
Abstract
microRNAs (miRNAs) are important regulators of gene expression. After excised from primary miRNA transcript by dicer-like1 (DCL1, an RNAse III enzyme), miRNAs bind and guide their effector protein named argonaute 1 (AGO1) to silence the expression of target RNAs containing their complementary sequences in plants. miRNA levels and activities are tightly controlled to ensure their functions in various biological processes such as development, metabolism and responses to abiotic and biotic stresses. Studies have identified many factors that involve in miRNA accumulation and activities. Characterization of these factors in turn greatly improves our understanding of the processes related to miRNAs. Here, we review recent progress of mechanisms underlying miRNA expression and functions in plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
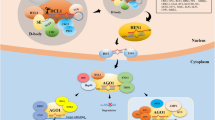
microRNAs (miRNAs) are ~20–25 nucleotide (nt) endogenous small RNA molecules, which repress gene expression at post-transcriptional levels [1–4]. miRNAs are released as a duplex from their primary transcripts (pri-miRNAs) that contain stem-loop structures by RNase III enzymes [1–4]. In the miRNA duplex, miRNA (guide strand) associates with argonaute (AGO) proteins to inhibit gene expression through cleavage and/or translational inhibition of target RNAs, while miRNA* (passenger strand) is often degraded [1–4]. Since plant miRNAs were first reported in 2002 [5–7], hundreds of miRNAs have been identified with deep-sequencing and genetic approaches [8]. They regulate many developmental processes including root initiation, leaf development, vascular development, flower development, phase transition and seed development [9–13]. Additionally, miRNAs are involved in diverse responses to stresses such as drought, salt, cold, oxidative, nutrient deficiency and biotic stresses [14–16]. The framework of miRNA biogenesis and function has been established in Arabidopsis thaliana (Arabidopsis), a flowering plant (Fig. 1) [1–4]. In Arabidopsis, dicer-like1 (DCL1; an RNAse III enzyme) excises the miRNA/miRNA* duplex from pri-miRNAs in nucleus [5, 6]. Then, the small RNA methyltransferase hua enhancer1 (HEN1) adds a methyl group to the 3′ end of the miRNA/miRNA* duplex to stabilize them [17]. Most miRNAs exit the nucleus and enter the cytoplasm with the assistance of hasty (HST) [18], a homolog of exportin 5. In Arabidopsis and rice, the major effector of miRNAs is AGO1, which has the endonuclease activity and is able to suppress gene expression through both target cleavage and translational inhibition [19–21]. Recent studies show that normal plant growth and physiology require tight control of miRNA levels and activities. In turn, the mechanisms controlling miRNA biogenesis, degradation and activity, have become an intense focus of research. This review aims to summarize rapid progress made in the regulation of miRNA accumulation and activity.

The framework of miRNA biogenesis and function. The transcription of pri-miRNAs is regulated by many transcription factors. Then, many protein factors are recruited to pri-miRNAs to form the processor complex of miRNAs through protein–protein and protein-RNA interactions. CDC5 and NOT2 do not interact with HYL1. Thus, whether CDC5 and NOT2 are in the D-body is unknown. After generation in nucleus, miRNA/miRNA* is methylated by HEN1 and exported into cytoplasm. miRNAs are loaded into AGO1 to direct target RNA cleavage or translational inhibition. It is not clear where the AGO1-miRNA assembly and miRNA methylation happens. Evidences suggest that translational inhibition by miRNA may occur at specific site of endoplasmic reticulum
Regulation of miRNA biogenesis
To date, many transcription factors and accessory factors involved in miRNA biogenesis have been identified. Studies on these components reveal that the abundance of miRNA is controlled through transcription, stability and pri-miRNA processing.
Transcriptional regulation of genes encoding miRNAs (MIR)
miRNAs are coded by endogenous genes (MIR) and many of them are conserved among different plant species [5, 22, 23]. To date, thousands of MIRs have been identified (http://www.mirbase.org/). MIRs are often located at intergenic regions and transcribed similarly as protein-coding genes [22, 24, 25]. Some MIRs are not independent transcription units. Instead, they are embedded in either intronic or exonic sequences of their host genes [26]. In addition, a few miRNAs are produced from transposable elements (TEs) in Arabidopsis and rice [27].
Most plant MIRs are transcribed by the DNA dependent RNA polymerase II (Pol II) to generate pri-miRNAs [24, 28]. Following transcription, a 5′ 7-methylguanosine cap and a 3′ polyadenylated tail are added to stabilize pri-miRNAs [24, 29, 30]. In the mutants deficient in cyclin-dependent kinase F; 1 (CDKF; 1) that regulates phosphorylation of the C-terminal domain of Pol II, pri-miRNAs lose their CAP structure and are reduced in abundance, indicating that the CAP structure stabilizes pri-miRNAs [31]. Protein factors also contribute to pri-miRNA stabilization. Dawdle (DDL), a forkhead-associated domain (FHA)-containing protein, is required for the accumulation of pri-miRNAs [32]. However, ddl does not affect the transcription of MIRs. The fact that DDL binds pri-miRNAs suggests that it might be a key regulator of pri-miRNA stability (Fig. 1) [32].
Like protein-coding genes, MIR promoters contain the TATA box and at least 21 cis-regulatory motifs, suggesting that MIR expression may subject to transcriptional regulation [24, 33, 34]. Indeed, this is demonstrated by the identification and characterization of some MIR transcription factors (Fig. 1). Lack of mediator (a multi-subunit complex), which is a conserved general transcriptional co-activator, reduces the occupancy of Pol II at MIR promoters and MIR promoter activities, resulting in decreased levels of pri-miRNAs and miRNAs [28]. These results suggest that mediator regulates MIR transcription through facilitating the recruitment of Pol II to MIR promoters [28]. Two homolog proteins, Not2a and Not2b, which contain a conserved NOT2_3_5 domain, are also involved in regulating MIR transcription in Arabidopsis [35]. NOT2 is a core member of the evolutionarily conserved carbon catabolite repression4 (CCR4)-NOT complex, which affects mRNA levels at both transcriptional and post-transcriptional levels [36]. NOT2b interacts with the Pol II C-terminal domain and is required for efficient MIR transcription [35]. NOT2a and NOT2b also influence the transcript levels of protein-coding genes, raising the possibility that NOT2 acts as a general transcription factor [35]. The cell division cycle 5 (CDC5) protein is a conserved DNA-binding protein in animals and plants [37]. CDC5 associates with both Pol II and MIR promoters. Consequently, lack of CDC5 impairs MIR promoter activity and the occupancy of Pol II at MIR promoters [38]. Thus, CDC5 acts as a positive transcription factor of MIRs [38]. However, whether CDC5 is able to regulate the transcription of protein-coding genes needs further investigation.
The transcription factors that regulate the transcription of individual miRNAs have also been characterized. Powerdress (PWR), a SANT-domain-containing protein with putative transcription factor and chromatin remodeling activities, promotes the recruitment of Pol II to the promoters of some MIR172 family members, while it shows no obvious effect on other MIRs [39]. Apetala2 (AP2), a transcription factor involved in seed development, stem cell maintenance, and floral organ identity, associates with the MIR156 and MIR172 loci. It acts oppositely in the transcription of MIR156 and MIR172, as lack of AP2 represses MIR156, but promotes MIR172 expression [40]. Further study shows that AP2 recruits two transcriptional repressor Leunig (LEG) and Seuss (SU) to the MIR172 loci to repress their expression [41]. The transcription factor FUSCA3 binds the MIR156A and MIR156C promoters and is required for the accumulation of pri-miR156a and pri-miR156c, suggesting that FUSCA3 is a positive transcription factor of MIR156A and MIR156C [42]. MIR expression can also be regulated by various stresses via specific transcription factors [43–46]. For example, copper deficiency induces the expression of MIR398B and MIR398C via the transcription factor squamosa promoter binding protein-like7 [47], while the expression of MYB2 (a transcription factor), which binds the MIR399F promoter, is induced to activate MIR399F transcription under phosphate starvation [48]. In addition, some MIRs show specific spatio-temporal expression pattern [43, 49, 50]. MIR165/166 expression is activated in the root endodermis by the transcription factor scarecrow, which is critical for the determination of the root xylem cell types [51].
Regulation of DCL1 activity
After transcription, pri-miRNAs are processed to precursor miRNAs (pre-miRNAs), which contain a stem-loop structure with 2-nt 3′ overhangs at the end of stem, and then to miRNA/miRNA* with 2-nt 3′ overhang and a 5′ phosphate at each strand by dicer-liker1 (DCL1, an RNAse III enzyme) in nucleus [52, 53]. Besides DCL1, its homolog DCL4 has also been shown to generate miRNAs from some pri-miRNAs [54]. In rice, the production of some 24-nt miRNAs requires the coordinative action of DCL1 and DCL3 [55]. This result suggests the potential divergence of miRNA biogenesis in different plant species.
Plant pri-miRNA hairpins are heterogeneous in length and structure with variable positioning of the miRNA/miRNA* duplex. The structures of pri-miRNAs play essential roles in regulating DCL1 activity [53, 56–60]. An imperfectly paired lower stem of ~15 bp below the miRNA/miRNA* duplex is crucial for the initial loop-distal cleavage of pri-miRNAs whereas the loop is crucial for efficient processing [53, 57, 58]. The loop of some pri-miRNAs such as pri-miR159a and pri-miR319a can be cleaved first and then miRNAs will be released from the stem with additional cuts. This maybe caused by their unusual long upper stem structures [59, 60]. A recent study shows that some pri-miRNAs can be bidirectionally processed by DCL1 due to their structure heterogeneity caused by multibranched terminal loops [56]. Although the base-to-loop processing results in the efficient production of miRNAs, the loop-to-base cleavage suppresses the generation of miRNAs from pri-miRNAs with multibranched terminal loops [56]. All these results suggest that the secondary structures of pri-miRNAs are crucial for miRNA maturation.
The efficient and precise pri-miRNA processing also needs assistance from many protein factors (Fig. 1). The zinc finger protein serrate (SE), the dsRNA-binding protein hyponastic leaves1 (HYL1) and the G-patch domain protein tough (TGH) interact with DCL1 and are required for miRNA accumulation [61–70]. In vitro biochemical assay shows that HYL1 and SE can enhance the accuracy and efficiency of pri-miRNA processing [71]. Consistent with this result, miscleaved products of pri-miRNAs are detected in hyl1 and mutations in the helicase and RNase III domains of DCL1, which are responsible for cleavage site selection and catalytic activity of DCL1, respectively, rescue the defects by hyl1 [72]. HYL1 is a double-stranded (ds) RNA-binding protein. Its N-terminal contains two RNA-binding domains, while its C-terminal harbors six repeats of 28 amino acids (aa) [73–75]. The two RNA-binding domains are sufficient for HYL1 function in miRNA biogenesis [74, 75]. Crystal structure analyses reveal that HYL1 probably binds the miRNA/miRNA* duplex region as a dimer to enable accurate pri-miRNA processing [74]. SE binds single-stranded RNAs (ssRNAs) through its N-terminal Domain [76–78]. The zinc finger domain of SE interacts with DCL1 and is required for the optimal DCL1 activity [77]. Crystal structure analyses show that the SE core forms a ‘walking man-like’ structure, in which the N-terminal alpha helices, the C-terminal non-canonical zinc finger domain and the novel middle domain resemble the leading leg, the lagging leg and the body, respectively [76]. This scaffold-like structure together with its RNA-binding capability suggests that SE may position a miRNA precursor toward the DCL1 catalytic site within the miRNA processing machinery [76].
The TGH is also an ssRNA-binding protein [67, 79]. TGH associates with both pre-miRNAs and pri-miRNAs in vivo [67], suggesting that it may bind the loop or bulge region of pre-miRNAs. Besides DCL1, TGH interacts with HYL1 and SE, demonstrating that it may be a component of the DCL1 complex [67]. Loss-of-function mutations in TGH reduce the DCL1 activity as well as the association of pri-miRNAs with the HYL1 complex, revealing that TGH can promote the cleavage efficiency and/or the recruitment of pri-miRNAs to DCL1 [67]. Another protein factor that improves the DCL1 activity is CDC5 [38]. CDC5 may modulate pri-miRNA processing through its interaction with DCL1, since it interacts with the helicase and dsRNA-binding domains of DCL1, which regulate the DCL1 activity [38]. Consistent with this notion, upon interaction with other proteins, the human dicer can change its conformation to obtain optimized activity [80]. CDC5 is a core component of the evolutionarily conserved MOS4-associated complex (MAC), which is required for proper plant development and immunity to bacterial infection. Besides CDC5, MAC contains MOS4, PRL1, MAC3 and MAC4 [37]. Whether other components of MAC act in the miRNA pathway needs further investigation [37]. It shall be noted that all these accessory factors described above may have other functions. For instance, SE has been shown to regulate alternative splicing [81]. Since all these proteins are either RNA or DNA-binding proteins, the identification of their substrates will help understand the crosstalk between miRNA pathway and other biological processes.
The effects of the accessory factors of DCL1 on individual miRNAs are variable [82]. Perhaps these protein factors may have specific spatial–temporal expression pattern, and, therefore, have more impacts on some miRNAs than others. For instance, CDC5 is highly expressed in the proliferating cells and may have greater influences on the miRNAs expressed in these cells [83]. Alternatively, they may need some protein partners for their optimal function. SICKLE (SIC), which is a proline-rich protein required for plant development and adaptation to abiotic stresses, colocalizes with HYL1 and is required for the accumulation of a subset of miRNAs [84], suggesting that it may act as a partner of HYL1 to regulate the processing of some pri-miRNAs [84]. Receptor for activated C kinase 1 (RACK1), which is a conserved protein and functions as a bridge or inhibitor of protein–protein interactions in all higher eukaryotes, is able to directly interact with SE [85]. Lack of RACK1 reduces the accumulation of miRNAs and the processing precision of some pri-miRNAs, suggesting that RACK1 maybe partnered with SE to regulate the DCL1 activity [85].
Regulation of DCL1 and HYL1 localization
HYL1, TGH, SE and DCL1 are localized in the specific subnuclear loci named Dicer-body (D-body) (Fig. 1). The D-bodies may be the site for pri-miRNA processing or storage since they associate with pri-miRNAs [67–70]. CDC5 and NOT2 also localize in the specific subnuclear loci containing DCL1 [35, 38]. Whether CDC5 and NOT2 are components of D-bodies is not known since they do not associate with HYL1 [35, 38]. Studies have revealed the correct localization of D-body may be critical for miRNA biogenesis. Two proteins, NOT2 and modifier of SNC2 (MOS2), have been shown to be required for the formation of the correct D-body pattern in Arabidopsis [35, 86]. MOS2 is an RNA-binding protein and interacts with pri-miRNAs in vivo. MOS2 does not interact with DCL1, HYL1, or SE [86]. In mos2, the levels of miRNAs are reduced, the localization of HYL1 in D-bodies is impaired and the recruitment of pri-miRNA to HYL1 is compromised, suggesting that MOS2 may facilitate the D-body formation and the recruitment of pri-miRNAs to the D-bodies [86]. Besides the association with Pol II, NOT2s directly interact with DCL1, which is conserved between rice and Arabidopsis [35]. Impairment of NOT2s results in increased numbers of DCL1-containing loci without altering the localization of HYL1, suggesting that it may have a role in D-body assembly [35].
Regulation of the levels of DCL1 and HYL1
The transcription of DCL1, HYL1 and SE are regulated to control miRNA processing. Several transcription factors have been shown to regulate their proper expression. Stabilized1 (STA1), an Arabidopsis pre-mRNA processing factor, is shown to promote the expression of DCL1 [87]. Histone acetyltransferase GCN5 shows a general repressive effect on miRNA production via inhibiting the transcription of HYL1 and SE [88]. In addition, the transcript levels of DCL1 can be post-transcriptionally regulated. miR162, a product of DCL1, is able to direct the cleavage of the DCL1 mRNA [89], whereas processing of pri-miR838, which resides in the DCL1 transcripts, results in a pre-mRNA that fails to produce the DCL1 protein [54, 89]. Additionally, the short interspersed elements (SINEs) transcribed from transposable elements mimic the structure of pri-miRNAs and are shown to sequester HYL1 from pri-miRNA processing [90].
Regulation of DCL1 activity by protein phosphorylation and dephosphorylation
In addition to protein factors and pri-miRNA structures, the phosphorylation of HYL1 and DCL1 also affects pri-miRNA processing. The forkhead-associated domain (FHA) of DDL is a conserved protein motif that interacts with phosphothreonine-containing proteins in prokaryotes and eukaryotes [32]. DDL interacts with the predicted phosphothreonine-containing helicase and RNAse III domains of DCL1 [91]. Mutations in the phosphothreonine-binding cleft of DDL abolish the DDL-DCL1 interaction, suggesting that DDL may use a canonical phosphothreonine recognition mechanism to interact with DCL1 [91]. Indeed, DCL1 is phosphorylated in vivo [92]. The fact that lack of DDL impairs miRNA maturation indicates that the interaction of DDL with phosphorylated DCL1 may play important roles in pri-miRNA processing [32]. C-terminal domain phosphatase-like 1 (CPL1) is a protein phosphatase and can dephosphorylate a serine motif in the C-terminal heptad repeat domain (CTD) of RNA polymerase II [93]. CPL1 has been shown to maintain the hypophosphorylated state of HYL1, which is phosphorylated and requires dephosphorylation for its optimal activity [93]. In the absence of CPL1, the dephosphorylation of HYL1 is impaired, leading to inaccurate and less efficient pri-miRNA processing [93]. Furthermore, SE physically interacts with CPL1. Lack of SE disrupts the CPL1-HYL1 interaction and dephosphorylation of HYL1, suggesting that SE functions as a scaffold to mediate CPL1 interaction with HYL1 [93].
The effect of splicing on pri-miRNA processing
Like protein-coding genes, pri-miRNAs often contain introns [5, 22, 23]. Splicing or alternative splicing of introns may have crucial roles in regulating miRNA maturation since they can alter the stem-loop structures of pri-miRNAs [4, 94, 95]. An example is MIR400, which resides in At1g32583, a host protein-coding gene. The heat stress-induced alternative splicing keeps the stem-loop of pri-miR400 in the host gene, which prevents pri-miR400 from processing and reduces miR400 accumulation [96]. A possible explanation for this observation is that pri-miR400 residing in the host mRNA may adapt an inhibitory structure to the access of DCL1 [96]. In addition, the processing of pri-miR162a and pri-miR842-miR846 is reduced in Arabidopsis when alternative splicing changes their stem-loop structures [97, 98]. In contrast, the excision of intron from the stem-loop is required for the production of natural antisense miRNAs in rice [97].
In addition to altering pri-miRNA structure, splicing itself or the recruitment of splicing machinery may enhance pri-miRNA processing. For instance, the processing efficiency of pri-mi163 and pri-miR161 can be improved by the splicing of the 3′ introns following their stem-loops [99, 100]. Furthermore, some proteins involved in mRNA splicing have been shown to play essential roles in miRNA maturation. The cap-binding protein 20 (CBP20) and 80 are the components of the cap-binding complex, which binds the cap structure of mRNAs and ensure the correct splicing of the first intron [101]. cbp80 or cbp20 reduces the abundance of miRNAs and increases the accumulation of both unspliced and spliced pri-miRNAs, demonstrating that CBP80 and CBP20 function in pri-miRNA processing independent of their role in splicing [102–104]. SE interacts with CBP20, indicating that CBP20/80 may be a part of the processing complex [35, 105]. Whether CBP80/CBP20 affects the accuracy of pri-miRNA processing is unknown. Besides CBP80/20, CDC5, another component involved in miRNA biogenesis, has also been shown to promote splicing of some mRNAs. It is not clear if CDC5 affects pri-miRNA splicing.
MiRNA stability and degradation
Two recent modeling analyses suggest that mutual degradation of miRNAs and targets may sharp their expression boundary [106, 107]. Although the predications are not be experimentally verified, they underline the importance of miRNA degradation. In fact, studies have revealed multiple mechanisms governing miRNA stability and degradation in plants.
Methylation and uridylation of miRNAs
To ensure proper levels of miRNA, plants evolve multiple mechanisms to regulate miRNA stability and degradation. Plant miRNAs are stabilized by a 2′-O-methylation modification at the 3′ terminal ribose, which is added by HUA1 enhancer1 (HEN1), an Mg2+-dependent methyltransferase (MTase) [17, 108]. miRNAs in hen1 contain 1–8 untemplated uredines (uridylation) at 3′ end and/or are truncated from 3′ end, demonstrating that methylation protects miRNAs from uridylation and degradation [17, 109–111]. HEN1 functions as a monomer to recognize ~22 nt dsRNAs with the 2 nt overhang at each end [108, 112]. Since HEN1 acts on the miRNA/miRNA* duplexes, methylation likely occurs before AGO1 loading. However, it is unclear whether methylation occurs in cytoplasm or nucleus, as HEN1 localizes at both compartments [110].
Uridylation is a critical regulatory mechanism destabilizing miRNAs in plants. In Arabidopsis, HEN1 SUPPRESSOR1 (HESO1), a terminal uridyl transferase, is responsible for the uridylation of the majority of miRNAs [113, 114]. heso1 increases overall abundance of miRNAs whereas the overexpression of HESO1 reduces miRNA accumulation in hen1, demonstrating that uridylation triggers degradation of miRNAs in higher plants [113]. In addition, heso1 increases the abundance of 3′ truncated miRNAs in hen1, suggesting that uridylation may trigger miRNA degradation through a mechanism other than 3′-to-5′ truncation [113, 114]. HESO1 interacts with AGO1 and uridylates AGO1-bound miRNAs in vitro [115]. Furthermore, uridylation of miRNAs is impaired in hen1 when AGO1 is mutated [111, 115]. These results demonstrate that miRNA uridylation occurs at the AGO1 complex and the necessity for methylation, which prevents miRNAs from AGO1-associated HESO1 activity and, therefore, ensures the function of the AGO1-miRNA complex (Fig. 2). In the alga Chlamydomonas reinhardtii (C. reinhardtii), MUT68, a terminal nucleotidyl transferase, adds U-tails to the 3′ terminus of miRNAs [116]. Impairment of MUT68 results in increased abundance of miRNAs. Furthermore, uridylation stimulates in vitro degradation of miRNAs by RRP6, which is the peripheral exosome subunit and degrades RNAs from 3′-to-5′ [116]. These results demonstrate that uridylation may triggers degradation of miRNAs through different mechanisms in various plant species.

Model of uridylation-triggered miRNA degradation. Under normal condition, miRNAs are methylated by HEN1, which protects miRNAs from uridylation at the AGO1 complex. Methylated miRNAs maybe subject to SDN degradation, resulting in 3′ truncated miRNAs. HESO1 may uridylate unmethylated miRNAs and 3′ truncated miRNAs to trigger their degradation. Unmethylated miRNA can also be degraded through unknown 3′-to-5′ trimming activity
Degradation of miRNAs by exoribonucleases in plants
The degradation of miRNAs is crucial to maintain the balance of miRNA levels and function. Enzymes responsible for miRNA turnover have been identified in Arabidopsis. In Arabidopsis, a family of 3′-to-5′ exoribonucleases including small RNA degrading nuclease 1, 2 and 3 (SDN1, SDN2 and SDN3) are involved in mature miRNA turnover [117]. Inactivation of SDN proteins results in increased miRNA abundance and impaired plant development [117]. An in vitro nuclease activity assay shows that SDN1 prefers to degrade short single-stranded RNAs, but not small RNA duplexes or pre-miRNAs [117]. Additionally, SDN1 can degrade 2′-O-methylation miRNAs, but not 3′ uridylated miRNAs, raising the possibility that SDN1 and HESO1 cooperate to regulate the degradation of 2′-O-methylation miRNAs in wild-type Arabidopsis (Fig. 2) [117]. Recently, the expression of a short tandem target mimic (STTM), which contains two short sequences mimicking miRNA target sites, which are resistant to miRNA-mediated cleavage, triggers the degradation of targeted miRNAs by SDNs [118]. In the green Alga, inactivation of exosome components, RRP6, leads to increased accumulation of miRNAs, demonstrating that miRNAs can be degraded from 3′-to-5′ by exosome [116]. In Drosophila, Nibbler, a 3′-to-5′ exoribonuclease, interacts with AGO1 to trim AGO1-bound miRNAs from 3′ end [119, 120]. It is possible that Nibbler homologs of plants also function in miRNA trimming.
Regulation of miRNA activity
AGO proteins
miRNAs mainly function through their effector protein AGO, which cleaves target RNA and/or inhibit translation. AGO contains four major functional domains: the N-terminal domain, the PAZ domain, the middle (MID) domain and the PIWI domain [121, 122]. Among them, the PAZ domain binds to the 3′ end of miRNAs, whereas MID domain interacts with the 5′ phosphate of miRNA [121, 122]. The PIWI domain adapts a structure similar to that of RNAse H and acts as a slicer to cleave target at a position opposite to the 10th and 11th nucleotides of miRNAs [123]. However, not all AGOs have the endonuclease activity since some key amino acids in the catalytic center are mutated in some AGOs in both plants and animals [124, 125]. Arabidopsis encodes 10 AGOs [121]. Each of them seems to bind a subset of small RNAs and has different functions. This is partially determined by the 5′ nucleotide of small RNAs [126, 127]. For instance, AGO1 has a preference on miRNAs with 5′ U [126, 127]. In addition, AGOs display specific spatial–temporal expression patterns, which may also contribute to their functional divergence [128]. In Arabidopsis, AGO1 is the major effector protein for miRNAs [21] while AGO7 and AGO10 specifically bind miR390 and miR165/166, respectively [127].
The levels and activity of AGO1 are regulated to ensure its proper function. AGO1 is a target of miR168 [129, 130]. Thus, AGO1 itself is subject to feedback regulation. Overexpression of an F-box protein FBW2 reduces AGO1 protein levels but not transcripts [131]. In contrast, fbw2 increases AGO1 protein levels, demonstrating that FBW2 is a negative regulator of AGO1 [131]. AGO1 can also be degraded through the autophagy pathway when the AGO1-miRNA assembly is disrupted [132]. Sequestering miRNAs from AGO1 can also regulate their functions. For example, AGO10 binds miR165/miR166 in shoot apical meristem (SAM) to prevent the formation of the AGO1-miR165/166 complex, which limits the function of miR165/miR166 and ensure the proper development of SAM [133].
AGO1 loading
The loading of miRNA into AGO1 is partially determined by structures of miRNA/miRNA* duplex, protein factors and 5′ nucleotides [126, 127, 134]. Analyses of an artificial miRNA/miRNA* duplex reveal that the strand with a lower 5′-end thermostability is preferentially loaded into AGO1 as the miRNA strand in Nicotiana benthamiana, suggesting that like in animals, the thermostability at the 5′ end of duplex strands plays important roles in miRNA loading [134]. HYL1 and CPL1 have been shown to facilitate the miRNA strand selection as their mutations increase the levels of miRNA* relative to Col, which is presumably caused by AGO1-binding [93, 134]. Loss-of-function mutations in squint (SQN), a cyclophilin 40 (CYP40) protein, reduce miRNA activity without altering their levels, indicating that SQN may be a protein partner of AGO1 [135]. In fact, heat shock protein 90 (HSP90) and (CyP40) form a complex with AGO1 to assist the miRNA loading in a cell-free system [136–138]. In addition, upon ATP-hydrolysis by HSP90, HSP90-CYP40 and miRNA* are removed from the AGO1-miRNA complex, which maybe due to the conformation alteration of AGO1 caused by HSP90 chaperone activity [136, 137]. Most miRNA*s are degraded upon disassociation from AGO1, which does not depend on the slicer activity of AGO1 [134, 139]. However, some miRNA*s can be loaded into other AGOs, and, therefore, be stabilized and become functional.
miRNA-mediated target cleavage
Plant miRNAs need high complementarity to recognize their substrate [19, 129, 140, 141]. A recent study shows that besides complementarity, the context of miRNA binding site and expression levels may also contribute to the target recognition in Arabidopsis [142]. In plants, target cleavage is considered as a predominant pathway for miRNA-mediated repression of gene expression [1], as AGO1 with mutations in the catalytic motif fails to complement ago1 [143]. Target cleavage by AGO proteins generates a 5′ RNA fragment (5′ fragment) with a 3′ hydroxyl group and a 3′ RNA fragment (3′ fragment) with a 5′ phosphate [144]. AGO1 slicing can trigger the decay of the target mRNAs by exonucleases without the requirement for 3′ deadenylation or 5′ decapping. In the mutants of XRN4 (A cytoplasmic 5′–3′ exoribonuclease) and FIERY1 (A putative regulator of XRN4), the levels of 3′ fragments are increased, revealing that 3′ fragments are degraded by XRN4 in plants [145]. In c. reinhardtii, the 3′ end of 5′ fragments is adenylated by the terminal nucleotidyl transferase MUT68, which triggers exosome-mediated 3′-to-5′ degradation of 5′ fragments [146]. In animals and higher plants, 5′ fragments are uridylated at 3′ end [146]. In Arabidopsis, HESO1 is a major enzyme uridylating 5′ fragments [115]. Lack of HESO1 increases the abundance of 5′ fragments, demonstrating that uridylation induces the degradation of 5′ fragments [115]. However, the proportion of 3′ truncated 5′ fragments is increased in heso1 relative to wild-type plants [115]. This result suggests that uridylated 5′ fragments may be degraded through a mechanism other than 3′-to-5′ trimming. Indeed, lack of exosome components RRP6L and CSL4 does not alter the abundance of 5′ fragments in Arabidopsis [115]. 5′ fragments are also subjected to 5′-to-3′ degradation, as xrn4 increases the accumulation of 5′ fragments [115]. However, it is not clear whether the 5′ trimming of 5′ fragments happens before or after uridylation.
miRNA-mediated target cleavage can be regulated by the competing endogenous RNAs. In Arabidopsis, a non-coding RNA named IPS1 contains a non-cleavable sequence with complementarity to miR399. IPS1 can be bound by AGO1-399 but cannot be cleaved, and, therefore, sequester AGO1-399, resulting in the accumulation of target PHO2 mRNA [147]. Bioinformatic analyses have identified the presence of endogenous mimic targets of miRNAs. It is worth to test if these mimic target transcripts can regulate the activity of the corresponding miRNAs.
miRNA-mediated translational inhibition
Several studies suggest that translational inhibition is also a common mechanism employed by plant miRNAs to repress gene expression [148–150]. In plants, a subset of miRNAs and a fraction of AGO1 are associated with polyribosomes, agreeing with a role of miRNA in translational inhibition [151, 152]. Researches have put insight into the mechanisms governing miRNA-mediated translational inhibition. A recent study shows that in plants, AGO1-miRNA can sterically inhibit the recruitment or movement of ribosomes after binding the 5′ untranslated region (UTR) or the open reading frame of target RNAs [153]. This result indicates that miRNAs can inhibit translation initiation or elongation of target RNAs in plants. In plants, a correlation between miRNA-mediated translational inhibition and the processing bodies (P-body) has been suggested. P-bodies are distinct cytoplasmic loci consisting of many enzymes involved in mRNA degradation [154]. In plants, a portion of AGO1 localizes in the P-bodies and the P-body components varicose (VCS) and SUO (a GW-repeat containing protein) are required for miRNA-mediated translational inhibition, suggesting that P-body may be a site for the storage of translational-repressed target mRNAs [149, 155]. In animal P-body, target transcripts of miRNAs can be destabilized through deadenylation and decapping, which is a slicer-independent mechanism [156, 157]. However, the causal relationship between translational repression and decay of target RNAs has not been established in plants. Mutations in VCS and decapping1 (DCP1; a P-body component required for mRNA decapping) reduce the levels of some target transcripts, indicating the presence of slicer-independent degradation of targets [158]. However, the upregulation of target transcripts in vcp and dcp1 may attribute to the decreased miRNA levels [158]. Consistent with this, VCS can regulate protein levels of some targets without affecting transcription [149].
Some miRNAs appear to repress gene expression through both translational inhibition and target cleavage. This raises a question of how translation inhibition vs. target cleavage is determined. A recent study shows that miRNAs display stronger translational inhibition in a transient in assay when their binding sites localize at the 5′ coding region [159], while another study reveals that miRNAs enhance translational repression in male germ cells of plants [160]. These results suggest that multiple factors help cells to select translational repression or target cleavage. In some cell types, the concurrence of translational inhibition and cleavage happens. A possible explanation is the selective interaction of AGO1 with protein factors. Alternatively, AGO1-miRNA-targets may be sorted into designated subcellular compartments specific for translational inhibition or target cleavage (Fig. 1). A considerable amount of AGO1 is associated with the endoplasmic reticulum (ER) [150]. In addition, lack of altered meristem program1 (AMP1), an integral ER membrane protein, impairs miRNA-mediated translational repression, but not target transcript cleavage, suggesting that ER may be a site for translational inhibition to occur [150]. Agreeing with this notion, amp1 reduces the exclusion efficiency of target mRNAs from membrane-bound polysomes [150].
Additional proteins involved in translational inhibition by miRNAs include the microtubule-severing enzyme katanin (KTN), 3-hydroxy-3-methylglutaryl CoA reductase (HMG1) and the sterol C-8 isomerase hydra1 (HYD1). KTN1 is required for the formation of the proper cortical microtubule array [161]. Disruption of KTN1 blocks miRNA-mediated translational inhibition, suggesting a role of microtubule in translational repression. This is consistent with the role of microtubules in ER organization and P-body dynamics [149]. HMG1 is essential for the biosynthesis of isoprenoids, which are substrates of multiple metabolic pathways such as membrane sterols and several plant hormones, while HYD1 is required for the synthesis of sterols [149, 162]. HMG1 and HYD1 positively affect both transcript and protein levels of target genes. The fact that the association of miRNAs with AGO1 is not impaired in hmg1 indicates that HGM1 acts downstream of miRNA biogenesis and loading [149, 162]. Given the function of HMG1 and HYD1 in sterol biogenesis, sterol may have a role in miRNA activity [149, 162]. As sterol is required for the correct localization of some integral membrane proteins, it is possible that specific membrane compartments involved in miRNA function may be impaired in hmg1 and hyd1 [149, 162].
miRNA-mediated DNA methylation
In additional to post-transcriptional repression, miRNA can inhibit gene expression at the transcriptional levels through directing DNA methylation. In rice, some 24-nt miRNAs can be sorted into AGO4, (an effector protein) and direct DNA methylation at the MIR and target loci in rice [55]. The production of these AGO4-associated miRNAs depends on DCL3 [55]. In contrast, AGO1-assoicated 24-nt miRNAs requires DCL1 for biogenesis. Based on these facts, it is proposed that the sorting of 24-nt miRNAs to AGOs is signaled by their biogenesis machinery [55]. It has been shown that AGO4 associates with a class of 24-nt small interfering RNAs (siRNAs) to trigger cytosine methylation through a process called RNA-directed DNA methylation [3]. It is likely that miRNAs trigger DNA methylation through a mechanism similar to that of siRNAs.
Perspective
To date, characterizations of various factors such as protein components and structure of pri-miRNAs and miRNAs have greatly improved our understanding of mechanisms related to miRNA biogenesis and function. However, challenges remain in plant miRNA pathway. miRNA expression is regulated through a combination of transcription, processing and turnover. A great challenge is to understand how plants integrate all these regulating mechanisms to control the levels of individual miRNAs in response to development and various stresses. Although factors involved in miRNA biogenesis have been identified, their functional mechanisms are still not clear. Many protein factors functioning in the miRNA pathway are involved in transcription, splicing, RNA decay and other processes, suggesting an interconnection between miRNA pathway and other biological processes. Further understanding of their relationship will improve our knowledge of regulatory networks of various biological processes. Translational inhibition has become a common but poorly understood mechanism used by plant miRNAs to repress gene expression. Elucidation of mechanisms governing miRNA-mediated translation inhibition needs to characterize RNA structures and additional protein factors involved in the process. Another interesting question is how cells select translation inhibition or target cleavage as the functional model for miRNAs. Finally, a practical challenge is how we optimize miRNA-based technology and use knowledge of miRNA to improve important agricultural trait.
References
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Baulcombe D (2004) RNA silencing in plants. Nature 431:356–363
Bologna NG, Voinnet O (2014) The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu Rev Plant Biol 65:473–503
Rogers K, Chen X (2013) Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 25:2383–2399
Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP (2002) MicroRNAs in plants. Genes Dev 16:1616–1626
Park W, Li J, Song R, Messing J, Chen X (2002) Carpel factory, a dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol 12:1484–1495
Llave C, Kasschau KD, Rector MA, Carrington JC (2002) Endogenous and silencing-associated small RNAs in plants. Plant Cell 14:1605–1619
Meyers BC, Souret FF, Lu C, Green PJ (2006) Sweating the small stuff: microRNA discovery in plants. Curr Opin Biotechnol 17:139–146
Chen X (2009) Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol 25:21–44
Martin RC, Liu PP, Goloviznina NA, Nonogaki H (2010) microRNA, seeds, and Darwin?: diverse function of miRNA in seed biology and plant responses to stress. J Exp Bot 61:2229–2234
Poethig RS (2009) Small RNAs and developmental timing in plants. Curr Opin Genet Dev 19:374–378
Moreno-Risueno MA, Van Norman JM, Benfey PN (2012) Transcriptional switches direct plant organ formation and patterning. Curr Top Dev Biol 98:229–257
Chuck G, Candela H, Hake S (2009) Big impacts by small RNAs in plant development. Curr Opin Plant Biol 12:81–86
Seo JK, Wu J, Lii Y, Li Y, Jin H (2013) Contribution of small RNA pathway components in plant immunity. Mol Plant-Microbe Interact : MPMI 26:617–625
Sunkar R, Li YF, Jagadeeswaran G (2012) Functions of microRNAs in plant stress responses. Trends Plant Sci 17:196–203
Khraiwesh B, Zhu JK, Zhu J (2012) Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta 1819:137–148
Yu B, Yang Z, Li J, Minakhina S, Yang M, Padgett RW, Steward R, Chen X (2005) Methylation as a crucial step in plant microRNA biogenesis. Science 307:932–935
Park MY, Wu G, Gonzalez-Sulser A, Vaucheret H, Poethig RS (2005) Nuclear processing and export of microRNAs in Arabidopsis. Proc Natl Acad Sci USA 102:3691–3696
Baumberger N, Baulcombe DC (2005) Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc Natl Acad Sci USA 102:11928–11933
Mourrain P, Beclin C, Elmayan T, Feuerbach F, Godon C, Morel JB, Jouette D, Lacombe AM, Nikic S, Picault N, Remoue K, Sanial M, Vo TA, Vaucheret H (2000) Arabidopsis SGS2 and SGS3 genes are required for posttranscriptional gene silencing and natural virus resistance. Cell 101:533–542
Wu L, Zhang Q, Zhou H, Ni F, Wu X, Qi Y (2009) Rice MicroRNA effector complexes and targets. Plant Cell 21:3421–3435
Nozawa M, Miura S, Nei M (2012) Origins and evolution of microRNA genes in plant species. Genome Biol Evol 4:230–239
Coruh C, Shahid S, Axtell MJ (2014) Seeing the forest for the trees: annotating small RNA producing genes in plants. Curr Opin Plant Biol 18C:87–95
Xie ZX, Allen E, Fahlgren N, Calamar A, Givan SA, Carrington JC (2005) Expression of Arabidopsis MIRNA genes. Plant Physiol 138:2145–2154
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36:D154–D158
Yang GD, Yan K, Wu BJ, Wang YH, Gao YX, Zheng CC (2012) Genomewide analysis of intronic microRNAs in rice and Arabidopsis. J Genet 91:313–324
Piriyapongsa J, Jordan IK (2008) Dual coding of siRNAs and miRNAs by plant transposable elements. RNA 14:814–821
Kim YJ, Zheng B, Yu Y, Won SY, Mo B, Chen X (2011) The role of Mediator in small and long noncoding RNA production in Arabidopsis thaliana. EMBO J 30:814–822
Jones-Rhoades MW, Bartel DP (2004) Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 14:787–799
Zhang BH, Pan XP, Wang QL, Cobb GP, Anderson TA (2005) Identification and characterization of new plant microRNAs using EST analysis. Cell Res 15:336–360
Hajheidari M, Farrona S, Huettel B, Koncz Z, Koncz C (2012) CDKF;1 and CDKD protein kinases regulate phosphorylation of serine residues in the C-terminal domain of Arabidopsis RNA polymerase II. Plant Cell 24:1626–1642
Yu B, Bi L, Zheng B, Ji L, Chevalier D, Agarwal M, Ramachandran V, Li W, Lagrange T, Walker JC, Chen X (2008) The FHA domain proteins DAWDLE in Arabidopsis and SNIP1 in humans act in small RNA biogenesis. Proc Natl Acad Sci USA 105:10073–10078
Megraw M, Baev V, Rusinov V, Jensen ST, Kalantidis K, Hatzigeorgiou AG (2006) MicroRNA promoter element discovery in Arabidopsis. RNA 12:1612–1619
Zhao X, Zhang H, Li L (2013) Identification and analysis of the proximal promoters of microRNA genes in Arabidopsis. Genomics 101:187–194
Wang L, Song X, Gu L, Li X, Cao S, Chu C, Cui X, Chen X, Cao X (2013) NOT2 proteins promote polymerase II-dependent transcription and interact with multiple MicroRNA biogenesis factors in Arabidopsis. Plant Cell 25:715–727
Collart MA, Panasenko OO (2012) The Ccr4–not complex. Gene 492:42–53
Palma K, Zhao Q, Cheng YT, Bi D, Monaghan J, Cheng W, Zhang Y, Li X (2007) Regulation of plant innate immunity by three proteins in a complex conserved across the plant and animal kingdoms. Genes Dev 21:1484–1493
Zhang S, Xie M, Ren G, Yu B (2013) CDC5, a DNA binding protein, positively regulates posttranscriptional processing and/or transcription of primary microRNA transcripts. Proc Natl Acad Sci USA 110:17588–17593
Yumul RE, Kim YJ, Liu X, Wang R, Ding J, Xiao L, Chen X (2013) POWERDRESS and diversified expression of the MIR172 gene family bolster the floral stem cell network. PLoS Genet 9:e1003218
Yant L, Mathieu J, Dinh TT, Ott F, Lanz C, Wollmann H, Chen X, Schmid M (2010) Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. Plant Cell 22:2156–2170
Grigorova B, Mara C, Hollender C, Sijacic P, Chen X, Liu Z (2011) LEUNIG and SEUSS co-repressors regulate miR172 expression in Arabidopsis flowers. Development 138:2451–2456
Wang F, Perry SE (2013) Identification of direct targets of FUSCA3, a key regulator of Arabidopsis seed development. Plant Physiol 161:1251–1264
Jeong DH, Park S, Zhai J, Gurazada SG, De Paoli E, Meyers BC, Green PJ (2011) Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell 23:4185–4207
Liu HH, Tian X, Li YJ, Wu CA, Zheng CC (2008) Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14:836–843
Zhao B, Liang R, Ge L, Li W, Xiao H, Lin H, Ruan K, Jin Y (2007) Identification of drought-induced microRNAs in rice. Biochemical and biophysical research communications 354:585–590
Liang G, He H, Yu D (2012) Identification of nitrogen starvation-responsive microRNAs in Arabidopsis thaliana. PLoS ONE 7:e48951
Yamasaki H, Hayashi M, Fukazawa M, Kobayashi Y, Shikanai T (2009) SQUAMOSA Promoter Binding Protein-Like7 Is a central regulator for copper homeostasis in Arabidopsis. Plant Cell 21:347–361
Baek D, Kim MC, Chun HJ, Kang S, Park HC, Shin G, Park J, Shen M, Hong H, Kim WY, Kim DH, Lee SY, Bressan RA, Bohnert HJ, Yun DJ (2013) Regulation of miR399f transcription by AtMYB2 affects phosphate starvation responses in Arabidopsis. Plant Physiol 161:362–373
Valoczi A, Varallyay E, Kauppinen S, Burgyan J, Havelda Z (2006) Spatio-temporal accumulation of microRNAs is highly coordinated in developing plant tissues. Plant J 47:140–151
Breakfield NW, Corcoran DL, Petricka JJ, Shen J, Sae-Seaw J, Rubio-Somoza I, Weigel D, Ohler U, Benfey PN (2012) High-resolution experimental and computational profiling of tissue-specific known and novel miRNAs in Arabidopsis. Genome Res 22:163–176
Carlsbecker A, Lee JY, Roberts CJ, Dettmer J, Lehesranta S, Zhou J, Lindgren O, Moreno-Risueno MA, Vaten A, Thitamadee S, Campilho A, Sebastian J, Bowman JL, Helariutta Y, Benfey PN (2010) Cell signalling by microRNA165/6 directs gene dose-dependent root cell fate. Nature 465:316–321
Margis R, Fusaro AF, Smith NA, Curtin SJ, Watson JM, Finnegan EJ, Waterhouse PM (2006) The evolution and diversification of Dicers in plants. FEBS Lett 580:2442–2450
Mateos JL, Bologna NG, Chorostecki U, Palatnik JF (2010) Identification of microRNA processing determinants by random mutagenesis of Arabidopsis MIR172a precursor. Curr Biol 20:49–54
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP (2006) A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 20:3407–3425
Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y (2010) DNA methylation mediated by a microRNA pathway. Mol Cell 38:465–475
Zhu H, Zhou Y, Castillo-Gonzalez C, Lu A, Ge C, Zhao YT, Duan L, Li Z, Axtell MJ, Wang XJ, Zhang X (2013) Bidirectional processing of pri-miRNAs with branched terminal loops by Arabidopsis Dicer-like1. Nat Struct Mol Biol 20:1106–1115
Song L, Axtell MJ, Fedoroff NV (2010) RNA secondary structural determinants of miRNA precursor processing in Arabidopsis. Curr Biol 20:37–41
Werner S, Wollmann H, Schneeberger K, Weigel D (2010) Structure determinants for accurate processing of miR172a in Arabidopsis thaliana. Curr Biol 20:42–48
Bologna NG, Mateos JL, Bresso EG, Palatnik JF (2009) A loop-to-base processing mechanism underlies the biogenesis of plant microRNAs miR319 and miR159. The EMBO journal 28:3646–3656
Cuperus JT, Montgomery TA, Fahlgren N, Burke RT, Townsend T, Sullivan CM, Carrington JC (2010) Identification of MIR390a precursor processing-defective mutants in Arabidopsis by direct genome sequencing. Proc Natl Acad Sci USA 107:466–471
Grigg SP, Canales C, Hay A, Tsiantis M (2005) SERRATE coordinates shoot meristem function and leaf axial patterning in Arabidopsis. Nature 437:1022–1026
Yang L, Liu ZQ, Lu F, Dong AW, Huang H (2006) SERRATE is a novel nuclear regulator in primary microRNA processing in Arabidopsis. Plant J 47:841–850
Lobbes D, Rallapalli G, Schmidt DD, Martin C, Clarke J (2006) SERRATE: a new player on the plant microRNA scene. EMBO Rep 7:1052–1058
Han MH, Goud S, Song L, Fedoroff N (2004) The Arabidopsis double-stranded RNA-binding protein HYL1 plays a role in microRNA-mediated gene regulation. Proc Natl Acad Sci USA 101:1093–1098
Vazquez F, Gasciolli V, Crete P, Vaucheret H (2004) The nuclear dsRNA binding protein HYL1 is required for microRNA accumulation and plant development, but not posttranscriptional transgene silencing. Curr Biol 14:346–351
Kurihara Y, Takashi Y, Watanabe Y (2006) The interaction between DCL1 and HYL1 is important for efficient and precise processing of pri-miRNA in plant microRNA biogenesis. RNA 12:206–212
Ren G, Xie M, Dou Y, Zhang S, Zhang C, Yu B (2012) Regulation of miRNA abundance by RNA binding protein TOUGH in Arabidopsis. Proc Natl Acad Sci USA 109:12817–12821
Fang Y, Spector DL (2007) Identification of nuclear dicing bodies containing proteins for microRNA biogenesis in living Arabidopsis plants. Curr Biol 17:818–823
Song L, Han MH, Lesicka J, Fedoroff N (2007) Arabidopsis primary microRNA processing proteins HYL1 and DCL1 define a nuclear body distinct from the Cajal body. Proc Natl Acad Sci USA 104:5437–5442
Fujioka Y, Utsumi M, Ohba Y, Watanabe Y (2007) Location of a possible miRNA processing site in SmD3/SmB nuclear bodies in Arabidopsis. Plant Cell Physiol 48:1243–1253
Dong Z, Han MH, Fedoroff N (2008) The RNA-binding proteins HYL1 and SE promote accurate in vitro processing of pri-miRNA by DCL1. Proc Natl Acad Sci USA 105:9970–9975
Liu C, Axtell MJ, Fedoroff NV (2012) The helicase and RNaseIIIa domains of Arabidopsis Dicer-Like1 modulate catalytic parameters during microRNA biogenesis. Plant Physiol 159:748–758
Lu C, Fedoroff N (2000) A mutation in the Arabidopsis HYL1 gene encoding a dsRNA binding protein affects responses to abscisic acid, auxin, and cytokinin. Plant Cell 12:2351–2366
Yang SW, Chen HY, Yang J, Machida S, Chua NH, Yuan YA (2010) Structure of Arabidopsis HYPONASTIC LEAVES1 and its molecular implications for miRNA processing. Structure 18:594–605
Wu F, Yu L, Cao W, Mao Y, Liu Z, He Y (2007) The N-terminal double-stranded RNA binding domains of Arabidopsis HYPONASTIC LEAVES1 are sufficient for pre-microRNA processing. Plant Cell 19:914–925
Machida S, Chen HY, Adam Yuan Y (2011) Molecular insights into miRNA processing by Arabidopsis thaliana SERRATE. Nucleic Acids Res 39:7828–7836
Iwata Y, Takahashi M, Fedoroff NV, Hamdan SM (2013) Dissecting the interactions of SERRATE with RNA and DICER-LIKE 1 in Arabidopsis microRNA precursor processing. Nucleic Acids Res 41:9129–9140
Ren G, Yu B (2012) Critical roles of RNA-binding proteins in miRNA biogenesis in Arabidopsis. RNA Biol 9:1424–1428
Ren G, Yu B (2012) Post-transcriptional control of miRNA abundance in Arabidopsis. Plant Signal Behav 7:1443–1446
Lau PW, Guiley KZ, De N, Potter CS, Carragher B, MacRae IJ (2012) The molecular architecture of human Dicer. Nat Struct Mol Biol 19:436–440
Raczynska KD, Stepien A, Kierzkowski D, Kalak M, Bajczyk M, McNicol J, Simpson CG, Szweykowska-Kulinska Z, Brown JW, Jarmolowski A (2014) The SERRATE protein is involved in alternative splicing in Arabidopsis thaliana. Nucleic Acids Res 42:1224–1244
Laubinger S, Zeller G, Henz SR, Buechel S, Sachsenberg T, Wang JW, Ratsch G, Weigel D (2010) Global effects of the small RNA biogenesis machinery on the Arabidopsis thaliana transcriptome. Proc Natl Acad Sci USA 107:17466–17473
Lin Z, Yin K, Zhu D, Chen Z, Gu H, Qu LJ (2007) AtCDC5 regulates the G2 to M transition of the cell cycle and is critical for the function of Arabidopsis shoot apical meristem. Cell Res 17:815–828
Zhan X, Wang B, Li H, Liu R, Kalia RK, Zhu JK, Chinnusamy V (2012) Arabidopsis proline-rich protein important for development and abiotic stress tolerance is involved in microRNA biogenesis. Proc Natl Acad Sci USA 109:18198–18203
Speth C, Willing EM, Rausch S, Schneeberger K, Laubinger S (2013) RACK1 scaffold proteins influence miRNA abundance in Arabidopsis. Plant J 76:433–445
Wu X, Shi Y, Li J, Xu L, Fang Y, Li X, Qi Y (2013) A role for the RNA-binding protein MOS2 in microRNA maturation in Arabidopsis. Cell Res 23:645–657
Ben Chaabane S, Liu R, Chinnusamy V, Kwon Y, Park JH, Kim SY, Zhu JK, Yang SW, Lee BH (2013) STA1, an Arabidopsis pre-mRNA processing factor 6 homolog, is a new player involved in miRNA biogenesis. Nucleic Acids Res 41:1984–1997
Kim W, Benhamed M, Servet C, Latrasse D, Zhang W, Delarue M, Zhou DX (2009) Histone acetyltransferase GCN5 interferes with the miRNA pathway in Arabidopsis. Cell Res 19:899–909
Xie Z, Kasschau KD, Carrington JC (2003) Negative feedback regulation of Dicer-Like1 in Arabidopsis by microRNA-guided mRNA degradation. Curr Biol 13:784–789
Pouch-Pelissier MN, Pelissier T, Elmayan T, Vaucheret H, Boko D, Jantsch MF, Deragon JM (2008) SINE RNA induces severe developmental defects in Arabidopsis thaliana and interacts with HYL1 (DRB1), a key member of the DCL1 complex. PLoS Genet 4:e1000096
Machida S, Yuan YA (2013) Crystal structure of Arabidopsis thaliana Dawdle forkhead-associated domain reveals a conserved phospho-threonine recognition cleft for dicer-like 1 binding. Mol Plant 6:1290–1300
Engelsberger WR, Schulze WX (2012) Nitrate and ammonium lead to distinct global dynamic phosphorylation patterns when resupplied to nitrogen-starved Arabidopsis seedlings. Plant J 69:978–995
Manavella PA, Hagmann J, Ott F, Laubinger S, Franz M, Macek B, Weigel D (2012) Fast-forward genetics identifies plant CPL phosphatases as regulators of miRNA processing factor HYL1. Cell 151:859–870
Kruszka K, Pacak A, Swida-Barteczka A, Stefaniak AK, Kaja E, Sierocka I, Karlowski W, Jarmolowski A, Szweykowska-Kulinska Z (2013) Developmentally regulated expression and complex processing of barley pri-microRNAs. BMC Genom 14:34
Szarzynska B, Sobkowiak L, Pant BD, Balazadeh S, Scheible WR, Mueller-Roeber B, Jarmolowski A, Szweykowska-Kulinska Z (2009) Gene structures and processing of Arabidopsis thaliana HYL1-dependent pri-miRNAs. Nucleic Acids Res 37:3083–3093
Yan K, Liu P, Wu CA, Yang GD, Xu R, Guo QH, Huang JG, Zheng CC (2012) Stress-induced alternative splicing provides a mechanism for the regulation of microRNA processing in Arabidopsis thaliana. Mol Cell 48:521–531
Jia F, Rock CD (2013) MIR846 and MIR842 comprise a cistronic MIRNA pair that is regulated by abscisic acid by alternative splicing in roots of Arabidopsis. Plant Mol Biol 81:447–460
Hirsch J, Lefort V, Vankersschaver M, Boualem A, Lucas A, Thermes C, d’Aubenton-Carafa Y, Crespi M (2006) Characterization of 43 non-protein-coding mRNA genes in Arabidopsis, including the MIR162a-derived transcripts. Plant Physiol 140:1192–1204
Bielewicz D, Kalak M, Kalyna M, Windels D, Barta A, Vazquez F, Szweykowska-Kulinska Z, Jarmolowski A (2013) Introns of plant pri-miRNAs enhance miRNA biogenesis. EMBO Rep 14:622–628
Schwab R, Speth C, Laubinger S, Voinnet O (2013) Enhanced microRNA accumulation through stemloop-adjacent introns. EMBO Rep 14:615–621
Raczynska KD, Simpson CG, Ciesiolka A, Szewc L, Lewandowska D, McNicol J, Szweykowska-Kulinska Z, Brown JW, Jarmolowski A (2010) Involvement of the nuclear cap-binding protein complex in alternative splicing in Arabidopsis thaliana. Nucleic Acids Res 38:265–278
Gregory BD, O’Malley RC, Lister R, Urich MA, Tonti-Filippini J, Chen H, Millar AH, Ecker JR (2008) A link between RNA metabolism and silencing affecting Arabidopsis development. Dev Cell 14:854–866
Laubinger S, Sachsenberg T, Zeller G, Busch W, Lohmann JU, Ratsch G, Weigel D (2008) Dual roles of the nuclear cap-binding complex and SERRATE in pre-mRNA splicing and microRNA processing in Arabidopsis thaliana. Proc Natl Acad Sci USA 105:8795–8800
Kim S, Yang JY, Xu J, Jang IC, Prigge MJ, Chua NH (2008) Two cap-binding proteins CBP20 and CBP80 are involved in processing primary MicroRNAs. Plant Cell Physiol 49:1634–1644
Gruber JJ, Zatechka DS, Sabin LR, Yong J, Lum JJ, Kong M, Zong WX, Zhang Z, Lau CK, Rawlings J, Cherry S, Ihle JN, Dreyfuss G, Thompson CB (2009) Ars2 links the nuclear cap-binding complex to RNA interference and cell proliferation. Cell 138:328–339
Levine E, McHale P, Levine H (2007) Small regulatory RNAs may sharpen spatial expression patterns. PLoS Comput Biol 3:e233
Muraro D, Mellor N, Pound MP, Help H, Lucas M, Chopard J, Byrne HM, Godin C, Hodgman TC, King JR, Pridmore TP, Helariutta Y, Bennett MJ, Bishopp A (2014) Integration of hormonal signaling networks and mobile microRNAs is required for vascular patterning in Arabidopsis roots. Proc Natl Acad Sci USA 111:857–862
Yang ZY, Ebright YW, Yu B, Chen XM (2006) HEN1 recognizes 21-24 nt small RNA duplexes and deposits a methyl group onto the 2′ OH of the 3′ terminal nucleotide. Nucleic Acids Res 34:667–675
Boutet S, Vazquez F, Liu J, Beclin C, Fagard M, Gratias A, Morel JB, Crete P, Chen X, Vaucheret H (2003) Arabidopsis HEN1: a genetic link between endogenous miRNA controlling development and siRNA controlling transgene silencing and virus resistance. Curr Biol 13:843–848
Li J, Yang Z, Yu B, Liu J, Chen X (2005) Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr Biol 15:1501–1507
Zhai J, Zhao Y, Simon SA, Huang S, Petsch K, Arikit S, Pillay M, Ji L, Xie M, Cao X, Yu B, Timmermans M, Yang B, Chen X, Meyers BC (2013) Plant microRNAs display differential 3′ truncation and tailing modifications that are ARGONAUTE1 dependent and conserved across species. Plant Cell 25:2417–2428
Huang Y, Ji L, Huang Q, Vassylyev DG, Chen X, Ma JB (2009) Structural insights into mechanisms of the small RNA methyltransferase HEN1. Nature 461:823–827
Ren G, Chen X, Yu B (2012) Uridylation of miRNAs by hen1 suppressor1 in Arabidopsis. Curr Biol 22:695–700
Zhao Y, Yu Y, Zhai J, Ramachandran V, Dinh TT, Meyers BC, Mo B, Chen X (2012) The Arabidopsis nucleotidyl transferase HESO1 uridylates unmethylated small RNAs to trigger their degradation. Curr Biol 22:689–694
Ren G, Xie M, Zhang S, Vinovskis C, Chen X, Yu B (2014) Methylation protects microRNAs from an AGO1-associated activity that uridylates 5′ RNA fragments generated by AGO1 cleavage. Proc Natl Acad Sci USA 111:6365–6370
Ibrahim F, Rymarquis LA, Kim EJ, Becker J, Balassa E, Green PJ, Cerutti H (2010) Uridylation of mature miRNAs and siRNAs by the MUT68 nucleotidyltransferase promotes their degradation in Chlamydomonas. Proc Natl Acad Sci USA 107:3906–3911
Ramachandran V, Chen X (2008) Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science 321:1490–1492
Yan J, Gu Y, Jia X, Kang W, Pan S, Tang X, Chen X, Tang G (2012) Effective small RNA destruction by the expression of a short tandem target mimic in Arabidopsis. Plant Cell 24:415–427
Han BW, Hung JH, Weng Z, Zamore PD, Ameres SL (2011) The 3′-to-5′ exoribonuclease Nibbler shapes the 3′ ends of microRNAs bound to Drosophila Argonaute1. Curr Biol 21:1878–1887
Liu N, Abe M, Sabin LR, Hendriks GJ, Naqvi AS, Yu Z, Cherry S, Bonini NM (2011) The exoribonuclease Nibbler controls 3′ end processing of microRNAs in Drosophila. Curr Biol 21:1888–1893
Vaucheret H (2008) Plant argonautes. Trends Plant Sci 13:350–358
Tolia NH, Joshua-Tor L (2007) Slicer and the argonautes. Nat Chem Biol 3:36–43
Rivas FV, Tolia NH, Song JJ, Aragon JP, Liu J, Hannon GJ, Joshua-Tor L (2005) Purified Argonaute2 and an siRNA form recombinant human RISC. Nat Struct Mol Biol 12:340–349
Hutvagner G, Simard MJ (2008) Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol 9:22–32
Liu X, Lu T, Dou Y, Yu B, Zhang C (2014) Identification of RNA silencing components in soybean and sorghum. BMC Bioinformatics 15:4
Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H, Long C, Chen S, Hannon GJ, Qi Y (2008) Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133:116–127
Montgomery TA, Howell MD, Cuperus JT, Li D, Hansen JE, Alexander AL, Chapman EJ, Fahlgren N, Allen E, Carrington JC (2008) Specificity of argonaute7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 133:128–141
Mallory A, Vaucheret H (2010) Form, function, and regulation of argonaute proteins. Plant Cell 22:3879–3889
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP (2002) Prediction of plant microRNA targets. Cell 110:513–520
Vaucheret H, Vazquez F, Crete P, Bartel DP (2004) The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev 18:1187–1197
Earley K, Smith M, Weber R, Gregory B, Poethig R (2010) An endogenous F-box protein regulates argonaute1 in Arabidopsis thaliana. Silence 1:15
Derrien B, Baumberger N, Schepetilnikov M, Viotti C, De Cillia J, Ziegler-Graff V, Isono E, Schumacher K, Genschik P (2012) Degradation of the antiviral component argonaute1 by the autophagy pathway. Proc Natl Acad Sci USA 109:15942–15946
Zhu H, Hu F, Wang R, Zhou X, Sze SH, Liou LW, Barefoot A, Dickman M, Zhang X (2011) Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 145:242–256
Eamens AL, Smith NA, Curtin SJ, Wang MB, Waterhouse PM (2009) The Arabidopsis thaliana double-stranded RNA binding protein DRB1 directs guide strand selection from microRNA duplexes. RNA 15:2219–2235
Smith MR, Willmann MR, Wu G, Berardini TZ, Moller B, Weijers D, Poethig RS (2009) Cyclophilin 40 is required for microRNA activity in Arabidopsis. Proc Natl Acad Sci USA 106:5424–5429
Iki T, Yoshikawa M, Meshi T, Ishikawa M (2012) Cyclophilin 40 facilitates HSP90-mediated RISC assembly in plants. EMBO J 31:267–278
Iki T, Yoshikawa M, Nishikiori M, Jaudal MC, Matsumoto-Yokoyama E, Mitsuhara I, Meshi T, Ishikawa M (2010) In vitro assembly of plant RNA-induced silencing complexes facilitated by molecular chaperone HSP90. Mol Cell 39:282–291
Earley KW, Poethig RS (2011) Binding of the cyclophilin 40 ortholog SQUINT to Hsp90 protein is required for SQUINT function in Arabidopsis. J Biol Chem 286:38184–38189
Kwak PB, Tomari Y (2012) The N domain of Argonaute drives duplex unwinding during RISC assembly. Nat Struct Mol Biol 19:145–151
Mallory AC, Reinhart BJ, Jones-Rhoades MW, Tang G, Zamore PD, Barton MK, Bartel DP (2004) MicroRNA control of PHABULOSA in leaf development: importance of pairing to the microRNA 5′ region. EMBO J 23:3356–3364
Liu Q, Wang F, Axtell MJ (2014) Analysis of complementarity requirements for plant MicroRNA targeting using a Nicotiana benthamiana quantitative transient assay. Plant Cell 26:741–753
Li J, Reichel M, Millar AA (2014) Determinants beyond both complementarity and cleavage govern microR159 efficacy in Arabidopsis. PLoS Genet 10:e1004232
Carbonell A, Fahlgren N, Garcia-Ruiz H, Gilbert KB, Montgomery TA, Nguyen T, Cuperus JT, Carrington JC (2012) Functional analysis of three Arabidopsis argonautes using slicer-defective mutants. Plant Cell 24:3613–3629
Llave C, Xie Z, Kasschau KD, Carrington JC (2002) Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 297:2053–2056
Souret FF, Kastenmayer JP, Green PJ (2004) AtXRN4 degrades mRNA in Arabidopsis and its substrates include selected miRNA targets. Mol Cell 15:173–183
Ibrahim F, Rohr J, Jeong WJ, Hesson J, Cerutti H (2006) Untemplated oligoadenylation promotes degradation of RISC-cleaved transcripts. Science 314:1893
Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, Garcia JA, Paz-Ares J (2007) Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet 39:1033–1037
Schwab R, Palatnik JF, Riester M, Schommer C, Schmid M, Weigel D (2005) Specific effects of microRNAs on the plant transcriptome. Dev Cell 8:517–527
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320:1185–1190
Li S, Liu L, Zhuang X, Yu Y, Liu X, Cui X, Ji L, Pan Z, Cao X, Mo B, Zhang F, Raikhel N, Jiang L, Chen X (2013) MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis. Cell 153:562–574
Lanet E, Delannoy E, Sormani R, Floris M, Brodersen P, Crete P, Voinnet O, Robaglia C (2009) Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant Cell 21:1762–1768
Reynoso MA, Blanco FA, Bailey-Serres J, Crespi M, Zanetti ME (2012) Selective recruitment of mRNAs and miRNAs to polyribosomes in response to rhizobia infection in Medicago truncatula. Plant J 73:289–301
Iwakawa HO, Tomari Y (2013) Molecular insights into microRNA-mediated translational repression in plants. Mol Cell 52:591–601
Liu J, Valencia-Sanchez MA, Hannon GJ, Parker R (2005) MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol 7:719–723
Yang L, Wu G, Poethig RS (2012) Mutations in the GW-repeat protein SUO reveal a developmental function for microRNA-mediated translational repression in Arabidopsis. Proc Natl Acad Sci USA 109:315–320
Behm-Ansmant I, Rehwinkel J, Doerks T, Stark A, Bork P, Izaurralde E (2006) mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev 20:1885–1898
Wu L, Fan J, Belasco JG (2006) MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA 103:4034–4039
Motomura K, Le QT, Kumakura N, Fukaya T, Takeda A, Watanabe Y (2012) The role of decapping proteins in the miRNA accumulation in Arabidopsis thaliana. RNA Biol 9:644–652
Li JF, Chung HS, Niu Y, Bush J, McCormack M, Sheen J (2013) Comprehensive protein-based artificial microRNA screens for effective gene silencing in plants. Plant Cell 25:1507–1522
Grant-Downton R, Kourmpetli S, Hafidh S, Khatab H, Le Trionnaire G, Dickinson H, Twell D (2013) Artificial microRNAs reveal cell-specific differences in small RNA activity in pollen. Curr Biol 23:R599–R601
Stoppin-Mellet V, Gaillard J, Vantard M (2006) Katanin’s severing activity favors bundling of cortical microtubules in plants. Plant J 46:1009–1017
Brodersen P, Sakvarelidze-Achard L, Schaller H, Khafif M, Schott G, Bendahmane A, Voinnet O (2012) Isoprenoid biosynthesis is required for miRNA function and affects membrane association of argonaute 1 in Arabidopsis. Proc Natl Acad Sci USA 109:1778–1783
Acknowledgments
This work was supported by a National Science Foundation Grant MCB-1121193 (to B. Y).
Author information
Authors and Affiliations
Corresponding author
Additional information
M. Xie and S. Zhang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Xie, M., Zhang, S. & Yu, B. microRNA biogenesis, degradation and activity in plants. Cell. Mol. Life Sci. 72, 87–99 (2015). https://doi.org/10.1007/s00018-014-1728-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1728-7