsible for ICD development are generation of reactive oxygen species (ROS) and ER stress [72]. ROS, produced in response to various stimuli, affects ER homeostasis, which as a cause leads to overload of ER with unfolded proteins. This triggers death pathways and induces mitochondrial apoptosis. As a bystander effect signaling cascades facilitate exposure of DAMPs on the surface of the cell and into the extracellular milieu [72]. In this regard exposure of dying cells to antioxidants, which scavenge ROS diminishes their immunogenicity [74]. These changes occur usually in the very beginning of death and lead to exposure/release of DAMPs already in pre-apoptotic or early or mid-apoptotic stages [57]. ROS generation and ER stress might be also a marker for necrosis (at least of regulated one) but whether this terminates in ICD or not still has to be addressed [103]. Accidental necrosis in this regard is not studied, but remains very interesting, especially if we take into account that necrotic cells used as cancer vaccines very often failed to show immunogenicity [6, 18, 29, 31, 40, 45, 72, 89].
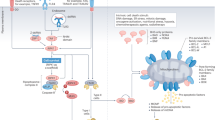
Significant advances have been made in past 20 years to identify molecular mediators released from dying/dead cells and responsible for immunogenicity of death and elucidate their mechanism of action as well as receptors responsible for their recognition by immune cells (Table 1; Fig. 1).

Factors defining immunogenicity of accidental necrotic cells. Necrotic cells release “eat-me” signals, “find-me” signals, DAMPs, ROS and proinflammatory cytokines which alert professional antigen presenting cells, DCs, cause their migration, maturation and activation. When cellular peptidases are inactivated in necrotic cells, accumulation of proteasomal degradation products, oligopeptides, occur and they form complexes with chaperones. Recruited DCs pick up antigen released from necrotic cells, load it on MHC-I, deliver to antigen-specific CD8+ T cells, trigger their activation and induce Ag-specific adaptive immune responses
ATP
First evidence for ATP having anti-tumor effect came from the study of Rapaport and Fontaine [78] which found that intra-peritoneal injection of ATP triggered anti-tumor immune response. Apoptotic as well as necrotic cells secrete or release ATP as a “find-me” signal into extracellular milieu and alarm the cells of the immune system [26, 44]. It is recognized by monocytes through P2Y2 receptors which control monocyte recruitment and initiation of immune response [26]. ATP is responsible for NLRP3-mediated IL-1β secretion by dendritic cells in adaptive anti-tumor immunity [37]. On the other hand, in the presence of ectonucleotidases (CD39, CD73) ATP can be hydrolyzed into adenosine which exerts pronounced immunosuppressive function [10, 76]. These results indicate that ATP might have context dependent opposite influence on the immune system activation during the cell death.
Calreticulin
Calreticulin (CRT) is an ER protein involved in regulation of Ca2+ homeostasis, assembly of MHC class I molecules and chaperone function, as well as in proliferation and migration of the cells [34, 36, 38]. Besides these functions CRT has been shown to serve as an “eat-me” signal after exposure on the cell surface and is implicated in the clearance of apoptotic cells [32]. It was also shown that ecto-CRT defines immunogenicity of cell death during apoptosis as well as necroptosis and is responsible for generation of adaptive immune responses in anticancer therapy [47, 72]. Whether CRT is exposed on the surface of the cells undergoing accidental necrosis and plays a role in immunogenicity or not still needs to be tested.
HSPs
Heat shock proteins are intracellular proteins constitutively expressed in the cells or induced during stress response. They localize in cytoplasm or in endoplasmic reticulum and play a role in protein folding. When released from dying/dead cells HSPs (HSP60, HSP70, HSP72, HSP90, GP96) interact with different receptors, including CD91, TLR2, TLR4 and exert certain immunostimulatory effects such as DC maturation, recruitment of innate immune cells, NK cell activation. HSPs possess specific function of peptide chaperones, deliver antigens for MHC class I-based direct presentation or cross-presentation and thereby mediate CD8+ T cell activation and cellular immunity (discussed in following review [98]). It is worth to mention that some studies using recombinant HSPs still suffer with the contamination of LPS despite the fact that this contamination was addressed experimentally. Therefore, such results must be considered carefully when the immune response generation ability of HSPs is discussed [13, 96].
HMGB1
High mobility group box 1 protein (HMGB1) initially was discovered as a nuclear protein which binds to DNA and plays a role in chromatin formation and modulation of gene expression [15, 69]. In acetylated form it can be actively secreted by monocytes/macrophages as a cytokine during sepsis [105] or by tumor cells during induction of apoptosis [4]. Alternatively HMGB1 is passively released when cellular integrity is lost, during accidental as well as controlled secondary necrosis [4, 88]. HMGB1 engages receptor for advanced glycosylation end product (RAGE), TLR2 or TLR4 [4, 21] and triggers inflammatory response, including cytokine secretion (TNF-α, IL-1, IL-6), migration of inflammatory cells or expression of co-stimulatory molecules [3, 81, 88]. Recent studies have shown that reduced HMGB1 acts as a chemoattractant, whereas disulphide bond-possessing molecule had a proinflammatory cytokine function and oxidized HMGB1 is inactive [104, 109]. Moreover, oxidation of HMGB1 in apoptotic cells is responsible for induction of tolerance [49]. These studies raise the question whether HMGB1 under conditions, when extended necrosis and ROS production occurs has proinflammatory effect or not. This might explain our previous studies showing that necrotic cells in vitro or in vivo do not always trigger CD8+ T cell activation and successful adaptive immune responses [30, 31]. In line with this observation, a recent study has shown that HMGB1 binds to TIM3 receptor on dendritic cells and negatively regulates nucleic acid-mediated anti-tumor immune responses in vivo [22].
S100 proteins, cytokines and uric acid
S100A8 S100A9, members of S100/calgranulin protein family, is released from dead cells and has an immunostimulatory effect. Like HMGB1 they interact with RAGE, attract innate immune cells and mediate inflammation [97]. These molecules are also involved in inflammation associated with cancer and promote carcinogenesis [35].
IL-1α, IL-6, IL-33 are inflammatory cytokines released during necrosis. They signal via IL-1R, ST2 or IL-6R and gp130, respectively, and possess strong proinflammatory activity [25, 66, 102].
Uric acid is the product of nucleic acid degradation and in the form of monosodium urate has a potential to cause dendritic cell maturation and neutrophil attraction. It is released from necrotic cells and acts through an unidentified mechanism [93].
Nucleic acids and ribonucleoproteins
Induction of necrosis and rupture of plasma membrane exposes RNPs, genomic or mitochondrial DNA and mRNA to the extracellular environment [20, 48, 116]. These DAMPs can interact with various pattern recognition receptors (PRRs), possess potent proinflammatory effect and can activate innate immune cells including macrophages neutrophils. Whether they lead to CD8+ T cell-mediated immune response generation is still not clear.
F-actin
It was shown that CD8α+ dendritic cells express DNGR-1 (CLEC9A), potential DAMP receptor, which responds to necrotic cells [83]. Engagement of this receptor by necrotic cells caused activation of antigen-specific CD8+ T cells. Recent study has identified F-actin, an evolutionarily conserved ancient protein to serve as a ligand responsible for this effect [1]. Whether it binds directly to DNGR-1 or in association with other molecules still remains to be identified. In addition, more studies are required to address the role of F-actin in necrosis—in a condition not always triggering adaptive immune response.
Antigenic source in necrotic cells for MHC-I-based presentation
Immunogenicity of the necrotic cells is greatly influenced by another important factor, an antigenic source in the dying/dead cells. Therefore, we will discuss this topic from a necrotic cell death perspective. Ag-specific CD8+ T cell activation and adaptive immune response generation require not only DAMPs/PAMPs signaling but presentation of specific antigenic epitope as peptide–MHC Class I complexes by professional APCs—DCs. DCs are specific cells which take up antigenic material, process it, generate Ag peptides, load on MHC-I and present on their surface in a context of co-stimulatory molecules in a process designated as a cross-presentation. This primes CD8+ T cells and generates effector immune responses. One of the major antigenic sources, which are used by DCs for processing and presentation, are dying/dead cells, including necrotic cells. Cross-presentation is influenced by several different factors and particularly, by the form and the amount of antigen present in Ag source, the localization of antigen, cellular machinery present in cells of antigenic source. These all define whether antigen will be available for CD8+ T cell presentation or not.
Until now three different forms of antigen are found to be utilized for generation of peptide–MHC-I complexes which include proteins [8, 71, 92], protein degradation products/oligopeptides [13, 58, 59] and defective ribosomal products (DRiPs) [63, 110, 111]. It was shown that particulate protein antigen is presented more effectively than soluble form [60] and 50,000-fold more soluble antigen is required to achieve CD8+ T cell activation than as a cell-associated form. It was established for cell-associated model antigen Ovalbumin (OVA) that as few as 0.2 ng/mouse was sufficient for OT-I T cell proliferation in vivo. For soluble ovalbumin this threshold was in a range of 10 µg protein per mouse [60]. Similarly, immunogenicity of oligopeptides/protein degradation products is enhanced when associated with chaperones [59, 98] and few hundred pictograms of peptides in complex with HSPs are sufficient to induce antigen-specific immune responses.
Majority of the cellular proteins are degraded by ubiquitin–proteasome pathway [39]. This is a key mechanism for removal of damaged, unfolded or defective molecules and generation of peptide pool [53]. Proteasomal hydrolysis might be involved in degradation of DRiPs as well. This step generates oligopeptides with the size of 2–30 amino acids [52] majority of which are targeted and degraded to amino acids. Minor amount of proteasomal degradation products are further processed and loaded on MHC-I. It was shown that in most cases proteasome generates mature C-terminus of an epitope [19, 75], which do not need further cleavage for binding to MHC-I. Inhibition of proteasome using specific inhibitors also revealed that it is almost exclusively single peptidase with carboxypeptidase activity in the cytosol targeting proteins. In opposite, proteasomal degradation products have extended N-termini and for generation of mature epitope their further processing and trimming by aminopeptidases is necessary. This can occur either by cytosolic aminopeptidases [leuzin aminopeptidase (LAP) [11], bleomycin hydrolase (BH), puromycine-sensitive aminopeptidase (PSA) [99], thimet oligopeptidase 1 (TOP-1) [95] tripeptidyl peptidase II (TPPII) [113]], ER aminopeptidase (ERAP1, ERAP2) [86] or ER aminopeptidase associated with antigen processing (ERAAP) [91]. Treatment of cells with metal chelator ο-phenanthroline or metallopeptidase inhibitor bestatine drastically reduces trimming process [11]. Trimming of N-termini, therefore, is a second proteolytic step in MHC-I epitope generation.
Oligopeptides generated by proteasome in the cytosol might be toxic and their accumulation could affect proper functioning of the cells. Cells possess variety of peptidases including endopeptidases and exopeptidases which rapidly degrade oligopeptides to amino acids and thereby preclude their accumulation. Peptide half-life of a few seconds indicates [79] that this process is highly efficient and removes almost 99 % of proteasome products [77]. Minute amounts of the peptides which escape degradation might be utilized for MHC-I loading and presentation. Therefore, oligopeptide degradation is a last step which might define whether epitopes will be available for MHC-I loading and presentation or not. Several peptidases present in cytosol or ER were described to have peptide destruction ability, including thimet oligopeptidase 1 (TOP-1), dipeptidyl peptidase 3 (DPP-3), PSA, TPPII, ERAP1, ERAP2 [29, 80, 84, 85, 114, 115]. These three machineries in accidental necrotic cells might be of great importance since they might control availability of antigen for peptide–MHC-I complex formation and for cross-priming of CD8+ T cells and thereby define the immunogenicity of the necrotic cell death.
Immunogenicity of primary accidental necrosis and the role of antigenic source
Several studies including ours have shown that using freeze-thawed accidental necrotic cells as vaccines failed to generate antigen-specific CD8+ T cell-mediated immune responses [6, 18, 29, 31, 40, 45, 72, 89, 112]. Induction of accidental necrosis is associated with rupture of the cellular membrane and release of intracellular content. Therefore, compartmentalization of the antigen in certain structures (such as exosomes [107] or DRibbles [63]) and marking them with “eat-me” signals for uptake and presentation might not take place or might be very inefficient. Instead when three cycles of freeze-thawing is applied to the cells, part of the cytoplasmic protein antigen (e.g., model antigen ovalbumin) is released in extracellular environment in a soluble form. Part of the antigen is still associated with cell debris as a particulate protein [31] and can be easily pulled down in a centrifugation step. Loss of the antigen by the necrotic cells and reduction of the amount of particulate antigen might be one mechanism why freeze-thawing renders necrotic cells non-immunogenic under sterile conditions in vivo. Moreover, even mixture of freeze-thawed lysate with recombinant soluble OVA protein failed to generate Ag-specific adaptive immune responses (unpublished observation) suggesting that even if DAMPs are released during accidental necrosis they are not sufficient to induce immune response when non-efficient soluble form of the protein antigen is present. In line with this observation, soluble fraction of freeze-thawed cells containing antigen and mixed with PAMPs such as LPS or CpG was not enough to trigger adaptive immunity. These results are in accordance with the study showing that soluble antigen is less effective for CD8+ T cell immune response generation [60]. Particulate antigen associated with freeze-thawed necrotic debris was still able to induce immune response in vivo but in the presence of CpG only. In addition, single vaccination with particulate fraction was not sufficient and challenge of the mice was necessary to trigger CD8+ T cell-mediated immune responses. All these results suggest that accidental sterile necrosis induced by freeze-thawing of the cells lack immunogenicity and is not a proper death for generation of CD8+ T cell responses despite presence of abundant protein antigen.
Freeze-thawing induces rapid accidental death of the cells when cytosolic peptidases including proteasome or peptidases (e.g., TOP-1, DPP-3) stay relatively intact and preserve their biological activity [29]. That means, proteasomes might still process ubiquitinated proteins and/or DRiPs and generate oligopeptides/proteasomal degradation products. We have confirmed this in an experiment when high molecular weight fraction (F 9–23) of freeze-thawed cell lysates were mixed with OVA protein and cultured with OT-I cells. Only in this condition T cell proliferation was observed. Pretreatment of the fraction with lactacystin, specific inhibitor of a proteasome, reduced this effect indicating that this fraction converted non-immunogenic soluble OVA into immunogenic form which might be recognized by T cells and that proteasomal activity might be responsible for this phenomenon [29]. At the same time, freeze-thawed cells still failed to activate CD8+ T cells indicating that either, cells contained natural inhibitors of a proteasome, or proteasomal degradation products were somehow eliminated and peptide–MHC-I formation was precluded. Indeed additional fractionation (F 37–45) and functional studies identified at least five different peptidases including TOP-1, DPP-3, prolyl endopeptidase (PEP), Neurolysin, arginyl aminopeptidase present in freeze-thawed cells and at least TOP-1 and DPP-3 had an ability to abolish T cell proliferation by targeting proteasomal degradation products. When freeze-thawed fractions F37–45 were treated by trypsin or inactivated by heat, they lost inhibitory effect on T cell proliferation indicating that one mechanism how to generate immunogenic necrosis might be targeting of peptidases responsible for oligopeptide degradation [29].
When we induced accidental necrosis by heating of the cells, necrotic cells gained Ag-specific CD8+ T cell activating function both in vitro and in vivo [29]. Again inhibition of a proteasome or addition of recombinant TOP-1 or DPP-3 reduced T cell activation ability. These data suggest that at least one mechanism of immunogenicity of accidental necrotic cell death relies on a balance between proteasome activity and peptide destructive/removal forces. This balance defines whether oligopeptides generated by proteasome will be available for chaperoning, further trimming and loading on MHC-I or not. Importantly, high temperature and extended heating of the cell inactivates proteasome as well, but it seems that peptidases involved in degradation of oligopeptides are more susceptible to the heat. This explains why heated necrotic cells still retain proteasomal degradation products and have an ability to serve as an antigenic source for Ag-specific CD8+ T cell-mediated immune responses. It is perhaps worth to find the golden ratio between inactivation of peptidases and preservation of a proteasome function for development of successful immunogenic accidental necrosis. Alternatively gene silencing or specific inhibitors of peptidases might be considered to achieve this goal. In contrast to freeze-thawing, accidental necrosis induced by heating generates relatively less amount of soluble fraction which might be explained by denaturation of biopolymers and extensive aggregation. This might be an additional factor which might enhance the ability of necrotic cells to mount Ag-specific immune response, since denatured/aggregated antigens are processed and presented more efficiently.
Different studies suggest that cellular peptidases might play dual role in oligopeptide processing. They might serve as a generator of mature epitopes by trimming N-termini or cause destruction of extended peptides as well as mature epitopes. It was shown that PSA, TPPII, ERAP1, ERAP2, TOP-1 might possess such function and in a context dependent manner either enhance immunogenicity or not. TOP-1 which was initially discovered by Camargo et al. [17] was initially shown to be involved in processing of peptides for MHC-I-based presentation [95]. This study was additionally supported by the observation that TOP-1 together with nardilysin was involved in oligopeptide trimming and epitope generation [51]. Additional studies discovered that this enzyme has broad specificity which relies on the length, shape and the amino acid composition of peptides [12, 16, 94]. This broad specificity can also explain why TOP-1 plays a role in degradation of epitopes and abortion of CD8+ T cell responses in vitro and in vivo [29, 85, 115]. We have purified at least four other peptidases in non-immunogenic primary necrotic cells. Moreover, when tested, DPP-3 had inhibitory effect similar to TOP-1 indicating that peptide degradation plays important role in defining the immunogenicity of dead cells. Finally, further studies are needed to reveal the role of other peptidases and dissect the mechanism behind the immunogenicity of necrotic cell death.
Immunogenicity of secondary necrotic cells
When cells are treated by irradiation (both UV as well as γ-irradiation) or by chemicals they undergo apoptosis, although after certain time these cells also show the signs of necrosis. Such necrosis is defined as a secondary necrosis. Several studies [45, 83] including our unpublished observations have shown that secondary necrotic cells induced by UV irradiation are immunogenic and they can mount Ag-specific CD8+ T cell-mediated immune response in vivo or in vitro. Although the immunogenicity of secondary necrosis as a subsequent step of apoptosis might rely on the postulates of ICD including up-regulation of “eat-me” or “find-me” signals, release of DAMPs and ER stress, the role of different antigenic source in this process is still to be addressed.
Infection and non-sterile necrosis
Microbial infection including viruses, bacteria or fungi, trigger inflammatory responses as well as innate and adaptive immune system activation. In this scenario, activation is mediated through pathogen-associated molecular pattern molecules PAMPs, which are recognized by PRR expressed on immune cells [43]. This special feature of microbial agents introduces extra level of complexity in the response of immune system to non-sterile cell death, including non-sterile necrosis. In this aspect microbial infection can influence the course and immunogenicity of cell death (including necrosis) in many different ways: (1) microbes and PAMPs can trigger or modulate death pathways within infected cells and lead to necrosis induction; (2) extended inflammatory response (e.g., cytokine storm), developed against microbes or microbial products can be a cause of bystander necrosis of neighboring uninfected cells; (3) DAMPs released from dying/dead infected cells can be responsible for immune system activation per se and trigger (de novo) or further enhance (alternatively suppress) innate and adaptive immune response; (4) infection can modulate DAMPs and thereby alter the recognition of DAMPs released from dying/dead cells by immune system; (5) PAMPs might convert non-immunogenic death into ICD (e.g., non-sterile necrosis) thereby leading to adaptive immune response generation against cell-associated antigens [31]. Taken all abovementioned into account it is clear that cell–microbial and host–pathogen interactions are very complex from cell death point of view and extensive studies are needed for understanding of the mechanisms of such interactions.
Abbreviations
- DAMP:
-
Damage-associated molecular pattern molecules
- PAMP:
-
Pathogen-associated molecular pattern molecules
- IFN:
-
Interferon
- HMGB1:
-
High mobility group box 1 protein
- TNF:
-
Tumor necrosis factor
- TLR:
-
Toll-like receptor
- RIPK:
-
Receptor-interacting protein kinase
- APC:
-
Antigen presenting cell
- ICD:
-
Immunogenic cell death
- ER:
-
Endoplasmic reticulum
- ROS:
-
Reactive oxygen species
- CRT:
-
Calreticulin
- HSP:
-
Heat shock protein
- LPS:
-
Lipopolysaccharide
- RAGE:
-
Receptor for advanced glycosylation end product
- IL:
-
Interleukin
- RNP:
-
Ribonucleoproteins
- PRR:
-
Pattern recognition receptor
- DC:
-
Dendritic cell
- DRiP:
-
Defective ribosomal product
- OVA:
-
Ovalbumin
- LAP:
-
Leuzin aminopeptidase
- PSA:
-
Puromycine-sensitive aminopeptidase
- BH:
-
Bleomycin hydrolase
- TOP-1:
-
Thimet oligopeptidase 1
- TPPII:
-
Tripeptidyl peptidase II
- ERAP:
-
ER aminopeptidase
- ERAAP:
-
ER aminopeptidase associated with antigen processing
- DRibbles:
-
DRiPs-containing blebs
- DPP-3:
-
Dipeptidyl peptidase 3
- PEP:
-
Prolyl endopeptidase
- Ag:
-
Antigen
References
Ahrens S, Zelenay S et al (2012) F-Actin Is an Evolutionarily Conserved Damage-Associated Molecular Pattern Recognized by DNGR-1, a Receptor for Dead Cells. Immunity 36:635–645
Albert ML, Sauter B et al (1998) Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 392(6671):86–89
Andersson U, Wang H et al (2000) High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 192(4):565–570
Apetoh L, Ghiringhelli F et al (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13(9):1050–1059
Ashkenazi A, Dixit VM (1999) Apoptosis control by death and decoy receptors. Curr Opin Cell Biol 11(2):255–260
Bartholomae WC, Rininsland FH et al (2004) T cell immunity induced by live, necrotic, and apoptotic tumor cells. J Immunol 173(2):1012–1022
Bartlett DL, Liu Z et al (2013) Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer 12(1):103
Basta S, Stoessel R et al (2005) Cross-presentation of the long-lived lymphocytic choriomeningitis virus nucleoprotein does not require neosynthesis and is enhanced via heat shock proteins. J Immunol 175(2):796–805
Basu S, Binder RJ et al (2000) Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol 12(11):1539–1546
Beavis PA, Stagg J et al (2012) CD73: a potent suppressor of antitumor immune responses. Trends Immunol 33(5):231–237
Beninga J, Rock KL et al (1998) Interferon-gamma can stimulate post-proteasomal trimming of the N terminus of an antigenic peptide by inducing leucine aminopeptidase. J Biol Chem 273(30):18734–18742
Berti DA, Morano C et al (2009) Analysis of intracellular substrates and products of thimet oligopeptidase in human embryonic kidney 293 cells. J Biol Chem 284(21):14105–14116
Binder RJ, Srivastava PK (2005) Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat Immunol 6(6):593–599
Boldin MP, Varfolomeev EE et al (1995) A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem 270(14):7795–7798
Bustin M, Hopkins RB et al (1978) Immunological relatedness of high mobility group chromosomal proteins from calf thymus. J Biol Chem 253(5):1694–1699
Camargo AC, Gomes MD et al (1997) Structural features that make oligopeptides susceptible substrates for hydrolysis by recombinant thimet oligopeptidase. Biochem J 324(Pt 2):517–522
Camargo AC, Shapanka R et al (1973) Preparation, assay, and partial characterization of a neutral endopeptidase from rabbit brain. Biochemistry 12(9):1838–1844
Casares N, Pequignot MO et al (2005) Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 202(12):1691–1701
Cascio P, Hilton C et al (2001) 26S proteasomes and immunoproteasomes produce mainly N-extended versions of an antigenic peptide. EMBO J 20(10):2357–2366
Cavassani KA, Ishii M et al (2008) TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med 205(11):2609–2621
Chen G, Ward MF et al (2004) Extracellular HMGB1 as a proinflammatory cytokine. J Interferon Cytokine Res 24(6):329–333
Chiba S, Baghdadi M et al (2012) Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 13(9):832–842
Cho YS, Challa S et al (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137(6):1112–1123
Deretic V, Levine B (2009) Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5(6):527–549
Eigenbrod T, Park JH et al (2008) Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol 181(12):8194–8198
Elliott MR, Chekeni FB et al (2009) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461(7261):282–286
Gallucci S, Lolkema M et al (1999) Natural adjuvants: endogenous activators of dendritic cells. Nat Med 5(11):1249–1255
Galluzzi L, Vitale I et al (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19(1):107–120
Gamrekelashvili J, Kapanadze T et al (2013) Peptidases released by necrotic cells control CD8+ T cell cross-priming. J Clin Invest 123(11):4755–4768
Gamrekelashvili J, Kruger C et al (2007) Necrotic tumor cell death in vivo impairs tumor-specific immune responses. J Immunol 178(3):1573–1580
Gamrekelashvili J, Ormandy LA et al (2012) Primary sterile necrotic cells fail to cross-prime CD8(+) T cells. Oncoimmunology 1(7):1017–1026
Gardai SJ, McPhillips KA et al (2005) Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123(2):321–334
Garg AD, Krysko DV et al (2012) A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J 31(5):1062–1079
Garg AD, Nowis D et al (2010) Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta 1805(1):53–71
Gebhardt C, Riehl A et al (2008) RAGE signaling sustains inflammation and promotes tumor development. J Exp Med 205(2):275–285
Gelebart P, Opas M et al (2005) Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int J Biochem Cell Biol 37(2):260–266
Ghiringhelli F, Apetoh L et al (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15(10):1170–1178
Gold LI, Eggleton P et al (2010) Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J 24(3):665–683
Goldberg AL, Cascio P et al (2002) The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol Immunol 39(3–4):147–164
Goldszmid RS, Idoyaga J et al (2003) Dendritic cells charged with apoptotic tumor cells induce long-lived protective CD4+ and CD8+ T cell immunity against B16 melanoma. J Immunol 171(11):5940–5947
Holler N, Zaru R et al (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1(6):489–495
Inoue H, Tani K (2014) Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ 21(1):39–49
Iwasaki A, Medzhitov R (2004) Toll-like receptor control of the adaptive immune responses. Nat Immunol 5(10):987–995
Iyer SS, Pulskens WP et al (2009) Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA 106(48):20388–20393
Janssen E, Tabeta K et al (2006) Efficient T cell activation via a toll-interleukin 1 receptor-independent pathway. Immunity 24(6):787–799
Joubert PE, Albert ML (2012) Antigen cross-priming of cell-associated proteins is enhanced by Macroautophagy within the antigen donor cell. Front Immunol 3:61
Kaczmarek A, Vandenabeele P et al (2013) Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38(2):209–223
Kariko K, Ni H et al (2004) mRNA is an endogenous ligand for toll-like receptor 3. J Biol Chem 279(13):12542–12550
Kazama H, Ricci JE et al (2008) Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29(1):21–32
Kerr JF, Wyllie AH et al (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257
Kessler JH, Khan S et al (2011) Antigen processing by nardilysin and thimet oligopeptidase generates cytotoxic T cell epitopes. Nat Immunol 12(1):45–53
Kisselev AF, Akopian TN et al (1999) The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J Biol Chem 274(6):3363–3371
Kloetzel PM (2004) Generation of major histocompatibility complex class I antigens: functional interplay between proteasomes and TPPII. Nat Immunol 5(7):661–669
Kroemer G, Galluzzi L et al (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87(1):99–163
Kroemer G, Galluzzi L et al (2013) Immunogenic cell death in cancer therapy. Annu Rev Immunol 31:51–72
Kroemer G, Galluzzi L et al (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16(1):3–11
Krysko DV, Garg AD et al (2012) Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 12(12):860–875
Kunisawa J, Shastri N (2006) Hsp90alpha chaperones large C-terminally extended proteolytic intermediates in the MHC class I antigen processing pathway. Immunity 24(5):523–534
Lev A, Takeda K et al (2008) The exception that reinforces the rule: crosspriming by cytosolic peptides that escape degradation. Immunity 28(6):787–798
Li M, Davey GM et al (2001) Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J Immunol 166(10):6099–6103
Li MO, Sarkisian MR et al (2003) Phosphatidylserine receptor is required for clearance of apoptotic cells. Science 302(5650):1560–1563
Li Y, Wang L-X et al (2009) Cross-presentation of tumor associated antigens through tumor-derived autophagosomes. Autophagy 5(4):576–577
Li Y, Wang LX et al (2011) Tumor-derived autophagosome vaccine: mechanism of cross-presentation and therapeutic efficacy. Clin Cancer Res 17(22):7047–7057
Lieberman J (2003) The ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol 3(5):361–370
Liu K, Iyoda T et al (2002) Immune tolerance after delivery of dying cells to dendritic cells in situ. J Exp Med 196(8):1091–1097
Luthi AU, Cullen SP et al (2009) Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31(1):84–98
Matzinger P (1994) Tolerance, danger, and the extended family. Annu Rev Immunol 12:991–1045
Michaud M, Martins I et al (2011) Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334(6062):1573–1577
Muller S, Ronfani L et al (2004) Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med 255(3):332–343
Neufeld TP, Baehrecke EH (2008) Eating on the fly: function and regulation of autophagy during cell growth, survival and death in drosophila. Autophagy 4(5):557–562
Norbury CC, Basta S et al (2004) CD8+ T cell cross-priming via transfer of proteasome substrates. Science 304(5675):1318–1321
Obeid M, Tesniere A et al (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13(1):54–61
Oerlemans MI, Liu J et al (2012) Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol 107(4):270
Panaretakis T, Kepp O et al (2009) Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 28(5):578–590
Paz P, Brouwenstijn N et al (1999) Discrete proteolytic intermediates in the MHC class I antigen processing pathway and MHC I-dependent peptide trimming in the ER. Immunity 11(2):241–251
Pellegatti P, Raffaghello L et al (2008) Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One 3(7):e2599
Princiotta MF, Finzi D et al (2003) Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 18(3):343–354
Rapaport E, Fontaine J (1989) Anticancer activities of adenine nucleotides in mice are mediated through expansion of erythrocyte ATP pools. Proc Natl Acad Sci USA 86(5):1662–1666
Reits E, Griekspoor A et al (2003) Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity 18(1):97–108
Rock KL, York IA et al (2004) Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat Immunol 5(7):670–677
Rovere-Querini P, Capobianco A et al (2004) HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep 5(8):825–830
Samuels MA (2007) The brain-heart connection. Circulation 116(1):77–84
Sancho D, Joffre OP et al (2009) Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458(7240):899–903
Saric T, Beninga J et al (2001) Major histocompatibility complex class I-presented antigenic peptides are degraded in cytosolic extracts primarily by thimet oligopeptidase. J Biol Chem 276(39):36474–36481
Saric T, Graef CI et al (2004) Pathway for degradation of peptides generated by proteasomes: a key role for thimet oligopeptidase and other metallopeptidases. J Biol Chem 279(45):46723–46732
Saveanu L, Carroll O et al (2005) Concerted peptide trimming by human ERAP1 and ERAP2 aminopeptidase complexes in the endoplasmic reticulum. Nat Immunol 6(7):689–697
Savill J, Dransfield I et al (2002) A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2(12):965–975
Scaffidi P, Misteli T et al (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418(6894):191–195
Scheffer SR, Nave H et al (2003) Apoptotic, but not necrotic, tumor cell vaccines induce a potent immune response in vivo. Int J Cancer 103(2):205–211
Seong SY, Matzinger P (2004) Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 4(6):469–478
Serwold T, Gaw S et al (2001) ER aminopeptidases generate a unique pool of peptides for MHC class I molecules. Nat Immunol 2(7):644–651
Shen L, Rock KL (2004) Cellular protein is the source of cross-priming antigen in vivo. Proc Natl Acad Sci USA 101(9):3035–3040
Shi Y, Evans JE et al (2003) Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425(6957):516–521
Sigman JA, Patwa TH et al (2005) Flexibility in substrate recognition by thimet oligopeptidase as revealed by denaturation studies. Biochem J 388(Pt 1):255–261
Silva CL, Portaro FC et al (1999) Thimet oligopeptidase (EC 3.4.24.15), a novel protein on the route of MHC class I antigen presentation. Biochem Biophys Res Commun 255(3):591–595
Somersan S, Larsson M et al (2001) Primary tumor tissue lysates are enriched in heat shock proteins and induce the maturation of human dendritic cells. J Immunol 167(9):4844–4852
Sparvero LJ, Asafu-Adjei D et al (2009) RAGE (receptor for advanced glycation endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 7:17
Srivastava P (2002) Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol 2(3):185–194
Stoltze L, Schirle M et al (2000) Two new proteases in the MHC class I processing pathway. Nat Immunol 1(5):413–418
Thorburn J, Horita H et al (2009) Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ 16(1):175–183
Uhl M, Kepp O et al (2009) Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8 + T cells. Cell Death Differ 16(7):991–1005
Vanden Berghe T, Kalai M et al (2006) Necrosis is associated with IL-6 production but apoptosis is not. Cell Signal 18(3):328–335
Vanden Berghe T, Linkermann A et al (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15(2):135–147
Venereau E, Casalgrandi M et al (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med 209(9):1519–1528
Wang H, Bloom O et al (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285(5425):248–251
Weber CK, Adler G (2003) Acute pancreatitis. Curr Opin Gastroenterol 19(5):447–450
Wolfers J, Lozier A et al (2001) Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med 7(3):297–303
Woo M, Hakem R et al (2000) Executionary pathway for apoptosis: lessons from mutant mice. Cell Res 10(4):267–278
Yang H, Lundback P et al (2012) Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med 18:250–259
Yewdell JW (2011) DRiPs solidify: progress in understanding endogenous MHC class I antigen processing. Trends Immunol 32(11):548–558
Yewdell JW, Anton LC et al (1996) Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J Immunol 157(5):1823–1826
Yoon TJ, Kim JY et al (2008) Anti-tumor immunostimulatory effect of heat-killed tumor cells. Exp Mol Med 40(1):130–144
York IA, Bhutani N et al (2006) Tripeptidyl peptidase II is the major peptidase needed to trim long antigenic precursors, but is not required for most MHC class I antigen presentation. J Immunol 177(3):1434–1443
York IA, Chang SC et al (2002) The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol 3(12):1177–1184
York IA, Mo AX et al (2003) The cytosolic endopeptidase, thimet oligopeptidase, destroys antigenic peptides and limits the extent of MHC class I antigen presentation. Immunity 18(3):429–440
Zhang Q, Raoof M et al (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464(7285):104–107
Acknowledgments
This work was supported by the Intramural Research Program of the NCI, NIH.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gamrekelashvili, J., Greten, T.F. & Korangy, F. Immunogenicity of necrotic cell death. Cell. Mol. Life Sci. 72, 273–283 (2015). https://doi.org/10.1007/s00018-014-1741-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1741-x