
Abstract
Bone formation and degradation are perfectly coordinated. In case of an imbalance of these processes diseases occur associated with exaggerated formation of new bone or bone loss as in osteoporosis. Most studies investigating osteoporosis either focus on osteoblast or osteoclast function and differentiation. Both processes have been suggested to be affected by reactive oxygen species (ROS). Besides a potentially harmful role of ROS, these small molecules are important second messengers. The family of NADPH oxidases produces ROS in a controlled and targeted manner, to specifically regulate signal transduction. This review will highlight the role of reactive oxygen species in bone cell differentiation and bone-loss associated disease with a special focus on osteoporosis and NADPH oxidases as specialized sources of ROS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bone is not a static structure but rather an organ with a relatively high turnover. Its constant formation and degradation is tightly regulated and maintains the proper function and adaptation to load of the skeleton. Three different cells contribute to this process: osteoblasts, osteoclasts and osteocytes. Formation of bone is realized by osteoblasts while osteoclasts degrade bone. An imbalance between bone formation and degradation results in overshooting bone formation as observed in osteopetrosis or loss of bone as most common in osteoporosis. The first and best characterized measure of bone quality is bone mineral density (BMD). Low bone mineral density is associated with a high total plasma oxidant status [1] and negatively correlates with plasma lipid oxidation as shown in osteoporotic postmenopausal women when compared with a healthy group [2]. A commonly used marker in the clinic to measure “oxidative stress” is plasma homocysteine. Indeed hyperhomocysteinemia is associated with a high level of reactive oxygen species (ROS) and an increased risk of osteoporosis in humans [3]. On the other hand, research of the recent years revealed that ROS are needed for differentiation and proper functionality of individual cell populations in the bone.
In biological samples, ROS usually comprise superoxide anion (O2 −) and hydrogen peroxide (H2O2) [4] and also nitric oxide (NO) could be considered as ROS. Cellular ROS sources are manifold-like small molecules (iron, flavins or thiols) or organelles like peroxisomes or mitochondria with ubiquinone and NADH dehydrogenase as enzymatic ROS producers. The ROS produced by the enzymes listed so far are considered byproducts of their virtual function. To avoid damage of the cell by those byproduct-ROS, antioxidant systems and ROS-degrading enzymes, like catalase, superoxide dismutase (SOD) and glutathione peroxidases evolved. This chemical-toxic view on ROS is contrasted by their contribution to cellular signal transduction, which is a consequence of their controlled formation by NADPH oxidases of the Nox family. These ROS generated by NADPH oxidases, specifically interfere with numerous signal transduction pathways and for example allow signaling through the transient inhibition of phosphatases [5]. Overactivation of NADPH oxidases, however, results in overt oxidative stress and thus these enzymes are not only exciting regulators of cellular function but also potential drug targets.
This review will highlight the role of ROS in bone disease with a special focus on osteoporosis and Nox-NADPH oxidases as specialized sources of ROS.
NADPH oxidases
NADPH oxidases are integral membrane proteins that enable the transport of electrons, accepted from NADPH at one side of the membrane to the other side, onto oxygen. All NADPH oxidases contain FADH and NADPH binding sites at their C-terminus. Seven members of the family of NADPH oxidases denominated according to the integral-membrane catalytically active core unit, Nox1 through Nox5, Duox-1 and Duox-2, have been described in mammals, with Nox2, the leukocyte NAPDH oxidase, being the best studied one. Nox2 is a 91 kDa glycosylated protein, which is highly expressed in neutrophils and macrophages, where it is essential for host defense.
Apart from pathogenes, several growth factors like angiotensin II, platelet derived growth factor (PDGF) or inflammatory cytokines like tumor necrosis factor alpha (TNFα) activate and induce Nox2 [6–8]. Nox2 requires the transmembrane protein p22phox for maturation and the cytosolic subunits p47phox, p67phox and Rac for activation. Kinases, such as protein kinase C (PKC), phosphorylate p47phox at multiple serine residues which facilitates the interaction of the protein with the plasma membrane and the transmembrane complex. Importantly, the translocation of p47phox to the membrane-bound subunit p22phox enables the recruitment of p67phox, which activity appears to be strictly dependent on the small GTPase Rac (for review see [9]). In addition to activating Nox-dependent ROS production, Rac has an essential role in cytoskeletal organization, making it a central molecule involved in cell migration and adhesion per se. Similar as Nox2, Nox1 and Nox3 also require p22phox and cytosolic subunits including Rac for their activity and are acutely activated through assembly of the subunits. Additionally Nox1 and 3 have been documented to form stable complexes with constitutively active substitutes of p67phox and p47phox, namely NoxA1 and NoxO1 [10, 11]. Due to their localization in the plasma membrane [12] Nox1 and 2 generate O2 − towards the extracellular space. It is still unclear to which extent the strong negatively charged O2 − passes the plasma membrane to induce intracellular effects. Probably, O2 − either spontaneously or catalyzed by extracellular superoxide dismutase (ecSOD) dismutes into H2O2, which then enters the cell through aquaporins [13] or freely passes the membrane of the same or the neighbor cell.
The second group of NADPH oxidases consists of the calcium-dependent Nox enzymes. These, namely Nox5, DUOX1 and DUOX2, are independent of Rac, but contain EF-hands that bind calcium (Ca2+) to activate the enzyme. DUOX1 and 2 are expressed predominantly in epithelial cells of the lung and in the thyroid gland. They are required for the thyroid-peroxidase-mediated oxidation of iodine which is involved in the formation of thyroxin (T4) and triiodothyronine (T3) [14, 15]. Due to gene deletion Nox5 is missing in rodents. In humans Nox5 can be found in endothelial and smooth muscle cells, macrophages, some lymphocytes and also in sperms where it contributes to the motility of the sperm tail [16]. Whether these NADPH oxidases are important for bone homeostasis is unclear and none of the members of this group has been studied in bone cells so far. Therefore, this group of NADPH oxidases is just shortly mentioned here for the sake of completeness.
The third group of NADPH oxidases only comprises Nox4, which has constitutive activity and, so far, no essential cytosolic interacting proteins or other activating mechanisms have been identified (with the exception of p22phox). Although an enhancement of Nox4 activity by Poldip2 has been described [17], most publications support the view that Nox4-dependent ROS formation is controlled by the expression level of the enzyme and not by its activity state. In particular TGFβ has been identified as a strong inducer of Nox4 expression [18]. In cells of mesenchymal origin, Nox4 is expressed at low levels in undifferentiated and at higher levels in differentiated cells [19]. Under physiological conditions Nox4 contributes rather to long-term adaptive signaling than to acute agonist-induced events (although exceptions may exist) and in particular, Nox4 has been suggested to drive the differentiating processes [20–22]. Nox4 predominately generates H2O2 through the subsequent transfer of two electrons through the membrane onto oxygen [23].
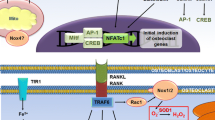
Some of the Nox enzymes exhibit a typical organ-specific expression pattern. Nox3 expression for example is mainly restricted to the inner ear, where it contributes to the formation of otolithes required for the sensing of gravity and linear acceleration [24]. Although otolithes and bones share some similarity, the restricted expression of Nox3 excludes it from interfering with bone metabolism. In contrast, Nox1, Nox2 as well as Nox4 have been found in bone cells like osteoclasts and osteoblasts as well as chondrocytes. A general overview of these three NADPH oxidases is provided in the scheme in Fig. 1.

General overview of the NADPH oxidases most relevant in bone remodeling. In bone the most relevant NADPH oxidases are Nox1, Nox2 and Nox4. Nox1 and Nox2 primarily generate superoxide anions. The activity of Nox1 and Nox2 is acutely inducible and requires an assembly with cytosolic subunits. Therefore, Nox1 and Nox2 mediate acute signal transduction which is important for proliferation. In case of an overactivation, the large amounts of O2 − formed by those NADPH oxidases contribute to inflammation. Nox4 in contrast, produces hydrogen peroxide constitutively independent from any cytosolic subunits. The level of H2O2 produced by this enzyme is relatively low and the cell adapts to Nox4-derived H2O2. Such adaptation leads to cellular quiescence or even differentiation. O2 − superoxide anion; H2O2 hydrogen peroxide
Bone homeostasis and bone turn over
Every year 10 % of the skeleton is turned over by two main players: osteoclasts remove old bone and osteoblasts build new bone. The most abundant cell in bone however, is the osteocyte which differentiates out of osteoblasts. This differentiation so far has not been attributed to be subject of redox signaling. One study suggests that the differentiation of induced pluripotent stem cells into osteocytes in vitro is enhanced in the presence of the antioxidant and sirtuin-inhibitor resveratrol [25]. As osteocytes live as long as 25 years they form a static pool and make up to 95 % of all bone cells in young but only 58 % in old individuals. This age associated loss of osteocytes appears to be related to an increase in reactive oxygen species. Interestingly, osteocyte death is dependent on the age of the bone, not on the age of the subject [26]. Most of the 19 papers on “osteocytes and reactive oxygen species” so far published deal with aging and oxidative stress rather that redox signaling in osteocytes. The source of age-related ROS in osteocytes so far is undefined. In summary, it appears that osteocyte death is a consequence of oxidative stress and that ROS may not play a role in osteocyte differentiation.
Bone turnover, bone mineral density and especially bone resorption under physiological conditions are tightly regulated by the RANK–RANKL–OPG (receptor activator of nuclear factor (NF)-κB–receptor–receptor activator of NF-κB ligand–osteoprotegerin) signaling pathway. The ligand RANKL is secreted from osteoblasts while its receptor RANK is expressed on osteoclasts. The decoy receptor OPG among others is also expressed and released by osteoblasts. OPG directly neutralizes RANKL as well as the pro-apoptotic actions of “TNFα-related apoptosis-inducing ligand” (TRAIL), not only in bone, but also in a variety of cancer cells [27]. When bound to its receptor RANK, RANKL activates the alternative NF-κB pathway (p52) which initiates the expression of inflammatory cytokines like tumor necrosis factor (TNF) α. TNFα stimulates synthesis of interleukin (IL)-1β and macrophage colony-stimulating factor (M-CSF) that promotes proliferation of macrophages. RANKL is essential to induce the differentiation from macrophages into osteoclast and to suppress osteoclast apoptosis. The resulting bone resorption by mature osteoclasts reduces bone mass. Bone resorption occurs after osteoclasts attached to the bone surface by αVβ3, the integrin vitronectin receptor. Through several Src-dependent signaling steps a ruffled border membrane is formed and lysosomal secretory vesicles fuse with this membrane part. H+ ions are pumped into the extracellular spaces formed by the ruffles and demineralize the matrix. Lysosomal proteases like cathepsin K subsequently degrade the remaining protein matrix scaffold [28].
The ratio of RANKL/OPG is a predictor for osteoporosis. Chronic increase of the parathyroid hormone (PTH) in case of chronic kidney disease is associated with bone loss. PTH not only directly stimulates osteoclasts, it also up-regulates RANKL and down-regulates OPG gene expressions thus increasing the RANKL/OPG ratio [29]. A side effect of osteolytic diseases can be vascular calcification. In fact an inverse correlation between osteoporotic bone remodeling and arterial and aortic valve calcification has been reported [30]. T-cells express RANKL and vascular smooth muscle cells (VSMCs) express RANK. Conversely, vascular dysfunction resulting in attenuated blood supply may lead to bone diseases as present in impaired fracture repair, osteoporosis or even osteonecrosis [31].
Bone formation is mediated by osteoblasts and osteoblastogenesis follows a totally different route: osteoblasts differentiate from mesenchymal stem cells under the control of bone morphogenic protein (BMP)-2 and Wnt/beta-catenin signaling [32, 33], therefore inflammatory pathways are not required for the process.
Bone formation and the function of osteoblasts
As pointed out above, RANKL produced by osteoblasts is needed for proper osteoclastogenesis. Accordingly, it seems logical to assume that osteoclastogenesis is dependent on osteoblastogenesis in vivo, as osteoblasts are a major source of RANKL. This however, is only a side-aspect of their function, as osteoblasts mainly maintain bone and facilitate bone formation.
Dysfunction of osteoblasts indirectly results in bone loss as exemplified for iron overload in post-menopausal women. With the cessation of the menstruation cycle iron load increases and serum ferritin more than doubles in menopause [34]. Ferritin and its ferroxidase activity suppress osteoblast-mediated bone formation and calcification, which is in part a consequence of an attenuated core-binding factor alpha 1-induced expression of osteoblast-specific genes, like alkaline phosphatase and osteocalcin [35]. Ferritin via its ferroxidase activity also generates hydrogen peroxide from the reaction of ferrous iron and dioxygen [36]. Through this mechanism, iron overload-induced ROS formation has been shown to result in enhanced bone resorption and bone loss in mice [37] probably due to reduced osteoblast activity. However, iron overload in vivo also increases the number of osteoclasts in the bone and iron overload enhances osteoclastogenesis in vitro. Importantly, both effects can be inhibited by the administration of the thiol-donating antioxidant N-acetyl-cysteine (NAC) [38].
ROS in osteoblasts dysfunction
A reduction in bone mass often results in low mechanic stability of the bone and thus in fractures. Copper/zinc superoxide dismutase (SOD1)-deficient mice exhibit a high level of intracellular ROS and display bone fragility resulting from low-turnover bone loss and impaired collagen cross-linking [39]. Whether or not impaired crosslinking of collagen is mediated by O2 − or H2O2 remains elusive in that specific publication, although the process can be facilitated by H2O2 [40]. Bone loss by mechanical unloading is a consequence of increased ROS formation in bone marrow and bone-forming cells. This, among others, is due to a decrease in SOD1 expression, and genetic knockout of SOD1 enhances unloading-induced bone loss. This is also exemplified by the fact that administration of vitamin C as a scavenger of ROS prevented bone loss during mechanical unloading [41]. With age the antioxidative capacity of many cells is reduced [42]. Accordingly mature osteoblasts are more sensitive to oxidative stress induced by 0.5–5 mM H2O2 as exemplified in MC3T3-E1 cells [43]. Such high concentrations however, are beyond the physiological range and rather represent severe oxidative stress that may occur upon serious infection than redox signaling.
One factor that induces osteoblast injury in vivo is TNFα. In rat primary osteoblasts TNFα suppressed cell viability, induced cellular apoptosis, suppressed Runx2 mRNA expression and inhibited alkaline phosphatase activity, which can be interpreted as sign of loss of osteoblast function. Furthermore, TNFα induced the formation of excessive amounts of nitric oxide (NO) by the inducible NO-synthase and increased ROS level by NADPH oxidase-activation and the induction of mitochondria dysfunction. Treatment of osteoblasts with molecular hydrogen (H2), which can act as reductant, reversed all effects of TNFα on osteoblasts potentially also by increasing SOD and catalase expression [44]. Choi and Lee published a series of studies on antimycin A-induced damage of the MC3T3-E1 osteoblastic cell line. Antimycin inhibits the mitochondrial electron transport chain and thereby, among other effects, increases intracellular ROS level. Antioxidants like apocynin or luteolin prevented the antimycin-induced decrease in PI3-K (phosphoinositide 3-kinase), Akt (protein kinase B) and CREB (cAMP-response element-binding protein) activity and thereby maintained the function of the osteoblast cell line [45–47].
In conclusion, it appears that deregulated ROS formation either as consequence of TNFα signaling or disruption of mitochondrial function impairs osteoblast function. This effect, however, appears not to be specific for osteoblasts and rather reflects oxidative stress and ROS-mediated cell damage.
NADPH oxidases in osteoblastogenesis
Bone morphogenic protein 2 (BMP-2) promotes differentiation of 2T3 osteoblast precursor cells into mature osteoblasts. BMP-2 elicits a rapid generation of ROS concomitant with an increased activation of NADPH oxidase. NAC and the unspecific flavin and NADPH oxidase inhibitor diphenylene iodonium (DPI) inhibit BMP-2-induced ROS production and NADPH oxidase activity respectively. This increase in ROS formation is needed for BMP-2-induced alkaline phosphatase expression and thus for maintenance of the osteoblast phenotype. It appears that, unlike osteoclasts cell lines, 2T3-cells express mainly the NADPH oxidase Nox4. NAC and DPI as well as a dominant negative Nox4 interrupt the positive feedback loop by which BMP-2 stimulates its own expression [48]. The finding that Nox4 contributes to the differentiation of mesenchymal cells is not unique for the cell line reported here as for example the differentiation of mesenchymal fibroblasts into adipocytes strongly depends on the expression of Nox4 [49]. Down-regulation of Nox4 prevented the insulin-induced differentiation of adipocytes in part by controlling the expression of the MAPKinase phosphatase MKP-1 and by maintaining progenitor markers like the preadipocyte marker pref-1.
The main action of vitamin D is to increase intestinal calcium reabsorption. The hormone, however, also affects osteoblast proliferation, differentiation and bone mineralization, but these effects vary with the timing of treatment, dosage and origin of the osteoblasts. The effects of vitamin D on differentiation and mineralization are mostly stimulatory in human and rat osteoblasts, whereas murine osteoblasts have been reported to be inhibited by vitamin D [50]. Somjen et al. found that in the human osteoblast cell line SaOS2 vitamin D treatment increased the formation of ROS within 1 h and that it also rapidly increased proliferation as measured by the production of new DNA. Both vitamin D-induced ROS formation and synthesis of new DNA were prevented by the unspecific flavoprotein inhibitor DPI given in an unreported concentration [51]. As the compound, however, has a small pharmacological range, it cannot be excluded that the observations reflect toxicity. Nevertheless, it is interesting that vitamin D acutely induces the ROS formation in an osteoblastic cell line. This finding is supported by a study from Boyan et al. [52] showing that vitamin D, depending on osteoblasts maturation state, exerts its effects via specific and distinct membrane receptors which results in protein kinase C (PKC) activation. PKC is the most important activator of the organizing subunit of the NADPH oxidase Nox2. On this basis, it appears that vitamin D-induced ROS formation in osteoblasts contributes at least to proliferation but apparently not to differentiation of these cells.
RANKL was reported to be expressed preferentially by immature osteoblasts, and expression levels decreased during osteoblast maturation in human cells [53]. As more immature osteoblasts will necessarily lead to an enhanced formation of RANKL, it is logical to assume that vitamin D decreases bone mass. However, Vitamin D is being used for decades as treatment of postmenopausal osteoporosis as it increases bone density and reduces the rate of fractures [54]. The precise mechanism of how vitamin D facilitates these effects is still discussed controversially. Probably vitamin D and its metabolites improve the calcium balance and facilitate mineral deposition in bone matrix largely without direct effects on bone cells [55].
Statins, 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, have been used as a cholesterol-lowering drug to clinically treat hyperlipidemia. In recent years, evidence for a possible anti-osteoporotic effect of statins accumulated. Mechanistically, statin intake was linked to a reduction in oxidative stress and restoration of NO formation as also demonstrated in aged and ovariectomized rats [56]. In an in vitro model MC3T3-E1 cells were treated with 100 µM H2O2. In such high concentration, H2O2 acts as an inflammation-like stress signal and therefore, induces Nox4. H2O2-mediated increase in Nox4 expression was prevented by simvastatin, which, although interpreted differently by the authors, act as indirect antioxidants and therefore prevent stress-induced gene expression [57].
The number of publications on NADPH oxidases and osteoblasts is still limited. However, it appears that Nox4 is involved in osteoblast differentiation and Nox2 plays a role in proliferation of osteoblast precursor cells but additional research, particularly in knockout mice, will be needed to settle these findings. Figure 2 provides a summary of the effects of ROS on osteoblastogenesis mentioned in this section (Fig. 2).

Effects of NADPH oxidases on osteoblasts. Osteoblasts differentiate from mesenchymal stem cells upon stimulation with BMP-2 and other growth factors. The impacts of the different ROS sources on osteoblast biology are shown in the table. Nox NADPH oxidase, BMP-2 bone morphogenic protein 2, upright arrow in green boxes means more osteoblastogenesis, upright arrow in red boxes means more osteoblast dysfunction; NOS nitric oxide synthase (low represents a moderate increase in NO mediated by eNOS and nNOS, high represents iNOS-derived extensive and pro-inflammatory level of NO), the impact of NO on osteoblasts will be discussed in the last section of this review
Bone resorption and the function of osteoclasts
Mechanisms of osteoclastogenesis
Functional osteoclasts are large multinucleated cells. Their formation does not require them to be attached to bone matrix, but involves fusion of at least three precursor cells. Transcription factors like PU.1, MITF, c-Fos, and NFATc1 control the expression of several proteins, including Atp6v0d2, OC-STAMP and CD9, that are involved in the fusion and are required for osteoclastogenesis [28]. RANKL seems to be the most effective cytokine to induce activation of the above mentioned transcription factors and thus formation of osteoclasts. Like most TNF receptor (TNFR) family members, RANK (alternative name is TRANCE-R) binds to TNF receptor-associated factors (TRAFs) as adaptor molecules. Six TRAFs (TRAF1–TRAF6) have been reported [58] and especially TRAF6 is critical for osteoclastogenesis so that TRAF6-deficient mice show severe osteopetrosis due to impaired osteoclastogenesis [59]. High TRAF6- and subsequent p38 MAP kinase-activation is probably the key mechanism that distinguishes RANK from other receptors acting on the same pathway as CD40, IL-1R and Toll-like receptor which are less effective in promoting osteoclastogenesis [60]. ROS have been first identified to play a role in TRAF signaling in Hek293 cells. Upon TNFα treatment of those cells TRAF2 is recruited to the membrane and there initiates a cascade leading to mitochondrial ROS formation [61]. The increase in ROS formation was dependent on TRAF2 recruitment to the membrane and essential for the TNFα induced NFκB-activation.
ROS in osteoclastogenesis
Upon stimulation with RANKL ROS formation rapidly increases in osteoclast precursor cells. A summary of the role of ROS in osteoclastogenesis is provided in Fig. 3. One portion of the acute ROS formation might be a consequence of iron-mediated reactions. Iron is an essential component in protein complexes of the mitochondrial respiratory chain and Steap4 is responsible for the conversion of ferric iron into ferrous iron for cellular usage. Knocking down Steap4 expression in macrophages decreases cellular ferrous iron concentration and acute RANKL-induced ROS formation is reduced by 50 % in Steap4 depleted osteoclast precursors [62]. In addition to this, low iron in osteoclast precursors was associated with a dramatic decrease in the activity of the calmodulin-dependent kinase/CREB pathway and in PGC-1β expression. These pathways, however, are rather involved in proliferation than in differentiation. The importance of mitochondria in acute RANKL-induced ROS formation is further supported by the work of Srinivasan et al. who found that treatment of RAW 264.7 macrophages with the mitochondria-specific antioxidant MitoQ prevents RANKL-induced ROS formation [63].

Effects of NADPH oxidases on osteoclastogenesis. Upon stimulation with RANKL, macrophage-derived precursor cells differentiate into osteoclasts. The impacts of the different ROS sources on osteoclast biology are shown in the table. The arrows indicate the activity of the individual ROS sources in the individual steps. Nox NADPH oxidase, Mito mitochondria, RANKL receptor activator of nuclear factor (NF)-κB–receptor ligand, NOS nitric oxide synthase, the impact of NO on osteoblasts will be discussed in the last section of this review
NADPH oxidases in osteoclastogenesis
In 2005, Lee et al. [64] identified a crucial role for ROS in RANKL-induced osteoclast differentiation in murine bone marrow-derived macrophages. In that particular work, the authors found an acute increase in Nox1-derived ROS formation within 10 min upon RANKL stimulation which was mediated by a TRAF6-dependent recruitment of Rac1 to the membrane. Additionally, MAP-kinase activation upon RANKL stimulation was dependent on the formation of ROS. The exact mechanism of the TRAF6-induced Rac1 activation however remained elusive. Whether, however, Rac and eventually Nox1 activation are necessary steps in osteoclastogenesis are discussed controversially. In RAW264.7 macrophages Rac1 was down-regulated in the course of RANKL-induced osteoclastogenesis [65] and in that particular study the authors found that during RANKL-induced osteoclastogenesis Nox2 expression is rapidly disappearing, which was paralleled by a reciprocal up-regulation of Nox1. Nevertheless and in contrast to an important role of either Nox1 or Nox2, bone marrow-derived macrophages from knockout mice of these NADPH oxidases generated similar level of ROS in response to RANKL and still differentiated into osteoclasts in the same way as wild-type bone marrow derived macrophages [66]. Recently, Nakanishi et al. [67] provided some evidence that at least in rat bone marrow-derived macrophages Nox2 is essentially involved in the expression of RANK. In the light of the mitochondria-mediated ROS formation upon RANKL stimulation of osteoclast precursors it may be possible to understand why RANKL-mediated ROS formation is impaired in Nox1 and Nox2 deficient cells: ROS triggered ROS formation has been observed for several years where an activation of Nox1 and Nox2 is mediated by mitochondrial ROS as reviewed elsewhere [68].
Yang et al. [69] were the first to identify Nox4 as a NADPH oxidase expressed in bone marrow-derived osteoclasts and these authors also demonstrated that Nox2 is not involved in acute RANKL-induced ROS formation. Importantly, Nox4 expression increased in the course of osteoclastogenesis. Unlike bone marrow-derived osteoclast precursors, it appears that this effect is missing in the RAW264.7 macrophage cell line so that Nox1 substitutes Nox4 in these cells. However, both studies found the same pattern of Nox expression in bone marrow-derived macrophages primed with M-CSF: Nox2 expression is up to 1,000 times higher than that of Nox1, and Nox4 is not detectable under basal conditions. Upon stimulation of bone marrow-derived macrophages with RANKL and thereby formation of, or differentiation into osteoclasts, Nox4 is up-regulated and becomes detectable [66, 70].
Using Nox4-deficient mice, we observed that Nox4 is one of the main factors needed for osteoclast differentiation [20]. In that particular work a higher bone density in Nox4-deficient mice was observed when compared to wild-type littermates. Nox4 expression was increased upon ovariectomy-induced osteoporosis in wildtype mice and bone loss in that model was prevented by a Nox4 inhibitor as well as by acute genetic deletion of Nox4. Eventually, we identified a small nucleotide polymorphism (SNP) associated with an elevated Nox4 expression and lower bone density in humans. As an underlying mechanism a lack of Ca2+ increase was discovered in Nox4 deficient cells which resulted in a reduced activation of NFATc1 as well as lower activation of the MAP-kinase Jnk and thereby less AP-1 activity upon RANKL stimulation in Nox4 deficient cells when compared to wildtype. Additionally, a previous study from our group showed that Nox4 maintains Nrf2 expression as well as its downstream target hemeoxygenase-1 in endothelial cells [71]. The connection between endothelial cells and osteoclasts is not obvious; however, both cell types originate from similar or even the same progenitors and share some central characteristics. Indeed, Van Phan et al. [72] found that the product of heme oxygenase-1, carbon monoxide protects against ovariectomy-induced bone loss by inhibiting osteoclastogenesis and Hyeon et al. [73] found that Nrf2 deficiency promotes RANKL-induced osteoclast differentiation.
Despite the importance of Nox4 for signaling in the course of differentiation, the enzyme does not seem to be involved in acute TRAF6-mediated RANKL-induced signaling. The increase in Nox4 expression rather appears to be a separate, later event in the course of differentiation. In contrast, Nox1 and Nox2 as mediators of the first oxidative flash after RANKL stimulation of osteoclast precursors seem to be mediators of acute TRAF6 signaling, but are not necessarily involved in the process of osteoclast differentiation per se. It is even possible that they can substitute each other or can even be replaced by mitochondria.
In conclusion, early RANKL-induced ROS formation mediated by mitochondria, Nox1 and/or Nox2 contributes to the acute activation of the CREB pathway which, however, appears not to be essential for osteoclastogenesis but rather mediates progenitor cell proliferation. In the course of RANKL-induced differentiation, Nox4 is up-regulated and mediates an increase in intracellular calcium and MAP-kinase activation which eventually via NFATc1 and AP-1 activation results in osteoclast formation.
ROS and loss of bone
ROS in estrogen-deficiency-induced osteoporosis
Aging is the most important risk factor for bone loss in both, men and women. Age-induced bone loss, however, seems to be related to reduced levels of sexual hormones in both genders rather than just to aging. The rapid drop in estrogens during female menopause makes bone loss however, more severe and obvious and the whole process occurs faster, to a greater degree and twenty years earlier in women than in men. Aging-induced bone loss is inversely related to estrogen levels [74] and in aging mice decreased bone formation parallels an increased ROS formation [75]. Interestingly, gonadectomy in mice and rats increases ROS level and induced bone loss and supplementation with the sexual steroids was sufficient to prevent this effect [75, 76]. The authors of both studies found that enzymatic antioxidants like SOD, GPx, GST decreased in ovariectomized animals when compared to controls. Additionally, together with a reduction in the thiol antioxidant defenses Lean et al. [77] found a crucial role of H2O2 in bone loss of ovarectomized mice as treatment of these animals with polyethylglycol-bound catalase inhibited bone loss. The same group reported that TNFα mediates bone loss in high ROS situations [78]. Depletion of glutathione by buthionine sulfoximine (BSO) administration as well as ovariectomy induced both a severe loss of bone mass, which was inhibited by administration of soluble TNFα receptors and abrogated in mice deleted for TNFα gene expression. However, in ovariectomized mice treated with soluble TNFα receptors, thiol antioxidant defenses in bone remained low, despite inhibition of bone loss. In summary, it may be attractive to speculate that a reduced antioxidative capacity and an increased ROS formation, especially of H2O2, together with TNFα are responsible for aging- and estrogen depletion-induced osteoporosis.
Besides aging, diabetes [79], dyslipidemia [80], smoking [81], alcohol [82] and inactivity are risk factors for osteoporosis as well.
ROS in ethanol-induced osteoporosis
Bones of young alcoholics have a similar weight as those of postclimacteric women [83]. This correlation led to the concept that ethanol-induced bone loss is a consequence of lower estrogen concentration. Chen et al. [84] found that 17β-estradiol prevents ethanol-induced induction of RANKL mRNA expression in osteoblasts and thereby prevented bone resorption and the decrease in tibial trabecular and total bone mineral density. In osteoblasts ethanol induced an increase in intracellular ROS formation through the induction of Nox1, Nox2 and Nox4. In rats, ethanol-induced Nox expression in the bones was inhibited by 17-β-estradiol, N-acetylcysteine and DPI [85]. Interestingly, 17β-estradiol also reduces NFκB activity. This results in a reduced expression of p47phox and thereby prevents the activation of Nox2 [86]. The importance of the cytosolic NADPH oxidase organizing subunit p47phox recently has been supported by a study were the authors found ethanol-induced bone loss to be completely prevented in p47phox knock out mice [87]. Conversely, the ethanol-induced ROS formation activated the Erk/STAT3 pathway and promoted RANKL expression and treatment with DPI at least prevented ethanol-induced RANKL expression [88]. Chen et al. infused rats intragastrically with ethanol 12 g/kg per day for 14 h from 6:00 PM to 8:00 AM during the dark cycle for 4 weeks. That is a total alcohol of 0.86 g/kg bodyweight per hour during infusion with a 12 h brake. Although blood alcohol was not measured by the authors, given that at least in humans only 0.1 g/kg is degraded per hour, the rats are constantly drunk, and such an experiment mimics severe alcohol abuse. Thus, the experiment can be criticized for potential indirect effects of the treatment. It is hard to assume that rats move normally under this alcohol concentration and therefore, loss of bone mass might in part be a consequence of mechanical unloading. Another possible reason for bone loss upon alcohol abuse is the permanent loss of electrolytes such as Na+, Mg2+, K+ and Ca2+ which are needed to maintain bone mineralization. It should be noted that, however, moderate ethanol consumption may even have positive effects on bone density and that therefore the effects of alcohol abuse on bone loss are just secondary. A recent study analyzing 13512 US Americans did not detect a negative, but rather a positive correlation between bone density and mid-level alcohol consumption. Even binge drinking was not associated with decreased bone density in men or women [89].
All above mentioned risk factors for osteoporosis are associated with inflammation and increased formation of ROS. Therefore, it is attractive to speculate that reduction in ROS-formation might be therapeutic to prevent skeletal diseases. Indeed, dietary supplementation with phyto-flavonoids revealed promising results in 3-month-old rats [90]. However, these study subjects are hardly representative for elderly people or alcoholics. Therefore, a better understanding of ROS related processes in osteoporosis is necessary before the therapeutic use of NADPH oxidase inhibitors or antioxidants.
NADPH oxidase in rheumatoid arthritis-induced bone loss
Rheumatoid arthritis is associated with high levels of ROS and local bone loss as consequence of the inflammation and systemic bone loss due to anti-inflammatory therapy with steroids. In the synovia of rheumatoid arthritis patients the level of IL-17 is particularly high and IL-17 has been shown to induce the expression of RANKL on osteoblasts and thereby in a co-culture system stimulates osteoclastogenesis [91]. Interestingly, IL-17 stimulated the proliferation of human mesenchymal stem cells in a TRAF6, Nox1 and Erk1/2 dependent manner [92].
Nox2 in leukocytes is an important mediator of the inflammatory response and leukocyte-derived ROS are important for bacterial killing in infectious arthritis. The situation in rheumatoid arthritis, however, is different as the inflammatory response is a consequence of an immune complex response of the acquired immune system and thus largely T cell-dependent. Paradoxically, loss of Nox2 induces a basal inflammatory state and inactivating mutations in p47phox have been linked to arthritis in humans. Utilizing p47phox mutant rats and mice, Holmdahl et al. identified an overaction of the T-cell response as the underlying mechanism and thus a more severe phenotype of the arthritis, per se. It is important to emphasize that the tissue destruction in this model is only to a minor extent mediated by Nox enzymes and release of proteases; stimulation of T-killer cells and many other factors contribute to the disease [93, 94]. On this basis, it is not surprising that variable findings were reported with respect to the role of the NADPH oxidase in this disease: In a model of antigen-induced bone loss by intra-articular injection of bovine serum albumin (BSA) in p47phox-mutant mice, no contribution of the cytosolic subunit of Nox2, p47phox, was observed in inflammation-mediated bone loss [95]. The authors studied mice that carry a point mutation in p47phox, causing a truncated and nonfunctional p47phox protein along with wildtypes as control. Upon BSA-induced arthritis a locally increased ROS formation in the distal but not in the proximal part of the femur was detected. However, no difference was observed in the grade of synovitis or joint destruction and loss of BMD was similar in wildtype mice and p47phox mutant animals. Interestingly, as mentioned above, the same animals in another study applying a collagen II-induced rheumatoid arthritis model had a more severe phenotype than the wildtype controls after 3 months, due to an increased T-cell activation [93]. Although these findings are interesting with respect to the immuno-genetic mechanism of arthritis, they do not illuminate the contribution of Nox-derived ROS to bone disease in humans. Cedergren et al. found that synovial fluid neutrophils from patients with rheumatoid arthritis, but not from non-rheumatoid arthritis patients, showed high baseline intracellular ROS production. Blood neutrophils from arthritis patients in remission are in a primed state as revealed by more rapid oxidative response after collagen-bead challenge and a more pronounced response after fMLF stimulation compared to a group of healthy blood donors [96].
In conclusion, bone loss during rheumatoid arthritis is probably not a consequence of p47phox-dependent Nox2 activation. It rather appears that other mechanisms, such as the excessive formation of NO by invading macrophages, induce bone loss in this disease.
Nitric oxide and bone
Both osteoclastogenesis and rheumatoid arthritis are associated with an increased NO formation. As NO can be scavenged by NADPH oxidase-derived O2 − to form the highly reactive peroxynitrite (ONOO−) a short chapter on NO is included here. It is known for 20 years that NO exerts biphasic effects on bone cell activity: High concentrations of NO inhibit bone resorption by attenuating osteoclast formation and differentiation and by attenuating the resorptive function of mature osteoclasts. In contrast, lower NO concentrations potentiate cytokine-induced bone resorption and may be essential for normal osteoclast function [97]. Nitric oxide (NO) mediates cellular signaling by activating soluble guanylyl cyclase (sGC) that converts guanosine-5′-triphosphate (GTP) to cyclic guanosine-3,5′-monophosphate (cGMP). Membrane-bound GCs produce cGMP in response to natriuretic peptides in osteoblasts, but comparison of TRAP activity with immunostaining for sGC beta1-subunit revealed that sGC beta1-subunit is only expressed in a sub-population of osteoclasts at least of the rat alveolar bone [98]. Therefore, it is more likely that NO affects osteoblasts than osteoclasts, or acts independent from cGC for example by S-nitrosation of target proteins.
The fact that every third postmenopausal woman develops osteoporosis and that physical inactivity also causes bone loss suggests that a factor induced by estrogens and mechano-stimulation protects from bone loss. Indeed estrogen [99] as well as shear stress [100] up-regulate endothelial NOS expression in osteoblasts and contribute to their proper function. Accordingly, eNOS−/− mice demonstrate a marked delay in postnatal bone formation, reduced bone volume and defects in osteoblast maturation and activity [101]. Interestingly, bodyweight is positively associated with bone density [102]. As body fat mass directly controls the circulating level of leptin, this hormone raised some interest as a potential candidate responsible for protective effects of fat on bone tissue [103]. Peripheral leptin increases bone formation mediated by osteoblasts [104]. As shown previously, leptin can induce the expression of nNOS in vessels and endothelial cells [105]. It is attractive to speculate, that leptin via the induction of a NO-synthase also in osteoblasts contributes to bone formation.
Another possible osteoblast-independent mechanism for the inhibition of bone loss activity by NO is the modification of cathepsin K. Cathepsin K is highly expressed in osteoclasts and degrades bone collagen. NO has been shown to inhibit the activity of purified cathepsin K by oxidative thiol modifications as well as S-nitrosation [106]. In the light of the positive effect of NO on bone formation, organic nitrates have been successfully used in the treatment of osteoporosis [107].
The calcium-activated NO synthases nNOS and eNOS are constitutively expressed in bone as well as in bone precursor cells. In contrast, the inducible NO synthase iNOS is expressed upon stimulation with pro-inflammatory cytokines such as IL-1, TNF, IFNγ and endotoxin in a variety of cell types, whereas glucocorticoids and the anti-inflammatory cytokines IL-3, IL-10 and TGF-β are inhibitory to their function [97]. Inducible NOS contributes to extremely high NO concentrations that might be a reason for bone loss in rheumatoid arthritis [108]. Experiments in iNOS-/- mice indicated that this particular isoform is the one involved in inflammatory-induced bone loss. Histomorphometric analysis of bones from normal animals with inflammation-induced bone loss showed a profound depression of bone formation and evidence of osteoblast apoptosis. These effects were missing in iNOS knockout animals, which suggests that iNOS-derived NO may contribute to inflammation-induced osteoporosis by suppressing bone formation and promoting osteoblast apoptosis and thereby reduces bone formation [109].
Concluding remarks
Bone is an organ that is renewed constantly. For a healthy skeleton it is essential that bone formation and bone degradation are perfectly coordinated. In case of an imbalance of bone formation or degradation, diseases occur associated with bone loss i.e. osteoporosis or excessive formation of new bone as present in osteopetrosis. Osteoporosis is a major health burden as every third woman and every fourth man develop the disease in their life. Beside high therapy costs, osteoporosis is associated with pain and reduced quality of life. Osteoporosis is mainly attributed to an overshooting activity of osteoclasts and almost all risk factors of osteoporosis are associated with increased levels of ROS. Therefore, a long standing interest in ROS-mediated osteoclastogenesis exists. NADPH oxidases as one of the most important source of ROS have been studied quite deeply for their role in osteoclastogenesis. Within the years a differentiated picture has evolved illustrating specific contributions of individual NADPH oxidases in the different phases of osteoclastogenesis. First approaches of targeted inhibition of individual NADPH oxidases like Nox4 in osteoporosis treatment reveal promising results. Further research on this topic will increase the target and organ specificity of potential drugs to prevent negative side effects of NADPH oxidase inhibitors.
Abbreviations
- Steap:
-
Six-transmembrane epithelial antigen of prostate
- IL:
-
Interleukin
- TNF:
-
Tumour necrosis factor
- IFN:
-
Interferon
- TGF:
-
Transforming growth factor
- NAC:
-
N-acetyl-cysteine
- RANK:
-
Receptor activator of nuclear factor (NF)-κB–receptor
- RANKL:
-
Receptor activator of NF-κB ligand
- OPG:
-
Osteoprotegerin
- BMP-2:
-
Bone morphogenic protein 2
References
Altindag O, Erel O, Soran N et al (2008) Total oxidative/anti-oxidative status and relation to bone mineral density in osteoporosis. Rheumatol Int 28(4):317–321. doi:10.1007/s00296-007-0452-0
Sendur OF, Turan Y, Tastaban E et al (2009) Antioxidant status in patients with osteoporosis: a controlled study. Joint Bone Spine 76(5):514–518. doi:10.1016/j.jbspin.2009.02.005
LeBoff MS, Narweker R, LaCroix A et al (2009) Homocysteine levels and risk of hip fracture in postmenopausal women. J Clin Endocrinol Metabol 94(4):1207–1213. doi:10.1210/jc.2008-1777
Schröder K (2014) NADPH oxidases in redox regulation of cell adhesion and migration. Antioxid Redox Signal 20(13):2043–2058. doi:10.1089/ars.2013.5633
Schröder K, Kohnen A, Aicher A et al (2009) NADPH oxidase Nox2 is required for hypoxia-induced mobilization of endothelial progenitor cells. Circ Res 105(6):537–544. doi:10.1161/CIRCRESAHA.109.205138
Adachi T, Togashi H, Suzuki A et al (2005) NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepatology 41(6):1272–1281. doi:10.1002/hep.20719
Lee C, Lin C, Lee I et al (2011) Activation and induction of cytosolic phospholipase A2 by TNF-α mediated through Nox2, MAPKs, NF-κB, and p300 in human tracheal smooth muscle cells. J Cell Physiol 226(8):2103–2114. doi:10.1002/jcp.22537
Wilkinson-Berka JL, Rana I, Armani R et al (2013) Reactive oxygen species, Nox and angiotensin II in angiogenesis: implications for retinopathy. Clin Sci 124(10):597–615. doi:10.1042/CS20120212
Miyano K, Sumimoto H (2007) Role of the small GTPase Rac in p22phox-dependent NADPH oxidases. Biochimie 89(9):1133–1144. doi:10.1016/j.biochi.2007.05.003
Banfi B, Clark RA, Steger K et al (2003) Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem 278(6):3510–3513. doi:10.1074/jbc.C200613200
Ueno N, Takeya R, Miyano K et al (2005) The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J Biol Chem 280(24):23328–23339. doi:10.1074/jbc.M414548200
Helmcke I, Heumüller S, Tikkanen R et al (2009) Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid Redox Signal 11(6):1279–1287. doi:10.1089/ARS.2008.2383
Al Ghouleh I, Frazziano G, Rodriguez AI et al (2013) Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc Res 97(1):134–142. doi:10.1093/cvr/cvs295
Katsuyama M, Matsuno K, Yabe-Nishimura C (2012) Physiological roles of NOX/NADPH oxidase, the superoxide-generating enzyme. J Clin Biochem Nutr 50(1):9–22. doi:10.3164/jcbn.11-06SR
Bedard K, Krause K (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1):245–313. doi:10.1152/physrev.00044.2005
Bedard K, Jaquet V, Krause K (2012) NOX5: from basic biology to signaling and disease. Free Radic Biol Med 52(4):725–734. doi:10.1016/j.freeradbiomed.2011.11.023
Lyle AN, Deshpande NN, Taniyama Y et al (2009) Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res 105(3):249–259. doi:10.1161/CIRCRESAHA.109.193722
Sturrock A, Cahill B, Norman K et al (2006) Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 290(4):L661–L673. doi:10.1152/ajplung.00269.2005
Clempus RE, Sorescu D, Dikalova AE et al (2007) Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol 27(1):42–48. doi:10.1161/01.ATV.0000251500.94478.18
Goettsch C, Babelova A, Trummer O et al (2013) NADPH oxidase 4 limits bone mass by promoting osteoclastogenesis. J Clin Invest 123(11):4731–4738. doi:10.1172/JCI67603
Hecker L, Vittal R, Jones T et al (2009) NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 15(9):1077–1081. doi:10.1038/nm.2005
Li J, Stouffs M, Serrander L et al (2006) The NADPH oxidase NOX4 drives cardiac differentiation: role in regulating cardiac transcription factors and MAP kinase activation. Mol Biol Cell 17(9):3978–3988. doi:10.1091/mbc.E05-06-0532
Takac I, Schröder K, Zhang L et al (2011) The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem 286(15):13304–13313. doi:10.1074/jbc.M110.192138
Paffenholz R, Bergstrom RA, Pasutto F et al (2004) Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev 18(5):486–491. doi:10.1101/gad.1172504
Kao C, Tai L, Chiou S et al (2010) Resveratrol promotes osteogenic differentiation and protects against dexamethasone damage in murine induced pluripotent stem cells. Stem Cells Dev 19(2):247–258. doi:10.1089/scd.2009.0186
Manolagas SC, Parfitt AM (2010) What old means to bone. Trends Endocrinol Metab 21(6):369–374. doi:10.1016/j.tem.2010.01.010
Lane D, Matte I, Laplante C et al (2013) Osteoprotegerin (OPG) activates integrin, focal adhesion kinase (FAK), and Akt signaling in ovarian cancer cells to attenuate TRAIL-induced apoptosis. J Ovarian Res 6(1):82. doi:10.1186/1757-2215-6-82
Boyce BF (2013) Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res 92(10):860–867. doi:10.1177/0022034513500306
Huang JC, Sakata T, Pfleger LL et al (2004) PTH Differentially Regulates Expression of RANKL and OPG. J Bone Miner Res 19(2):235–244. doi:10.1359/JBMR.0301226
Hjortnaes J, Butcher J, Figueiredo J et al (2010) Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur Heart J 31(16):1975–1984. doi:10.1093/eurheartj/ehq237
McCarthy I (2006) The physiology of bone blood flow: a review. J Bone Joint Surg Am 88((suppl_2)):4. doi:10.2106/JBJS.F.00890
Zaidi M (2007) Skeletal remodeling in health and disease. Nat Med 13(7):791–801. doi:10.1038/nm1593
Pinzone JJ, Hall BM, Thudi NK et al (2009) The role of Dickkopf-1 in bone development, homeostasis, and disease. Blood 113(3):517–525. doi:10.1182/blood-2008-03-145169
Jian J, Pelle E, Huang X (2009) Iron and menopause: does increased iron affect the health of postmenopausal women? Antioxid Redox Signal 11(12):2939–2943. doi:10.1089/ARS.2009.2576
Zarjou A, Jeney V, Arosio P et al (2010) Ferritin ferroxidase activity: a potent inhibitor of osteogenesis. J Bone Miner Res 25(1):164–172. doi:10.1359/jbmr.091002
Yang X, Chen-Barrett Y, Arosio P et al (1998) Reaction paths of iron oxidation and hydrolysis in horse spleen and recombinant human ferritins. Biochemistry 37(27):9743–9750. doi:10.1021/bi973128a
Tsay J, Yang Z, Ross FP et al (2010) Bone loss caused by iron overload in a murine model: importance of oxidative stress. Blood 116(14):2582–2589. doi:10.1182/blood-2009-12-260083
Jia P, Xu YJ, Zhang ZL et al (2012) Ferric ion could facilitate osteoclast differentiation and bone resorption through the production of reactive oxygen species. J Orthop Res 30(11):1843–1852. doi:10.1002/jor.22133
Nojiri H, Saita Y, Morikawa D et al (2011) Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J Bone Miner Res 26(11):2682–2694. doi:10.1002/jbmr.489
Monnier VM, Glomb M, Elgawish A et al (1996) The mechanism of collagen cross-linking in diabetes: a puzzle nearing resolution. Diabetes 45(Suppl 3):S67–S72
Morikawa D, Nojiri H, Saita Y et al (2013) Cytoplasmic reactive oxygen species and SOD1 regulate bone mass during mechanical unloading. J Bone Miner Res 28(11):2368–2380. doi:10.1002/jbmr.1981
Dernbach E, Urbich C, Brandes RP et al (2004) Antioxidative stress-associated genes in circulating progenitor cells: evidence for enhanced resistance against oxidative stress. Blood 104(12):3591–3597. doi:10.1182/blood-2003-12-4103
Fatokun AA, Stone TW, Smith RA (2008) Responses of differentiated MC3T3-E1 osteoblast-like cells to reactive oxygen species. Eur J Pharmacol 587(1–3):35–41. doi:10.1016/j.ejphar.2008.03.024
Cai W, Zhang M, Yu Y et al (2013) Treatment with hydrogen molecule alleviates TNFα-induced cell injury in osteoblast. Mol Cell Biochem 373(1–2):1–9. doi:10.1007/s11010-012-1450-4
Choi EM (2011) Luteolin protects osteoblastic MC3T3-E1 cells from antimycin A-induced cytotoxicity through the improved mitochondrial function and activation of PI3K/Akt/CREB. Toxicol In Vitro 25(8):1671–1679. doi:10.1016/j.tiv.2011.07.004
Choi EM, Lee YS (2012) Protective effect of apocynin on antimycin A-induced cell damage in osteoblastic MC3T3-E1 cells. J Appl Toxicol 32(9):714–721. doi:10.1002/jat.1689
Choi EM, Lee YS (2011) Involvement of PI3K/Akt/CREB and redox changes in mitochondrial defect of osteoblastic MC3T3-E1 cells. Toxicol In Vitro 25(5):1085–1088. doi:10.1016/j.tiv.2011.03.022
Mandal CC, Ganapathy S, Gorin Y et al (2011) Reactive oxygen species derived from Nox4 mediate BMP2 gene transcription and osteoblast differentiation. Biochem J 433(2):393–402. doi:10.1042/BJ20100357
Schröder K, Wandzioch K, Helmcke I et al (2009) Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler Thromb Vasc Biol 29(2):239–245. doi:10.1161/ATVBAHA.108.174219
van Driel M, van Leeuwen Johannes P T M (2014) Vitamin D endocrine system and osteoblasts. Bonekey Rep 3:493. doi:10.1038/bonekey.2013.227
Somjen D, Katzburg S, Grafi-Cohen M et al (2011) Vitamin D metabolites and analogs induce lipoxygenase mRNA expression and activity as well as reactive oxygen species (ROS) production in human bone cell line. J Steroid Biochem Mol Biol 123(1–2):85–89. doi:10.1016/j.jsbmb.2010.11.010
Boyan BD, Bonewald LF, Sylvia VL et al (2002) Evidence for distinct membrane receptors for 1 alpha,25-(OH)(2)D(3) and 24R,25-(OH)(2)D(3) in osteoblasts. Steroids 67(3–4):235–246
Atkins GJ, Kostakis P, Pan B et al (2003) RANKL expression is related to the differentiation state of human osteoblasts. J Bone Miner Res 18(6):1088–1098. doi:10.1359/jbmr.2003.18.6.1088
Tilyard MW, Spears GF, Thomson J et al (1992) Treatment of postmenopausal osteoporosis with calcitriol or calcium. N Engl J Med 326(6):357–362. doi:10.1056/NEJM199202063260601
Eisman JA, Bouillon R (2014) Vitamin D: direct effects of vitamin D metabolites on bone: lessons from genetically modified mice. Bonekey Rep 3:499. doi:10.1038/bonekey.2013.233
Yin H, Shi Z, Yu Y et al (2012) Protection against osteoporosis by statins is linked to a reduction of oxidative stress and restoration of nitric oxide formation in aged and ovariectomized rats. Eur J Pharmacol 674(2–3):200–206. doi:10.1016/j.ejphar.2011.11.024
Huang W, Shang W, Li D et al (2012) Simvastatin protects osteoblast against H2O2-induced oxidative damage via inhibiting the upregulation of Nox4. Mol Cell Biochem 360(1–2):71–77. doi:10.1007/s11010-011-1045-5
Walsh MC, Choi Y (2003) Biology of the TRANCE axis. Cytokine Growth Factor Rev 14(3–4):251–263
Lomaga MA, Yeh WC, Sarosi I et al (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 13(8):1015–1024
Kadono Y, Okada F, Perchonock C et al (2005) Strength of TRAF6 signalling determines osteoclastogenesis. EMBO Rep 6(2):171–176. doi:10.1038/sj.embor.7400345
Chandel NS, Schumacker PT, Arch RH (2001) Reactive oxygen species are downstream products of TRAF-mediated signal transduction. J Biol Chem 276(46):42728–42736. doi:10.1074/jbc.M103074200
Zhou J, Ye S, Fujiwara T et al (2013) Steap4 plays a critical role in osteoclastogenesis in vitro by regulating cellular iron/reactive oxygen species (ROS) levels and cAMP response element-binding protein (CREB) activation. J Biol Chem 288(42):30064–30074. doi:10.1074/jbc.M113.478750
Srinivasan S, Koenigstein A, Joseph J et al (2010) Role of mitochondrial reactive oxygen species in osteoclast differentiation. Ann N Y Acad Sci 1192:245–252. doi:10.1111/j.1749-6632.2009.05377.x
Lee NK, Choi YG, Baik JY et al (2005) A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 106(3):852–859. doi:10.1182/blood-2004-09-3662
Sasaki H, Yamamoto H, Tominaga K et al (2009) NADPH oxidase-derived reactive oxygen species are essential for differentiation of a mouse macrophage cell line (RAW264.7) into osteoclasts. J Med Invest 56(1–2):33–41
Sasaki H, Yamamoto H, Tominaga K et al (2009) Receptor activator of nuclear factor-kappaB ligand-induced mouse osteoclast differentiation is associated with switching between NADPH oxidase homologues. Free Radic Biol Med 47(2):189–199. doi:10.1016/j.freeradbiomed.2009.04.025
Nakanishi A, Hie M, Iitsuka N et al (2013) A crucial role for reactive oxygen species in macrophage colony-stimulating factor-induced RANK expression in osteoclastic differentiation. Int J Mol Med 31(4):874–880. doi:10.3892/ijmm.2013.1258
Daiber A (2010) Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta 1797(6–7):897–906. doi:10.1016/j.bbabio.2010.01.032
Yang S, Madyastha P, Bingel S et al (2001) A new superoxide-generating oxidase in murine osteoclasts. J Biol Chem 276(8):5452–5458. doi:10.1074/jbc.M001004200
Yang S, Zhang Y, Ries W et al (2004) Expression of Nox4 in osteoclasts. J Cell Biochem 92(2):238–248. doi:10.1002/jcb.20048
Schröder K, Zhang M, Benkhoff S et al (2012) Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 110(9):1217–1225. doi:10.1161/CIRCRESAHA.112.267054
van Phan T, Sul O, Ke K et al (2013) Carbon monoxide protects against ovariectomy-induced bone loss by inhibiting osteoclastogenesis. Biochem Pharmacol 85(8):1145–1152. doi:10.1016/j.bcp.2013.01.014
Hyeon S, Lee H, Yang Y et al (2013) Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic Biol Med 65:789–799. doi:10.1016/j.freeradbiomed.2013.08.005
Gurney EP, Nachtigall MJ, Nachtigall LE et al (2014) The Women’s Health Initiative trial and related studies: 10 years later: a clinician’s view. J Steroid Biochem Mol Biol 142C:4–11. doi:10.1016/j.jsbmb.2013.10.009
Almeida M, Han L, Martin-Millan M et al (2007) Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem 282(37):27285–27297. doi:10.1074/jbc.M702810200
Muthusami S, Ramachandran I, Muthusamy B et al (2005) Ovariectomy induces oxidative stress and impairs bone antioxidant system in adult rats. Clin Chim Acta 360(1–2):81–86. doi:10.1016/j.cccn.2005.04.014
Lean JM, Jagger CJ, Kirstein B et al (2005) Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology 146(2):728–735. doi:10.1210/en.2004-1021
Jagger CJ, Lean JM, Davies JT et al (2005) Tumor necrosis factor-alpha mediates osteopenia caused by depletion of antioxidants. Endocrinology 146(1):113–118. doi:10.1210/en.2004-1058
Gower BA, Casazza K (2013) Divergent effects of obesity on bone health. J Clin Densitom 16(4):450–454. doi:10.1016/j.jocd.2013.08.010
Parhami F, Garfinkel A, Demer LL (2000) Role of lipids in osteoporosis. Arterioscler Thromb Vasc Biol 20(11):2346–2348. doi:10.1161/01.ATV.20.11.2346
Iqbal J, Sun L, Cao J et al (2013) Smoke carcinogens cause bone loss through the aryl hydrocarbon receptor and induction of Cyp1 enzymes. Proc Natl Acad Sci 110(27):11115–11120. doi:10.1073/pnas.1220919110
Mikosch P (2014) Alcohol and bone. Wien Med Wochenschr 164(1–2):15–24. doi:10.1007/s10354-013-0258-5
Saville PD (1965) Changes in bone mass with age and alcoholism. J Bone Joint Surg Am 47:492–499
Chen J, Haley RL, Hidestrand M et al (2006) Estradiol protects against ethanol-induced bone loss by inhibiting up-regulation of receptor activator of nuclear factor-kappaB ligand in osteoblasts. J Pharmacol Exp Ther 319(3):1182–1190. doi:10.1124/jpet.106.109454
Chen J, Lazarenko OP, Shankar K et al (2011) Inhibition of NADPH oxidases prevents chronic ethanol-induced bone loss in female rats. J Pharmacol Exp Ther 336(3):734–742. doi:10.1124/jpet.110.175091
Sumi D, Hayashi T, Matsui-Hirai H et al (2003) 17beta-estradiol inhibits NADPH oxidase activity through the regulation of p47phox mRNA and protein expression in THP-1 cells. Biochim Biophys Acta 1640(2–3):113–118
Mercer KE, Sims CR, Yang CS et al (2014) Loss of functional NADPH oxidase 2 protects against alcohol-induced bone resorption in female p47phox−/− mice. Alcohol Clin Exp Res 38(3):672–682. doi:10.1111/acer.12305
Chen J, Shankar K, Nagarajan S et al (2008) Protective effects of estradiol on ethanol-induced bone loss involve inhibition of reactive oxygen species generation in osteoblasts and downstream activation of the extracellular signal-regulated kinase/signal transducer and activator of transcription 3/receptor activator of nuclear factor-kappaB ligand signaling cascade. J Pharmacol Exp Ther 324(1):50–59. doi:10.1124/jpet.107.130351
Wosje KS, Kalkwarf HJ (2007) Bone density in relation to alcohol intake among men and women in the United States. Osteoporos Int 18(3):391–400. doi:10.1007/s00198-006-0249-0
Oršolić N, Goluža E, Đikić D et al (2013) Role of flavonoids on oxidative stress and mineral contents in the retinoic acid-induced bone loss model of rat. Eur J Nutr. doi:10.1007/s00394-013-0622-7
Kotake S, Udagawa N, Takahashi N et al (1999) IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 103(9):1345–1352. doi:10.1172/JCI5703
Huang H, Kim HJ, Chang E et al (2009) IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Differ 16(10):1332–1343. doi:10.1038/cdd.2009.74
Hultqvist M, Olofsson P, Holmberg J et al (2004) Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc Natl Acad Sci 101(34):12646–12651. doi:10.1073/pnas.0403831101
Gelderman KA, Hultqvist M, Pizzolla A et al (2007) Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest 117(10):3020–3028. doi:10.1172/JCI31935
Engdahl C, Lindholm C, Stubelius A et al (2013) Periarticular bone loss in antigen-induced arthritis. Arthritis Rheum 65(11):2857–2865. doi:10.1002/art.38114
Cedergren J, Forslund T, Sundqvist T et al (2007) Intracellular oxidative activation in synovial fluid neutrophils from patients with rheumatoid arthritis but not from other arthritis patients. J Rheumatol 34(11):2162–2170
Ralston SH (1997) The michael mason prize essay 1997, Nitric oxide and bone: what a gas! Br J Rheumatol 36(8):831–838
Korkmaz Y, Baumann MA, Schroder H et al (2004) Localization of the NO-cGMP signaling pathway molecules, NOS III-phosphorylation sites, ERK1/2, and Akt/PKB in osteoclasts. J Periodontol 75(8):1119–1125. doi:10.1902/jop.2004.75.8.1119
Armour KE, Ralston SH (1998) Estrogen upregulates endothelial constitutive nitric oxide synthase expression in human osteoblast-like cells. Endocrinology 139(2):799–802. doi:10.1210/endo.139.2.5910
Zaman G, Pitsillides AA, Rawlinson SC et al (1999) Mechanical strain stimulates nitric oxide production by rapid activation of endothelial nitric oxide synthase in osteocytes. J Bone Miner Res 14(7):1123–1131. doi:10.1359/jbmr.1999.14.7.1123
Aguirre J, Buttery L, O’Shaughnessy M et al (2001) Endothelial nitric oxide synthase gene-deficient mice demonstrate marked retardation in postnatal bone formation, reduced bone volume, and defects in osteoblast maturation and activity. Am J Pathol 158(1):247–257. doi:10.1016/S0002-9440(10)63963-6
Felson DT, Zhang Y, Hannan MT et al (1993) Effects of weight and body mass index on bone mineral density in men and women: the Framingham study. J Bone Miner Res 8(5):567–573. doi:10.1002/jbmr.5650080507
Thomas T, Burguera B (2002) Is leptin the link between fat and bone mass? J Bone Miner Res 17(9):1563–1569. doi:10.1359/jbmr.2002.17.9.1563
Turner RT, Kalra SP, Wong CP et al (2013) Peripheral leptin regulates bone formation. J Bone Miner Res 28(1):22–34. doi:10.1002/jbmr.1734
Benkhoff S, Loot AE, Pierson I et al (2012) Leptin potentiates endothelium-dependent relaxation by inducing endothelial expression of neuronal NO synthase. Arterioscler Thromb Vasc Biol 32(7):1605–1612. doi:10.1161/ATVBAHA.112.251140
Percival MD, Ouellet M, Campagnolo C et al (1999) Inhibition of cathepsin K by nitric oxide donors: evidence for the formation of mixed disulfides and a sulfenic acid. Biochemistry 38(41):13574–13583
Jamal SA, Reid LS, Hamilton CJ (2013) The effects of organic nitrates on osteoporosis: a systematic review. Osteoporos Int 24(3):763–770. doi:10.1007/s00198-012-2262-9
Kaur H, Halliwell B (1994) Evidence for nitric oxide-mediated oxidative damage in chronic inflammation. Nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett 350(1):9–12
Van’t Hof RJ, Ralston SH (2001) Nitric oxide and bone. Immunology 103(3):255–261
Acknowledgments
The author thanks Ralf P. Brandes for critical reading the manuscript and making helpful suggestions. This work was supported by the Deutsche Forschungsgemeinschaft (SFB815/TP1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Schröder, K. NADPH oxidases in bone homeostasis and osteoporosis. Cell. Mol. Life Sci. 72, 25–38 (2015). https://doi.org/10.1007/s00018-014-1712-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1712-2