
Abstract
Sodium absorption by the distal part of the nephron, i.e., the distal convoluted tubule, the connecting tubule, and the collecting duct, plays a major role in the control of homeostasis by the kidney. In this part of the nephron, sodium transport can either be electroneutral or electrogenic. The study of electrogenic Na+ absorption, which is mediated by the epithelial sodium channel (ENaC), has been the focus of considerable interest because of its implication in sodium, potassium, and acid–base homeostasis. However, recent studies have highlighted the crucial role played by electroneutral NaCl absorption in the regulation of the body content of sodium chloride, which in turn controls extracellular fluid volume and blood pressure. Here, we review the identification and characterization of the NaCl cotransporter (NCC), the molecule accounting for the main part of electroneutral NaCl absorption in the distal nephron, and its regulators. We also discuss recent work describing the identification of a novel “NCC-like” transport system mediated by pendrin and the sodium-driven chloride/bicarbonate exchanger (NDCBE) in the β-intercalated cells of the collecting system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The kidney plays a critical role in almost all physiological processes, including blood pressure, cell volume and pH regulation, muscle contractility, and neuron excitability, as it keeps constant the concentration, or the body content, of the different ions and water. To ensure homeostasis, a very large amount of plasma and solute is filtered. The different renal epithelial cell types, which can achieve selective reabsorption or secretion of water and ions, then modify the composition of this ultrafiltrate. As a consequence, the daily excretion of water and ions into urine exactly matches the daily intake brought about by the diet. Since the daily intake of each substance can vary considerably from one individual to another, and from time to time, the amount of the different solutes or water absorbed or secreted by epithelial cells is tightly controlled. Schematically, three different zones of the nephron can be functionally distinguished. The proximal tubule (PT) achieves a massive reabsorption of water and solutes. The loop of Henle accounts for the creation and maintenance of a cortico-papillary gradient of solutes, and hence of osmoles, required for the concentration of final urine. The terminal part of the nephron, which includes the distal convoluted tubule (DCT), the connecting tubule (CNT), and the collecting duct (CD), is responsible for the fine-tuning of all electrolytes and water balances.
Among the different substances transported across the renal epithelium, the sodium ion is of particular importance. Indeed, sodium chloride is the main source of osmoles in the extracellular fluid, including the plasma, and therefore it is one of the critical determinants of blood pressure. The importance of the renal regulation of sodium balance for blood pressure regulation has been initially proposed by Arthur Guyton who observed that volume regulation, and the relationship between blood pressure and renal sodium handling, are abnormal in human individuals affected by hypertension [1]. In fact, a central component of the feedback system for long-term control of arterial pressure is the pressure-natriuresis mechanism, whereby an increase in renal perfusion pressure leads to a decrease in sodium reabsorption and increase in sodium excretion. Guyton’s theory was based upon a complex mathematical model of blood pressure regulation. However, according to his pioneering hypothesis, most of the genes mutated in patients with Mendelian syndromes of altered blood pressure have indeed all turned out to be involved in the control of renal NaCl absorption [2]. The finding that all known inherited and acquired forms of hypertension ultimately operate via the same common pathway has led to the proposal that common forms of hypertension should feature increased renal sodium reabsorption as well [2].
The sodium ion is not only important because it sets blood pressure. The main bioenergizer of animal cell membranes is generally the Na+/K+ P-ATPase, which converts the energy derived from metabolism into steep sodium and potassium gradients across the cell membrane [3, 4]. The inwardly directed sodium gradient is then used to energize the uptake of many other solutes into the cells via sodium-dependent secondary active cotransporters or exchangers. For example, along the renal tubule, sodium absorption drives the absorption of glucose, amino acids, phosphates, bicarbonate, and chloride. When the process that accounts for sodium absorption across the renal epithelium is electrogenic, sodium transport can also be the primary determinant of the transepithelial voltage difference (V te) that develops across several specific parts of the nephron. In the thick ascending limb (TAL) of Henle’s loop, the V te drives the passive absorption of various cations, particularly Ca2+ and Mg2+ [5]. In the CD, V te drives the secretion of both K+ and H+ [6]. Therefore, any change in the rate of sodium absorption in the different nephron segments not only affects sodium balance but can also lead to other electrolyte imbalances.
This interaction of sodium ions with other ions is particularly important in the distal nephron, which comprises the distal convoluted tubule (DCT), the connecting tubule (CNT), and the collecting duct (CD). There, the sodium ion can be absorbed along with chloride via electroneutral processes, or can be absorbed by the electrogenic epithelial sodium channel (ENaC), a mechanism which in turn drives potassium and proton secretion. Many physiological or pathophysiological conditions indicate that the respective proportion of electrogenic versus electroneutral sodium absorption is central for the coordinated (or independent) control of blood pressure, blood K+ concentration, and acid–base status. For instance, genetic or pharmacologic inactivation of the electroneutral transport in the DCT favors electrogenic sodium absorption by the CD and leads to the development of hypovolemia along with hypokalemia and metabolic alkalosis [7, 8], while its excessive activation provokes hyperkalemia and metabolic acidosis [8–10].
In summary, sodium transport in the distal nephron plays a central role in the control of fundamental physiological processes. Many excellent reviews have described in detail the properties, regulation, and roles of the electrogenic epithelial sodium channel ENaC [6, 11–14]. The purpose of the present review is to summarize the knowledge obtained recently about the different mechanisms of electroneutral NaCl transport processes identified to date in the distal nephron.
Electroneutral NaCl transport by the kidney: from thiazide diuretics to NCC
The discovery of the different ion transporters accounting for the renal absorption of Na+ is relatively recent. It was the conclusion of intense research efforts aimed at understanding the mechanisms of action of diuretics, and to identify the molecular basis of rare Mendelian diseases characterized by a phenotype mimicking the use of these drugs. Chlorothiazide is one of the oldest diuretics identified [15]. It was originally designed empirically, without the knowledge of renal ion transporters, by modifying acetazolamide, a carbonic anhydrase blocker with weak natriuretic properties [16]. Interestingly, the authors of the seminal article describing the effects of chlorothiazide administration to dogs [15] observed that this drug markedly differs from carbonic anhydrase blockers in that the excretion of sodium promoted by the drug is accompanied by chloruresis rather than by bicarbonaturia. Several years later, in vitro studies reached the conclusion that three modes of transepithelial chloride transport exist: (1) the first is the passive diffusion of the chloride anion through the paracellular pathway driven by transepithelial differences in concentration and electrical potential, like in the toad bladder [17], (2) the second involves electroneutral Cl−/HCO3 − exchange [18], and finally (3) J.L. Renfro [19] discovered that in the urinary bladder of the teleost Pseudopleuronectes americanus (i.e., the winter flounder) active Cl− transport can also be directly coupled to Na+. Importantly, a subsequent study performed by J.B. Stokes demonstrated for the first time that the NaCl cotransport system of the winter flounder’s urinary bladder is inhibitable by thiazide compounds [20–23]. However, the identification of the molecule targeted by thiazides in mammalian kidneys remained a matter of controversy for a long time because thiazides retain some of the properties of acetazolamide in that they are able to inhibit carbonic anhydrase and thereby a parallel counter-transport system for Na+/H+ and Cl−/HCO3 − exchange [24, 25]. In order to gain insight into the molecular nature of the NaCl absorptive pathway targeted by thiazides, several groups tried to use a tritiated derivative of metolazone, a thiazide-like compound. Beaumont et al. [26, 27] showed that [3H]-metolazone binds a “high affinity receptor” located at the apical membrane of distal convoluted tubule cells. The binding of [3H]-metolazone could be displaced by several different thiazide derivatives [26], or inhibited by Cl− [28]. In addition, the density of this receptor was regulated under physiological conditions known to modulate NaCl transport, like changes in dietary sodium or chronic diuretic administration [29, 30]. Ellison et al. [31] were able to solubilize and purify this [3H]-metolazone receptor from rabbit kidney cortex, and used this material to generate a monoclonal antibody that turned out to recognize specifically a unique 125-kDa protein. This protein, again, localized to the apical membrane of cells in the distal convoluted tubule [32]. However, the identification of the molecule accounting for thiazide-sensitive NaCl cotransport did not come from these elegant biochemical and physiological studies. Indeed, the major breakthrough came, again, from studies performed in fish. Indeed, Gamba et al. [33], using a functional expression cloning strategy, finally identified the molecule accounting for the NaCl cotransport activity of the urinary bladder of the winter flounder described originally by J.L. Renfo [19, 33]. The gene encoding the Na+/K+/2Cl− cotransporter NKCC1 of the rectal gland of the shark, Squalus acanthus, was subsequently cloned [34], and the superfamily of cation-chloride cotransporters SLC12 was defined. Both genes turned out to share remarkably high sequence homologies, thus enabling the identification of other members of this superfamily through the search of conserved sequences in the genomic databases (for review see Ref. [35]). Gamba et al. [36] isolated the cDNAs encoding the rat isoform of NaCl and Na+/K+/2Cl− cotransporters very shortly after the first cloning in fish. Finally, another branch of the SLC12 gene family, including four distinct KCl cotransporters, was subsequently identified [37–40].
The importance of the NaCl cotransporter (encoded by the SLC12A3 gene) of the DCT in renal sodium homeostasis is highlighted by the fact that NCC is a target of aldosterone, the main hormone controlling renal Na+ transport [41, 42]. Moreover, inactivating mutations of SLC12A3 in humans [43] cause Gitelman syndrome, an inherited recessive disease characterized by low blood pressure; even heterozygous inactivating mutations confer a low blood pressure or a protection against arterial hypertension [44]. In contrast, excessive activity of NCC is central to the phenotype of patients with familial hyperkalemic hypertension (FHHt) [9], also known as Gordon’s syndrome or Pseudohypoaldosteronism type II, a rare inherited disease characterized by hypertension that is highly sensitive to thiazide compounds. However, NCC activation in this disease is not caused by activating mutations in the SLC12A3 gene but by mutations in genes involved in regulatory pathways controlling NCC [9, 45–48]. The link between the molecular and the physiological regulators of NCC has since been intensively studied.
Regulation of NCC activity: from hormones to cellular pathways
In the adult kidney, NCC expression is exclusively restricted to the distal convoluted tubule (DCT) of the kidney [49–51]. NCC mRNA and protein are particularly abundant in the early DCT and decrease gradually along the late DCT in mouse, rat, and human. The rabbit DCT does not show a gradual decrease in NCC expression but a rather abrupt transition with the CNT cells [49]. DCT cells are mitochondria-rich cells with long basolateral infoldings. It is also associated with the highest Na+–K+-ATPase activity of any nephron segment [52], probably reflecting the high rate of transport activity of this segment.
Hormones controlling renal salt balance, particularly from the renin–angiotensin–aldosterone system, are known to regulate NCC and tune the intracellular NCC regulatory mechanisms to modify NaCl transport and balance. Other hormones such as vasopressin [53–56] or PTH [57] are known to regulate NCC. Finally, three different groups recently reported that NCC is regulated by insulin, making a potential bridge between hyperinsulinism and salt-sensitive hypertension [58–60].
NCC regulation by the renin–angiotensin–aldosterone system
From the discovery of the remarkable effects of thiazides on blood pressure, it has been obvious that NCC plays a critical role in renal sodium handling. Therefore, it was intuitively proposed that NCC might be regulated by hormones of the renin–angiotensin–aldosterone system (RAAS).
While the effects of the RAAS on ENaC are quite clear, its importance in NCC regulation is still not completely elucidated. The mineralocorticoid hormone aldosterone binds to the cytosolic mineralocorticoid receptor, translocates to the nucleus, and activates the transcription of its target genes. During NaCl restriction, the secretion of aldosterone increases, thereby activating both the transcription and protein abundance of the three subunits composing ENaC [61]. This in turn stimulates Na+ retention by the distal nephron. However, glucocorticoids (e.g., cortisol) have the same affinity for the mineralocorticoid receptor than aldosterone and are present in the blood at much higher concentrations than aldosterone. Thus, it is predicted that without any protective mechanism the mineralocorticoid receptor should mostly be activated by glucocorticoid, which would prevent any action of aldosterone. The 11β-hydroxysteroid dehydrogenase type 2 (11BHSD2) is an intracellular enzyme that degrades glucocorticoids but not aldosterone. Thus, 11BHSD2 prevents the mineralocorticoid receptor from being activated by glucocorticoids. All aldosterone-sensitive cells are thought to express 11BHSD2. However, 11BHSD2 is absent from the early DCT (the nephron segment characterized by high NCC expression) and is only detectable in the late DCT (a nephron segment characterized by low NCC expression) [62]. Therefore, it was initially proposed that aldosterone is not active in the DCT, and hence could not regulate NCC. However, several groups reported that aldosterone stimulates thiazide-sensitive Na+ reabsorption in the DCT [42, 63], an effect correlated with an increase in NCC abundance [41]. How the DCT cells are protected against illegitimate activation of the mineralo-corticoid receptor by glucocorticoid remains elusive. Further, during chronic exposure to primary aldosteronism, the kidney has the ability to decrease its sensitivity to aldosterone and thereby minimize Na+ retention by a phenomenon called “aldosterone escape”. Wang et al. [64] showed that NCC abundance is strongly repressed in rats during primary hyperaldosteronism while ENaC is continuously stimulated by a chronic administration of aldosterone. The authors proposed that the downregulation of NCC in this setting account for the escape. The mechanisms blocking NCC responsiveness to aldosterone are still unknown. Nevertheless, the absence of 11BHSD2 in DCT cells and the observation that NCC can be inhibited while aldosterone’s secretion is increased both suggest that the effects of aldosterone on NCC might not be direct but rather require some additional factors.
The second factor from the RAAS that affects NCC is angiotensin II (AngII), a vasoactive peptide produced by cleavage of angiotensin I by the angiotensin-converting enzyme (ACE). Angiotensin I itself is produced by renin from angiotensinogen. The production of AngII is stimulated during volume depletion to keep blood pressure constant by favoring renal NaCl retention and vascular vasoconstriction. Consequently, inhibition of AngII generation by ACE inhibitors and angiotensin II receptors antagonists are commonly used as anti-hypertensive drugs. In the kidney, angiotensin II stimulates most of Na+ transporters, among which NCC [65–69]. NCC is indeed targeted to the DCT apical membrane upon angiotensin II infusion [65]. Until recently, it was unclear whether most, if not all, effects of AngII require aldosterone. In fact, aldosterone’s secretion by the adrenals is stimulated by AngII. Thus, the increase in NCC expression observed during AngII treatment could be mediated by aldosterone rather than being a direct effect of AngII. However, a study has recently shown that locally produced AngII rather than circulating AngII plays a crucial role in the regulation of NCC [70]. The hypertensive response and NCC upregulation are indeed blunted in a mouse model devoid of renal ACE. The proposed model is that circulating AngII activates ACE in the proximal tubule, thus increasing the intra-renal production of AngII. This locally synthesized AngII then stimulates Na+ transporters along the entire distal nephron and particularly enhances NCC phosphorylation and abundance [70]. According to this paradigm, the effects of AngII cannot be mediated by aldosterone. Moreover, another study conducted in rats by Van der Lubbe et al. [71] showing that NCC expression is still increased by a chronic infusion of AngII in adrenalectomized rats further supports the possibility that AngII directly affects NCC expression or activity.
NCC regulation by sodium and potassium intake
NCC is not exclusively regulated by the RAAS. Recent studies have established that chronic and acute K+ loading decrease NCC activity [72, 73]. An increase in K+ intake stimulates K+ secretion by the distal nephron and a reduced NaCl reabsorption in the DCT through NCC is proposed as one of the mechanisms. Indeed, a decrease in NCC-mediated NaCl absorption increases Na+ and Cl− delivery to the CNT and the CD. This is then expected to stimulate electrogenic Na+ reabsorption via ENaC and thus promote potassium secretion by principal cells. The effects of acute K+ loading on NCC are independent of the accompanying anion since KHCO3 and KCl loading produce the same decrease in NCC [72]. The effects of K+ loading are also independent of plasma aldosterone levels, as mice that do not generate aldosterone (aldosterone synthase-deficient mice) are able to decrease NCC phosphorylation levels during acute K+ loading. The latter observation supports the existence of an unidentified kaliuretic factor, regulating NCC [74, 75].
The downregulation of NCC by K+ loading seems in contradiction with the stimulation of NCC by aldosterone, as an increase in K+ intake is the major stimulus for aldosterone secretion. NCC is therefore inversely regulated in two situations of elevated aldosterone, i.e., increased during NaCl restriction or and decreased during K+ loading. The difference between the two situations is the level of circulating, and therefore intra-renal, AngII. While AngII level is high during NaCl restriction, it is low during K+ load. AngII level is also reduced when Na+ intake increases and this is associated with a decrease in NCC expression [76]. NCC could therefore be regulated by AngII rather than by aldosterone. One study, however, supports the direct regulation of NCC by aldosterone. Using adrenalectomized rats submitted to a chronic aldosterone infusion, van der Lubbe and collaborators [71] showed that NCC activation by aldosterone is not inhibited in vivo by losartan, an angiotensin II receptor inhibitor. These contradictory results illustrate the complexity of NCC regulation, and more generally of the coordinated regulation of Na+, K+, and Cl− balance by the distal nephron. Many more studies will be required before a clear physiological model could be established.
The molecular mechanisms by which NCC expression and activity are regulated have started to be unraveled over the last few years. Two main mechanisms have been identified: phosphorylation/dephosphorylation and degradation of the co-transporter.
NCC regulation by phosphorylation
As mentioned above, NCC belongs to the SCL12 family of electroneutral cation-coupled chloride cotransporters, which contains two branches, i.e., the sodium-driven cotransporters (NCC, NKCC1, and NKCC2) and the potassium-driven cotransporters (KCC1-4). Many in vitro studies had shown that NKCC1 activity is modulated by phosphorylation (for review, see [77]) and five threonine residues (Thr175, Thr179, Thr184, Thr189, and Thr202 of shark NKCC1), located in the amino-terminal intra-cellular domain of the protein, were then identified as being subjected to phosphorylation and modulating NKCC1 activity [78, 79]. The phosphorylation of only one of these residues, Thr189, is absolutely required for the cotransporter activity. The phosphorylation of the other residues is modulatory; phosphorylation of Thr184 and Thr202, for example, increases the sensitivity of NKCC1 to changes in intracellular chloride concentration [78]. These five residues are conserved in NCC and NKCC2. These residues are Thr46, Thr50, Thr55, Thr60, and Ser73 in human NCC [80] and, for the sake of simplicity, we will rename them Thr1, Thr2, Thr3, Thr4, and Ser1 in the remaining review, as cDNAs from different species were used in the cited articles. An additional phosphorylation site, without any homology to NKCCs, was identified (Ser91 in human NCC, renamed Ser2 here) by Richardson and collaborators in cells submitted to intracellular chloride depletion [80].
The mutation of Thr4 of rat NCC, corresponding to Thr189 in shark NKCC1, to alanine abolishes sodium transport in Xenopus laevis oocytes [81], thus demonstrating that phosphorylation of this residue is essential for NCC activity. This was later confirmed in transfected HEK293 cells [80]. Pacheco-Alvarez further tested the functional importance of Thr3 and Ser1 (corresponding to Thr184 and Thr202 of shark NKCC1), and the results differ from what was obtained for NKCC1. While the mutation of Thr3 inhibits NCC activity only moderately (25 % decrease), like NKCC1, the mutation of Ser1 strongly reduces NCC activity (75 % decrease), when it had almost no effect on NKCC1 basal activity. These differences might result from species and/or conformational and/or amino-acid-sequence differences between NKCC1 and NCC. Importantly, the mutation of Thr4 markedly reduced the phosphorylation of Thr1, Thr3 and, to a lesser extent, Ser2 in HEK293 cells [80]. The abrogation of NCC activity by this mutation could therefore result from a loss of phosphorylation of these three residues in combination with the loss of Thr4 phosphorylation. This study prompted the development of antibodies recognizing the phosphorylated residues of the cotransporter and the use of NCC phosphorylation level as an index of NCC activity in vivo.
Like any transporter or channel, NCC activity can be regulated by modifying its transport capacity or its insertion at the plasma membrane. By performing immuno-electron microscopy on rat kidneys, the group of A. McDonough indeed showed that phosphorylated NCC is found only in the apical membrane while total NCC is found both in the apical membrane and in intracellular vesicles [82]. Whether phosphorylation of the five aforementioned residues affects one or the other or both is still a matter of debate, as reviewed in [83]. The group of G. Gamba showed in Xenopus laevis oocytes than the mutation of the phosphorylated residues to alanine does not affect the cell surface expression of the cotransporter [81]. In addition, a NCC cDNA bearing mutations in all three residues fails to be activated by intracellular chloride depletion, which strongly activates wild-type NCC [81]. These results thus suggest that phosphorylation regulates NCC activity and/or sensitivity to intracellular chloride concentration but not its insertion at the apical membrane. However, Richardson and collaborators showed that the transfection of a human cDNA bearing a mutation of the Thr4 residue into alanine in HEK293 cells prevents the insertion of NCC at the plasma membrane [84]. This study therefore suggests that phosphorylation of the N-terminal residues stimulates NCC activity only by increasing its insertion into the plasma membrane [82].
The kinases that phosphorylate NCC were once more identified by homology with NKCC1. In 2002, the group of E. Delpire identified the SPAK (Ste20-related proline-alanine-rich kinase) and OSR1 (oxidative stress response 1) kinases through a yeast two-hybrid screen using KCC3 as a bait [85]. It was then shown that both kinases can also bind NKCC1, NKCC2, and NCC. SPAK and OSR1 phosphorylate NCC-activating residue (Thr4) [80] as well as Thr1, Thr3, and Ser2. The kinase(s) responsible for NCC phosphorylation on Thr2 and Ser1 remain(s) to be identified.
SPAK and OSR1 are both expressed in the DCT but also expressed in the Thick Ascending Limb of Henle’s loop, consistent with a role in the regulation of NCC and NKCC2 [56]. The importance of SPAK for NCC phosphorylation in vivo was demonstrated by the characterization of several mouse models, in which SPAK is either knocked-out [86, 87] or bears a missense mutation that prevents its activation (see below; [88] ). In all cases, NCC phosphorylation is dramatically reduced (by 60–85 %), which results in the development of a Gitelman-like syndrome in mutant mice, with decreased blood pressure, hypokalemia, and hypocalciuria. Importantly, these studies show that OSR1 cannot compensate for the lack of SPAK and thus probably plays only a very minor role in NCC regulation in vivo. This is supported by the fact that NCC expression and phosphorylation are increased rather than decreased in OSR +/− mice, which display a 50 % reduction in OSR1 expression [89].
However, SPAK is, as its substrates, activated by phosphorylation and yeast two-hybrid screens identified WNK1 and WNK4 as responsible for SPAK phosphorylation [90, 91]. WNK1 and WNK4 belong to the WNK (With No lysine (K)) subfamily of serine-threonine kinases and became the focus of numerous studies related to NCC regulation when mutations in the WNK1 and WNK4 genes were found in patients affected by familial hyperkalemic hypertension (FHHt) [47]. This rare Mendelian disorder is characterized by moderate hypertension, hyperkalemia, and hyperchloremic metabolic acidosis. One of the trademarks of the disease is the sensitivity of patients to a very low dose of thiazides. FHHt was therefore believed to be the consequence of NCC activation. Consistent with this hypothesis, WNK1 and WNK4 are both expressed in the DCT [92, 93]. In addition, this hypothesis was confirmed by the characterization of two FHHt mouse models, expressing a mutated WNK4 cDNA, which display increased NCC expression and phosphorylation [9, 10].
The mechanisms by which WNK1 and WNK4 could regulate NCC phosphorylation have been quite extensively studied, mainly in vitro, but many results remain controversial, even though they were obtained in similar models. As mentioned above, WNK1 and WNK4 both bind and phosphorylate SPAK. Phosphopeptide mapping studies demonstrated that WNK1 phosphorylates SPAK at a residue located within the T-loop of the catalytic domain (Thr233 in human SPAK) and a serine residue located within a C-terminal non-catalytic region (Ser373 in SPAK) [91]. Further studies showed that phosphorylation of the T-loop residue is sufficient to activate SPAK, as its mutation into alanine impairs NCC phosphorylation both in vitro and in vivo [80, 88]. The role of the second phosphorylated residue remains unclear [91]. These studies strongly suggest the existence of a WNK1-SPAK-NCC phosphorylation cascade in the DCT, in which NCC is activated by phosphorylation by SPAK, itself activated by phosphorylation by WNK1. We confirmed this hypothesis in vivo, in a mouse model harboring an activation of WNK1 [94]. We observed an increased phosphorylation of SPAK near the apical membrane of DCT cells in the mutant mice, while it was more diffuse in the cytoplasm of control DCTs. This observation suggests that phosphorylation by WNK1 may be required for bringing the SPAK kinase closer to its substrate NCC, in the subapical compartment, thus allowing the phosphorylation and membrane insertion of the co-transporter.
The regulation of NCC phosphorylation by WNK4 appears more complex. Studies performed in Xenopus laevis oocytes showed that WNK4 inhibits NCC activity [48, 95] by reducing its membrane insertion through enhanced lysosomal degradation (see below). These in vitro studies were first confirmed by in vivo studies. A mouse transgenic mouse model overexpressing WNK4 indeed exhibits decreased NCC expression [9]. These results are, however, in contradiction with studies showing that WNK4 can phosphorylate SPAK in vitro, even if to a lesser extent than WNK1 [91, 96]. The situation became even more complex with the characterization of a WNK4 knock-out model and a new transgenic model of WNK4 overexpression. WNK4 −/− mice indeed display a dramatic reduction in NCC phosphorylation and expression and thus a Gitelman-like syndrome [97], similar to what is observed in SPAK mutant mice [86–88]. The group of S. Uchida very recently generated a new transgenic model of WNK4 overexpression: surprisingly, this model displays the exact opposite phenotype of the previous one, i.e., increased NCC expression and phosphorylation [98]. Taken together, these two in vivo studies suggest that WNK4 is an activator of NCC, rather than an inhibitor. Unfortunately, no clear explanation has been found yet for these contradictory results. One hypothesis is that WNK4 could exhibit positive or negative effects on NCC activity depending on the physiological situation and that the net effect could depend upon the expression level of WNK4 relative to WNK1, as they have been shown to interact through their carboxy-terminal domain and phosphorylate each other in vitro [99]. A recent study by Na and collaborators [100] supports the “dual effect” of WNK4 towards NCC. The authors characterized the sensitivity of WNK4 kinase activity to intracellular calcium concentration. The initial hypothesis was that WNK4 missense mutations identified in FHHt patients could modify this sensitivity. Most of the mutations are indeed located in an acidic motif, rich in negatively charged amino-acid residues, and result in an alteration of the negative charge [47]. The negatively charged acidic domain could act as a calcium-sensor and its mutations could modify its sensitivity to Ca2+ ions. Na and collaborators first showed that OSR1 phosphorylation by WNK4 is stimulated when Ca2+ concentration increases. This change in kinase activity is not observed when a WNK4 FHHt-mutant is used [100]. These data suggest that WNK4 could be switched from an inhibitory or at least from a “weak activator” mode to an activator mode when intracellular calcium concentration increases. WNK1 kinase activity could be similarly stimulated as the acidic domain is extremely conserved between members of the WNK1 family. These observations are supported by in vivo studies. SPAK phosphorylation is indeed increased in mouse models expressing a WNK4 mutant, which display all the clinical signs of FHHt [10]. Furthermore, the inactivation of SPAK in these mice corrects their blood pressure and biological phenotype [101].
An increase in intracellular calcium concentration is known to be induced by angiotensin II (angII), which could therefore change WNK4 kinase activity. This is particularly interesting in the context of the results obtained by San-Cristobal and collaborators, who showed that NCC inhibition by WNK4 is abrogated by angiotensin II in a SPAK-dependent manner in Xenopus oocytes [69]. Accordingly, SPAK phosphorylation is stimulated by angII in vitro and in vivo [69, 102]. In addition, the stimulation of NCC phosphorylation by angII is abrogated by WNK4 inactivation in mice [97]. The characterization of the activation status of WNK1 and WNK4 during angII treatment is hampered by the lack of antibodies recognizing the phosphorylated activated form of the kinases in the mouse kidney. Similarly, the implication of WNK1 in angII-mediated stimulation of NCC is hampered by the lack of a pertinent mouse model, as WNK1 knock-out leads to embryonic death, caused by cardiovascular development defects [103]. The study of Na and collaborators thus provides a mechanism by which angII could activate NCC through the WNK-SPAK cascade [100].
Several years ago, it was shown that aldosterone also activates NCC [41]. This again could be mediated by the WNK-SPAK cascade. The team of E.J. Hoorn indeed showed that SPAK phosphorylation and abundance are increased in adrenalectomized rats receiving chronic aldosterone infusion and losartan treatment, thus permitting the characterization of the effects of aldosterone alone on NCC regulation [71]. They also showed that WNK4 abundance is increased by this treatment.
In conclusion, we have gained a lot of information regarding the regulation of NCC abundance and/or activity by phosphorylation over the past decade. However, crucial questions remain, especially regarding the physiological situations in which the different kinases are stimulated and by which hormone(s). In particular, the observation that aldosterone could activate NCC is puzzling. Aldosterone is indeed secreted in response to sodium depletion or potassium load, two situations which require opposite regulations of NCC. While NCC needs to be activated during sodium depletion, it has to be downregulated during potassium load (see above). How aldosterone leads to opposite changes in NCC phosphorylation during these physiological challenges remains to be understood.
NCC regulation by degradation
Studies in Xenopus oocytes showed that WNK4 inhibits NCC activity by reducing its surface expression [48]. This could be achieved by stimulating the endocytosis or by attenuating the surface delivery rate of the cotransporter. Two groups first showed that clathrin-dependent endocytosis is not involved in WNK4-mediated inhibition of NCC [104, 105], thus favoring the second hypothesis. This was confirmed by direct measurements of NCC forward trafficking, which revealed that WNK4 inhibits the anterograde movement of cotransporters traveling to the plasma membrane from the trans-Golgi network [106]. This is achieved through an increased interaction of NCC with the lysosomal-targeting receptor sortilin [107] and the AP-3 adaptor complex, which facilitates cargo transport to lysosomes [106].
A second set of studies, however, showed that WNK4 could also stimulate NCC endocytosis. Like many other transporters, NCC surface expression is reduced by the phorbol ester TPA. Ko and collaborators [108] showed that TPA does not exert this effect through the classical PKC pathway but via activation of the Ras-guanyl-releasing protein RasGRP1, resulting in downstream activation of ERK1/2. Phosphorylated ERK1/2 then stimulates the ubiquitination and dynamin-dependent endocytosis of NCC. Interestingly, the team of H. Cai showed that WNK4 also stimulates ERK1/2 phosphorylation [109]. The in vivo relevance of this pathway was assessed in the rats fed a low- or high-NaCl diet. A low-NaCl diet decreases while a high-NaCl diet increases ERK1/2 phosphorylation [110]. Taken together, these studies suggest that WNK4 could stimulate NCC ubiquitination and endocytosis via an ERK1/2-dependent pathway.
The regulation of NCC surface expression by ubiquitination is reminiscent of that of the epithelial sodium (Na) channel ENaC. ERK1/2 phosphorylation indeed facilitates the interaction of the β- and γ-subunits of the channel with the ubiquitin ligase Nedd4-2, thereby promoting the ubiquitination, endocytosis, and proteosomal degradation of the channel [111]. It was recently demonstrated that NCC is also ubiquitinated by Nedd4-2 [112]. As for ENaC, sgk1 prevents NCC ubiquitination by phosphorylating and thus inhibiting Nedd4-2. The importance of NCC regulation through Nedd4-2-dependent degradation was confirmed in vivo in mice bearing a nephron-specific inactivation of the ubiquitin ligase, which display increased NCC expression [113]. This pathway once more links NCC to aldosterone. It is indeed well known that sgk-1 expression and phosphorylation are induced by aldosterone, thus leading to increased ENaC surface expression and activity. The inhibition of Nedd-4-2 dependent ubiquitination of NCC could therefore contribute to the activation of the cotransporter by aldosterone.
Identification of a NCC-like transport system in the renal β-intercalated cells
Overview of the mechanisms of chloride absorption by the connecting tubule and the cortical collecting duct
In contrast to the DCT, which is composed of a single cell type, the downstream segments (i.e., the CNT and CCD) are characterized by a cellular heterogeneity. They harbor a mixture of three main cell types: the principal/CNT cells (PCs), the α- and β-intercalated cells (ICs). Until recent studies, Na+ and Cl− transport in the CNT and CCD were thought to be achieved and regulated independently. Na+ reabsorption was thought to be exclusively achieved through ENaC working in tandem with the basolateral sodium pump (Na+/K+ P-ATPase), both expressed by CNT cells and principal cells of CCD. In this paradigm, Cl− transport does not occur through PCs but rather through the paracellular route or through ICs [114], where it is closely related to bicarbonate transport [115].
Non α-intercalated cells (i.e., β-ICs and non α- non β-ICs) express an electroneutral Na+-independent Cl−/HCO3 − exchanger at the apical membrane, which has been identified as pendrin (Pds), the product of the SlC26A4 gene [116]. Cl− absorption in the mouse CCD is eliminated with genetic ablation of Slc26a4 [117]. Conversely, Cl− absorption is increased in CCDs of mice overexpressing Pds in ICs [118]. Thus, in non α-ICs, apical uptake of Cl− occurs through pendrin, while basolateral efflux is likely mediated by the ClC-K Cl− channel (ClC-KB in humans, Clc-k2 in rodents) associated to barttin, a regulatory sub-unit [119, 120]. The potassium chloride cotransporter KCC4, located at the basolateral plasma membrane in intercalated cells [121, 122], also appears to facilitate Cl− exit in α-IC [121]. Whether KCC4 is also expressed in non α-IC and participate to Cl− absorption in these cells is currently unsettled.
In the paracellular reabsorptive process, Cl− transport is driven by the transepithelial voltage difference (V te) generated by electrogenic Na+ absorption through ENaC [123]. The contribution of the amiloride-sensitive (i.e., ENaC-dependent) component of Cl− absorption is variable between studies (see Table 1). In CCDs isolated from NaCl-restricted mice, even though amiloride eliminated both the V te and K+ secretion, it had no effect on transepithelial Cl− absorption [124], indicating that virtually all Cl− take the transcellular rather than the paracellular route. Cl− absorption was not observed in the CCD of pendrin-null mice treated with DOCP and supplemented with NaHCO3, indicating that Cl− absorption in the CDD under these conditions was completely dependent on pendrin [117]. However, in perfused CCDs isolated from deoxycorticosterone pivalate-treated rats, in the presence of vasopressin, amiloride, which completely eliminated the lumen-negative voltage, decreased chloride absorption by ~50 % [125]. We observed similar results in mice treated with deoxycorticosterone pivalate, ~50 % of the transepithelial Cl absorption was insensitive to amiloride (unpublished results). In contrast, in isolated and perfused CCDs from aldosterone-treated mice in the presence of angiotensin II in the bath solution, benzamil, a derivative of amiloride, reduced Cl− absorption by 66 % and reduced lumen-negative V te by 75 % [126]. Differences in relative contributions of the paracellular Cl− pathway between studies might result from differences in the physiological state of the tubules due to different in vivo and ex vivo conditions. To this regard, previous studies support the notion that chronic deoxycorticosterone treatment causes a decrease in the Cl− conductance of the paracellular pathway [127]. Recent studies proposed that claudins, transmembrane proteins of tight junctions, are modulators of the permeability properties of the paracellular pathway. Claudin-4, -7, and -8 are expressed in the collecting duct. Studies of ion permeability and selectivity using overexpression or knock-down of claudin-7 in cell cultures led to controversial results [128, 129]. Claudin-7-deficient mice have renal salt wasting and chronic dehydration, suggesting that claudin-7 is crucial for the barrier function of the tight junction [130]. Based on studies in cell culture, claudin-4 is thought to form a paracellular pore. siRNA knockdown of claudin-4 or claudin-8 in cultured mouse collecting duct cells significantly decreased the paracellular Cl− permeability without affecting the Na+ permeability [131]. Claudin-8 was not found to affect Cl− permeability by itself but rather to be necessary for the recruitment of claudin-4 to tight junctions [131]. Claudin-4-deficient mice have been recently generated. No conclusion regarding claudin-4 and its paracellular Cl− channel function in native tissue could be drawn from these mice as they develop lethal hydronephrosis and obstructive uropathy due to urothelial hyperplasia [132]. Acute aldosterone treatment modulates claudin-4 phosphorylation and increased paracellular Cl− conductance in cultured rat cortical collecting duct cells [133]. Aldosterone also upregulates claudin-8 transcription in the distal colon [134]. If the expression of the gene encoding claudin-8 is regulated similarly in the ASDN as in the distal colon, aldosterone is expected to upregulate claudin-8 and to increase paracellular Cl− conductance. Taken together, claudin-4 provides a potential molecular mechanism for coupling paracellular Cl− transport to Na+ reabsorption in the collecting duct in response to aldosterone stimulation. Abnormal increases in paracellular Cl− absorption across the tight junction in the collecting duct was first advanced to explain FHHt [135, 136]. The serine threonine kinase WNK4, especially the mutant WNK4 which produces FHHt, can phosphorylate claudin-4 [137] or claudin-7 [138] and promote paracellular Cl− permeability in cultured cells [137–139]. This is compatible with the original hypothesis that a gain-of-function in chloride shunt conductance could cause the syndrome. However, transgenic mouse models harboring the FHHt mutations reveal no difference in paracellular Cl− permeability of the collecting duct [10].
Identification of a novel electroneutral thiazide-sensitive NaCl transport system in the intercalated cells
Studies performed in the 1990s found that even though the expression of NCC is restricted to the DCT, approximately 50 % of Na+ and Cl− absorption in the rat CCD is blocked by thiazides, a compound that does not target ENaC [125, 140, 141]. Using an approach combining the use of different mouse models bearing a genetic ablation of sodium transporters along the distal nephron with physiological studies, we demonstrated that thiazide-sensitive NaCl absorption in the CCD results from the functional coupling of two bicarbonate transporters: pendrin and the Na+-driven Cl−/HCO3 − exchanger (NDCBE/SLC4A8) [124, 142]. Experiments conducted in isolated and perfused CCDs to access whether thiazides inhibit amiloride-resistant NaCl absorption by blocking NDCBE, and/or pendrin demonstrated that thiazides block NDCBE and pendrin in intact tubules [124]. The latter study was also consistent with the classical view that the apical epithelial Na+ channel ENaC and the apical K+ channel ROMK are responsible for Na+/K+ exchange in PCs of the CCD. Indeed, in CCDs isolated from Na+-depleted mice, thiazides completely abolished chloride absorption but did not affect V te and K+ secretion, while amiloride had the converse effects.
Pendrin/NDCBE-dependent NaCl absorption by intercalated cells is energized by a proton pump
The identification of an electroneutral NaCl absorption by two bicarbonate transporters in ICs raises several issues. Indeed, the luminal bicarbonate concentration in the CNT and CCD is expected to be very low due to avid reabsorption of bicarbonate in the proximal tubule and the loop of Henle. Hence, one can assume that the bicarbonate required for sustaining NDCBE activity comes from active bicarbonate secretion by pendrin. Moreover, chloride accumulation into the cells through pendrin is expected to favor sodium and bicarbonate uptake via NDCBE. Pendrin has been shown to be energized by an outwardly directed bicarbonate gradient, which results from primary active proton extrusion by the H+ V-ATPase [126, 143]. Moreover, ICs are thought to have very low Na+/K+ P-ATPase activity [144]. These considerations raise the question of the dependence of transepithelial NaCl absorption in β-ICs on either the Na+/K+ P-ATPase or the H+ V-ATPase. To address this issue, we tested the effect of ouabain, a blocker of the Na+/K+ P-ATPase, or bafilomycin A1, a blocker of the H+ V-ATPase, on NaCl absorption by ICs. The Na+ flux in CCDs was only partially inhibited by either amiloride or ouabain. The simultaneous application of both blockers did not lead to significant additive effects, demonstrating that ouabain alone is sufficient to block the amiloride-sensitive component of Na+ absorption (i.e., ENaC activity) but does not affect amiloride-resistant Na+ transport (i.e., Pds/NDCBE activity). Conversely, Cl− transport was not affected by application of amiloride, ouabain, or simultaneous application of both compounds. By contrast, basolateral application of bafilomycin A1 fully inhibited the amiloride-resistant component of Na+ and Cl− absorption. These experiments demonstrate that Na+ absorption by principal cells is primarily energized by the Na+/K+ P-ATPase, whereas NaCl transepithelial absorption by β-ICs is energized by the H+ V-ATPase.
The putative anion exchanger AE4/SLC4A9 is involved in NaCl absorption by intercalated cells
The aforementioned studies also indicate that basolateral NaCl exit from β-ICs is independent of the Na+/K+ P-ATPase. In the absence of the Na+/K+ P-ATPase, the parallel action of pendrin and NDCBE is predicted to lead to net accumulation of Na+ and HCO3 − into the cell. Thus, we tested whether Na+ transport across the basolateral membrane of β-ICs could be mediated by a bicarbonate-dependent sodium transporter. AE4, encoded by the SLC4A9 gene, has been reported to be specifically expressed in β-ICs [145]. The localization and transport characteristics of AE4 were to some extent controversial. First described as a 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS)-insensitive Na+-independent Cl−/HCO3 − exchanger, AE4 shares more similarities with Na+–HCO3 − cotransporters than with anion exchangers of the SLC4 superfamily [146, 147]. Subsequently, AE4 was reported to be rather DIDS sensitive [148]. Finally, others suggested that AE4 might mediate Cl−-independent Na+–HCO3 − cotransport rather than Cl−/HCO3 − exchange [147, 149].
The subcellular localization and function of AE4, and its potential role in Na+ extrusion across the basolateral membrane of ICs, were assessed using Slc4a9 disrupted mice. AE4 is exclusively detected at the basolateral membrane of β-ICs [144]. Experiments performed on isolated CCDs from Slc4a9 +/+ and Slc4a9 −/− mice demonstrated that AE4 mediates basolateral Na+–HCO3 − cotransport when expressed in its normal environment [144]. Furthermore, we also confirmed that it mediates sodium extrusion from renal β-ICs as AE4 inactivation, like NDCBE, blocked amiloride-resistant NaCl absorption by these cells [124, 144].
A new paradigm of ion transport by the collecting duct
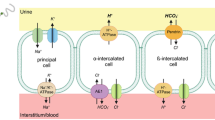
Based on these studies, we propose a new model for Na+, Cl−, and K+ transport in the CCD. Principal cells mediate Na+ reabsorption in exchange for K+ and this process is energized by the outwardly directed gradient of Na+ generated by the Na+/K+ P-ATPase. In intercalated cells, the H+ pump favors the generation of HCO3 − by extruding H+. This generates an outwardly directed HCO3 − gradient that in turn drives uphill accumulation of Cl− into the cell. Then, the outwardly directed Cl− gradient drives the uptake of 1 Na+ and 2 HCO3 − ions. The basolateral efflux of Cl− might occur through a Cl− channel or a KCl cotransporter while Na+(HCO3 −)n efflux occurs via AE4 (Fig. 1).

Electroneutral sodium chloride transport in the distal nephron. Two main electroneutral NaCl transport pathways are found in the distal nephron. a In the distal convoluted tubule (DCT), NaCl uptake is mediated by the NaCl cotransporter (NCC) at the apical pole of the DCT cells. Na+ transport is energized by the basolateral sodium/potassium ATPase (Na/K ATPase). Several channels and transporters are known to participate in NaCl transport. The potassium channel Kir4.1 (Kcnj10) recycles K+ across the basolateral membrane to support Na/K ATPase activity and the Cl− channel Clcnk2 and its regulatory subunit Barttin account for the chloride exit across the basolateral membrane. b In the β-intercalated cells of the connecting tubule and cortical collecting duct, a second electroneutral NaCl transport pathway has been identified. It involves the apical Cl−/HCO3 − exchanger pendrin (PDS) and the Na+-driven Cl−/HCO3 − exchanger (NDCBE). The basolateral Na+ exit is mediated by the NaHCO3 cotransporter Slc4a9 (AE4). The mechanism of chloride exit is still unknown but could involve the KCL cotransporter KCC4 and also, like in DCT cells, the Cl− channel Clcnk2 and its regulatory subunit Barttin. In this case, NaCl transport is energized by the basolateral vacuolar proton pump (vATPase) and not by the Na/K ATPase
Physiological relevance of NaCl absorption by intercalated cells
The role of pendrin in both the maintenance of chloride balance and the regulation of blood pressure is supported by expression and functional in vivo studies. Pendrin expression is primarily and inversely regulated by dietary chloride intake [150] and by factors associated with changes in distal chloride delivery [151]. Furthermore, and like NCC, pendrin expression is stimulated by components of the renin–angiotensin–aldosterone system. Accordingly, targeted inactivation of pendrin induces hypotension [152], which is aggravated when the animals are fed a NaCl-depleted diet [117]. Pendrin disruption also protects the mice against mineralocorticoid-induced hypertension [153]. Similarly, mice with disruption of the gene encoding the B1 subunit of the proton pump, which also exhibit very low level of pendrin expression, display a renal loss of NaCl causing hypovolemia and lower blood pressure [154]. Conversely, we recently published results showing that mice overexpressing pendrin in ICs develop salt-sensitive hypertension [118]. They exhibit a delayed increase in urinary NaCl and ultimately develop hypertension when exposed to a high-salt diet, indicating that a primary abnormality of renal chloride reabsorption can also lead to NaCl-sensitive hypertension.
The involvement of NDCBE in renal Na+ handling has not been assessed yet. In a previous study, we showed that NDCBE-deficient mice fed a Na+-depleted diet could not upregulate the amiloride-resistant, thiazide-sensitive NaCl reabsorption pathway in the cortical collecting duct [124]. As this system would tend to enhance Na+ retention, one can expect NDCBE dysfunction to be associated with volume depletion.
The key observations published up to now, which led to the conclusion that a luminal amiloride-resistant, thiazide-sensitive NaCl transport in the CCD exists and is accomplished by the parallel action of the Cl−/HCO3 − exchanger pendrin and the Na+-driven Cl−/2HCO3 − exchanger (NDCBE/Slc4a8), and is important in maintaining Na+ balance, are summarized in Table 1.
Crosstalk between β-intercalated and principal cells
During NaCl restriction, pendrin-null mice excrete more Na+ and Cl− than wild-type mice and therefore display an apparent vascular volume contraction and lower blood pressure. Higher natriuresis in pendrin-deficient mice after either dietary NaCl restriction or administration of aldosterone was associated with decreased ENaC expression [152]. It was also shown that in mice given furosemide and a high-salt diet, conditions known to increase ENaC function, ENaC-mediated current was lower in CCDs from pendrin-null mice than from wild-type mice [152]. It has therefore been proposed that pendrin could also work in tandem with ENaC to reabsorb NaCl. However, since pendrin and ENaC are expressed in two different types of cell, this should involve modulation of ENaC activity by an extra-cellular signal. Pech et al. [155] tested the hypothesis that this signal is mediated by luminal bicarbonate. They showed that in pendrin-null mice, increasing distal delivery of bicarbonate restores ENaC activity by increasing β- and γ-ENaC protein abundance and, more importantly, γ-ENaC proteolytic cleavage, a process associated with an increase in the channel activity [156]. The authors came to the conclusion that luminal alkalinization due to HCO3 − secretion by pendrin could enhance ENaC in PCs. This is in line with recent studies by Gueutin et al. [154]. In this study, isolated and perfused CCDs from mice lacking the B1 subunit of the v-H+-ATPase (Atp6v1b1 −/− mice), which were shown to develop hypovolemia, did not absorb NaCl and did not develop lumen-negative transepithelial voltage, indicating that ENaC and NDCBE/pendrin activities were both impaired [154]. Of interest, in these mice, pendrin expression was virtually suppressed and ENaC expression was decreased specifically in the cortex as in pendrin-null mice [152]. The authors described a new mechanism that can fully explain these observations; they demonstrated that blockade of the basolateral v-H+-ATPase in β-ICs leads to ATP release, which in turn triggers PGE2 release by acting on luminal calcium-coupled purinergic receptors, presumably P2Y2 receptors, resulting in inhibition of ENaC in neighboring PCs. In summary, these studies introduce a new paradigm of crosstalk between PC and IC and provide further evidence that both cell types are important in maintaining Na+ balance and thus blood pressure.
In conclusion, there are two thiazide-sensitive systems mediating electroneutral NaCl reabsorption in the distal nephron, and not one as originally thought. The first one consists of one co-transporter, NCC, and is present exclusively in the distal convoluted tubule. The second one consists of two exchangers, NDCBE and pendrin, in the β-intercalated cells of the connecting tubule and cortical collecting duct. If the inactivation of one of these systems can be compensated for by the other, as observed in Ncc −/− mice [124], the combined deletion of NCC and pendrin causes severe salt wasting, volume depletion, and renal failure [157]. This last observation highlights the crucial part played by the electroneutral Na–Cl reabsorption in the maintenance of NaCl balance and hence blood pressure. Recent studies introduce a new paradigm of crosstalk between PCs and ICs and provide evidence that β-ICs are important in maintaining Na+ balance and thus normal blood pressure also by controlling ENaC activity in neighboring PCs through release of paracrine factors.
References
Guyton AC (1991) Blood pressure control—special role of the kidneys and body fluids. Science 252(5014):1813–1816
Lifton RP, Gharavi AG, Geller DS (2001) Molecular mechanisms of human hypertension. Cell 104(4):545–556
Skou JC (1957) The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta 23(2):394–401
Skou JC (1998) Nobel Lecture. The identification of the sodium pump. Biosci Rep 18(4):155–169
Bourdeau JE, Burg MB (1979) Voltage dependence of calcium transport in the thick ascending limb of Henle’s loop. Am J Physiol Renal Physiol 236(4):F357–F364
Verrey F, Hummler E, Schild L et al (2008) Mineralocorticoid action in the aldosterone-sensitive distal nephron. In: Alpern RJ, Hebert SC (eds) The kidney: physiology and pathophysiology, 4th edn. MA Academic, Burlington, p 889–924
Gitelman HJ, Graham JB, Welt LG (1966) A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians 79:221–235
Velazquez H, Wright FS (1986) Control by drugs of renal potassium handling. Annu Rev Pharmacol Toxicol 26:293–309
Lalioti MD, Zhang J, Volkman HM et al (2006) Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38(10):1124–1132
Yang SS, Morimoto T, Rai T et al (2007) Molecular pathogenesis of pseudohypoaldosteronism type II: generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 5(5):331–344
Kellenberger S, Schild L (2002) Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82(3):735–767
Palmer LG, Patel A, Frindt G (2012) Regulation and dysregulation of epithelial Na+channels. Clin Exp Nephrol 16(1):35–43
Rossier BC, Stutts MJ (2009) Activation of the epithelial sodium channel (ENaC) by serine proteases. Annu Rev Physiol 71:361–379
Butterworth MB, Edinger RS, Frizzell RA et al (2009) Regulation of the epithelial sodium channel by membrane trafficking. Am J Physiol Renal Physiol 296(1):F10–F24
Beyer KH Jr, Baer JE, Russo HF et al (1958) Electrolyte excretion as influenced by chlorothiazide. Science 127(3290):146–147
Novello FC, Sprague JM (1957) Benzothiadiazine dioxides as novel diuretics. J Am Chem Soc 79:2028
Leslie BR, Schwartz JH, Steinmetz PR (1973) Coupling between Cl− absorption and HCO3− secretion in turtle urinary bladder. Am J Physiol 225(3):610–617
Frizzell RA, Koch MJ, Schultz SG (1976) Ion transport by rabbit colon. I. Active and passive components. J Membr Biol 27(3):297–316
Renfro JL (1977) Interdependence of Active Na+ and Cl− transport by the isolated urinary bladder of the teleost Pseudopleuronectes americanus. J Exp Zool 199(3):383–390
Costanzo LS, Windhager EE (1978) Calcium and sodium transport by the distal convoluted tubule of the rat. Am J Physiol Renal Physiol 235(5):F492–F506
Hansen LL, Schilling AR, Wiederholt M (1981) Effect of calcium, furosemide and chlorothiazide on net volume reabsorption and basolateral membrane potential of the distal tubule. Pflugers Arch: Eur J Physiol 389(2):121–126
Kunau RT Jr, Weller DR, Webb HL (1975) Clarification of the site of action of chlorothiazide in the rat nephron. J Clin Invest 56(2):401–407
Ellison DH, Velazquez H, Wright FS (1987) Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol 253(3 Pt 2):F546–F554
Goldfarb DS, Chan AJ, Hernandez D et al (1991) Effect of thiazides on colonic NaCl absorption: role of carbonic anhydrase. Am J Physiol 261(3 Pt 2):F452–F458
Stokes JB (1984) Sodium chloride absorption by the urinary bladder of the winter flounder. A thiazide-sensitive, electrically neutral transport system. J Clin Invest 74(1):7–16
Beaumont K, Vaughn DA, Fanestil DD (1988) Thiazide diuretic drug receptors in rat kidney: identification with [3H]metolazone. Proc Natl Acad Sci USA 85(7):2311–2314
Beaumont K, Vaughn DA, Healy DP (1989) Thiazide diuretic receptors: autoradiographic localization in rat kidney with [3H]metolazone. J Pharmacol Exp Ther 250(1):414–419
Tran JM, Farrell MA, Fanestil DD (1990) Effect of ions on binding of the thiazide-type diuretic metolazone to kidney membrane. Am J Physiol 258(4 Pt 2):F908–F915
Chen ZF, Vaughn DA, Beaumont K et al (1990) Effects of diuretic treatment and of dietary sodium on renal binding of 3H-metolazone. J Am Soc Nephrol 1(1):91–98
Morsing P, Velazquez H, Wright FS et al (1991) Adaptation of distal convoluted tubule of rats. II. Effects of chronic thiazide infusion. Am J Physiol 261(1 Pt 2):F137–F143
Ellison DH, Morrisey J, Desir GV (1991) Solubilization and partial purification of the thiazide diuretic receptor from rabbit renal cortex. Biochim Biophys Acta 1069(2):241–249
Ellison DH, Biemesderfer D, Morrisey J et al (1993) Immunocytochemical characterization of the high-affinity thiazide diuretic receptor in rabbit renal cortex. Am J Physiol 264(1 Pt 2):F141–F148
Gamba G, Saltzberg SN, Lombardi M et al (1993) Primary structure and functional expression of a cDNA encoding the thiazide-sensitive, electroneutral sodium-chloride cotransporter. Proc Natl Acad Sci USA 90(7):2749–2753
Xu JC, Lytle C, Zhu TT et al (1994) Molecular cloning and functional expression of the bumetanide-sensitive Na–K–Cl cotransporter. Proc Natl Acad Sci USA 91(6):2201–2205
Hebert SC, Gamba G, Kaplan M (1996) The electroneutral Na(+)–(K+)–Cl− cotransport family. Kidney Int 49(6):1638–1641
Gamba G, Miyanoshita A, Lombardi M et al (1994) Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium–(potassium)–chloride cotransporter family expressed in kidney. J Biol Chem 269(26):17713–17722
Gillen CM, Brill S, Payne JA et al (1996) Molecular cloning and functional expression of the K–Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem 271(27):16237–16244
Hiki K, D’Andrea RJ, Furze J et al (1999) Cloning, characterization, and chromosomal location of a novel human K+–Cl− cotransporter. J Biol Chem 274(15):10661–10667
Mount DB, Mercado A, Song L et al (1999) Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem 274(23):16355–16362
Payne JA, Stevenson TJ, Donaldson LF (1996) Molecular characterization of a putative K–Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem 271(27):16245–16252
Kim GH, Masilamani S, Turner R et al (1998) The thiazide-sensitive Na–Cl cotransporter is an aldosterone-induced protein. Proc Natl Acad Sci USA 95(24):14552–14557
Velazquez H, Bartiss A, Bernstein P et al (1996) Adrenal steroids stimulate thiazide-sensitive NaCl transport by rat renal distal tubules. Am J Physiol 270(1 Pt 2):F211–F219
Simon DB, Nelson-Williams C, Bia MJ et al (1996) Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na–Cl cotransporter. Nat Genet 12(1):24–30
Ji W, Foo JN, O’Roak BJ et al (2008) Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 40(5):592–599
Boyden LM, Choi M, Choate KA et al (2012) Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 482(7383):98–102
Louis-Dit-Picard H, Barc J, Trujillano D et al (2012) KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet 44(4):456–460 S451–S453
Wilson FH, Disse-Nicodeme S, Choate KA et al (2001) Human hypertension caused by mutations in WNK kinases. Science 293(5532):1107–1112
Wilson FH, Kahle KT, Sabath E et al (2003) Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na–Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci USA 100(2):680–684
Bachmann S, Velazquez H, Obermuller N et al (1995) Expression of the thiazide-sensitive Na–Cl cotransporter by rabbit distal convoluted tubule cells. J Clin Invest 96(5):2510–2514
Loffing J, Loffing-Cueni D, Valderrabano V et al (2001) Distribution of transcellular calcium and sodium transport pathways along mouse distal nephron. Am J Physiol Renal Physiol 281(6):F1021–F1027
Plotkin MD, Kaplan MR, Verlander JW et al (1996) Localization of the thiazide sensitive Na–Cl cotransporter, rTSC1 in the rat kidney. Kidney Int 50(1):174–183
Katz AI, Doucet A, Morel F (1979) Na–K-ATPase activity along the rabbit, rat, and mouse nephron. Am J Physiol 237(2):F114–F120
Ecelbarger CA, Kim GH, Wade JB et al (2001) Regulation of the abundance of renal sodium transporters and channels by vasopressin. Exp Neurol 171(2):227–234
Mutig K, Saritas T, Uchida S et al (2010) Short-term stimulation of the thiazide-sensitive Na+–Cl− cotransporter by vasopressin involves phosphorylation and membrane translocation. Am J Physiol Renal Physiol 298(3):F502–F509
Pedersen NB, Hofmeister MV, Rosenbaek LL et al (2010) Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int 78(2):160–169
Saritas T, Borschewski A, McCormick JA et al (2013) SPAK differentially mediates vasopressin effects on sodium cotransporters. J Am Soc Nephrol 24(3):407–418
Ko B, Cooke LL, Hoover RS (2011) Parathyroid hormone (PTH) regulates the sodium chloride cotransporter via Ras guanyl releasing protein 1 (Ras-GRP1) and extracellular signal-regulated kinase (ERK)1/2 mitogen-activated protein kinase (MAPK) pathway. Transl Res 158(5):282–289
Chavez-Canales M, Arroyo JP, Ko B et al (2013) Insulin increases the functional activity of the renal NaCl cotransporter. J Hypertens 31(2):303–311
Komers R, Rogers S, Oyama TT et al (2012) Enhanced phosphorylation of Na(+)–Cl− co-transporter in experimental metabolic syndrome: role of insulin. Clin Sci (Lond) 123(11):635–647
Sohara E, Rai T, Yang SS et al (2011) Acute insulin stimulation induces phosphorylation of the Na–Cl cotransporter in cultured distal mpkDCT cells and mouse kidney. PLoS One 6(8):e24277
Masilamani S, Kim GH, Mitchell C et al (1999) Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104(7):R19–R23
Bostanjoglo M, Reeves WB, Reilly RF et al (1998) 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na–Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9(8):1347–1358
Rozansky DJ, Cornwall T, Subramanya AR et al (2009) Aldosterone mediates activation of the thiazide-sensitive Na–Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 119(9):2601–2612
Wang XY, Masilamani S, Nielsen J et al (2001) The renal thiazide-sensitive Na–Cl cotransporter as mediator of the aldosterone-escape phenomenon. J Clin Invest 108(2):215–222
Sandberg MB, Riquier AD, Pihakaski-Maunsbach K et al (2007) ANG II provokes acute trafficking of distal tubule Na+–Cl(-) cotransporter to apical membrane. Am J Physiol Renal Physiol 293(3):F662–F669
Talati G, Ohta A, Rai T et al (2010) Effect of angiotensin II on the WNK-OSR1/SPAK-NCC phosphorylation cascade in cultured mpkDCT cells and in vivo mouse kidney. Biochem Biophys Res Commun 393(4):844–848
Brooks HL, Allred AJ, Beutler KT et al (2002) Targeted proteomic profiling of renal Na(+) transporter and channel abundances in angiotensin II type 1a receptor knockout mice. Hypertension 39(2 Pt 2):470–473
Gurley SB, Riquier-Brison AD, Schnermann J et al (2011) AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab 13(4):469–475
San-Cristobal P, Pacheco-Alvarez D, Richardson C et al (2009) Angiotensin II signaling increases activity of the renal Na–Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc Natl Acad Sci USA 106(11):4384–4389
Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK et al (2013) The absence of intrarenal ACE protects against hypertension. J Clin Invest 123(5):2011–2023
van der Lubbe N, Lim CH, Meima ME et al (2012) Aldosterone does not require angiotensin II to activate NCC through a WNK4-SPAK-dependent pathway. Pflugers Arch: Eur J Physiol 463(6):853–863
Sorensen MV, Grossmann S, Roesinger M et al (2013) Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83(5):811–824
Vallon V, Schroth J, Lang F et al (2009) Expression and phosphorylation of the Na+–Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297(3):F704–F712
Lee FN, Oh G, McDonough AA et al (2007) Evidence for gut factor in K+ homeostasis. Am J Physiol Renal Physiol 293(2):F541–F547
Rabinowitz L (1988) Model of homeostatic regulation of potassium excretion in sheep. Am J Physiol 254(2 Pt 2):R381–R388
Song J, Hu X, Shi M et al (2004) Effects of dietary fat, NaCl, and fructose on renal sodium and water transporter abundances and systemic blood pressure. Am J Physiol Renal Physiol 287(6):F1204–F1212
Gamba G (2005) Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev 85(2):423–493
Darman RB, Forbush B (2002) A regulatory locus of phosphorylation in the N terminus of the Na–K–Cl cotransporter, NKCC1. J Biol Chem 277(40):37542–37550
Vitari AC, Thastrup J, Rafiqi FH et al (2006) Functional interactions of the SPAK/OSR1 kinases with their upstream activator WNK1 and downstream substrate NKCC1. Biochem J 397(1):223–231
Richardson C, Rafiqi FH, Karlsson HK et al (2008) Activation of the thiazide-sensitive Na+–Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121(Pt 5):675–684
Pacheco-Alvarez D, Cristobal PS, Meade P et al (2006) The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem 281(39):28755–28763
Lee DH, Maunsbach AB, Riquier-Brison AD et al (2013) Effects of ACE inhibition and ANG II stimulation on renal Na–Cl cotransporter distribution, phosphorylation, and membrane complex properties. Am J Physiol Cell Physiol 304(2):C147–C163
Gamba G (2012) Regulation of the renal Na+–Cl− cotransporter by phosphorylation and ubiquitylation. Am J Physiol Renal Physiol 303(12):F1573–F1583
Richardson C, Sakamoto K, de los Heros P et al (2011) Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 124(Pt 5):789–800
Piechotta K, Lu J, Delpire E (2002) Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 277(52):50812–50819
McCormick JA, Mutig K, Nelson JH et al (2011) A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14(3):352–364
Yang SS, Lo YF, Wu CC et al (2010) SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21(11):1868–1877
Rafiqi FH, Zuber AM, Glover M et al (2010) Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med 2(2):63–75
Lin SH, Yu IS, Jiang ST et al (2011) Impaired phosphorylation of Na(+)–K(+)–2Cl(−) cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc Natl Acad Sci USA 108(42):17538–17543
Piechotta K, Garbarini N, England R et al (2003) Characterization of the interaction of the stress kinase SPAK with the Na+–K+–2Cl− cotransporter in the nervous system: evidence for a scaffolding role of the kinase. J Biol Chem 278(52):52848–52856
Vitari AC, Deak M, Morrice NA et al (2005) The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391(Pt 1):17–24
Delaloy C, Hadchouel J, Imbert-Teboul M et al (2006) Cardiovascular expression of the mouse WNK1 gene during development and adulthood revealed by a BAC reporter assay. Am J Pathol 169(1):105–118
Ohno M, Uchida K, Ohashi T et al (2011) Immunolocalization of WNK4 in mouse kidney. Histochem Cell Biol 136(1):25–35
Vidal-Petiot E, Elivra-Matelot E, Mutig K et al. (2014) WNK1-related familial hyperkalemic hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci USA 110(35):14366–14371
Yang CL, Angell J, Mitchell R et al (2003) WNK kinases regulate thiazide-sensitive Na–Cl cotransport. J Clin Invest 111(7):1039–1045
Wang Z, Yang CL, Ellison DH (2004) Comparison of WNK4 and WNK1 kinase and inhibiting activities. Biochem Biophys Res Commun 317(3):939–944
Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N et al (2012) Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109(20):7929–7934
Wakabayashi M, Mori T, Isobe K et al (2013) Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Rep 3(3):858–868
Thastrup JO, Rafiqi FH, Vitari AC et al (2012) SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem J 441(1):325–337
Na T, Wu G, Peng JB (2012) Disease-causing mutations in the acidic motif of WNK4 impair the sensitivity of WNK4 kinase to calcium ions. Biochem Biophys Res Commun 419(2):293–298
Chiga M, Rafiqi FH, Alessi DR et al (2011) Phenotypes of pseudohypoaldosteronism type II caused by the WNK4 D561A missense mutation are dependent on the WNK-OSR1/SPAK kinase cascade. J Cell Sci 124(Pt 9):1391–1395
van der Lubbe N, Lim CH, Fenton RA et al (2011) Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int 79(1):66–76
Zambrowicz BP, Abuin A, Ramirez-Solis R et al (2003) Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc Natl Acad Sci USA 100(24):14109–14114
Cai H, Cebotaru V, Wang YH et al (2006) WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int 69(12):2162–2170
Golbang AP, Cope G, Hamad A et al (2006) Regulation of the expression of the Na/Cl cotransporter by WNK4 and WNK1: evidence that accelerated dynamin-dependent endocytosis is not involved. Am J Physiol Renal Physiol 291(6):F1369–F1376
Subramanya AR, Liu J, Ellison DH et al (2009) WNK4 diverts the thiazide-sensitive NaCl cotransporter to the lysosome and stimulates AP-3 interaction. J Biol Chem 284(27):18471–18480
Zhou B, Zhuang J, Gu D et al (2010) WNK4 enhances the degradation of NCC through a sortilin-mediated lysosomal pathway. J Am Soc Nephrol 21(1):82–92
Ko B, Joshi LM, Cooke LL et al (2007) Phorbol ester stimulation of RasGRP1 regulates the sodium-chloride cotransporter by a PKC-independent pathway. Proc Natl Acad Sci USA 104(50):20120–20125
Zhou B, Wang D, Feng X et al (2012) WNK4 inhibits NCC protein expression through MAPK ERK1/2 signaling pathway. Am J Physiol Renal Physiol 302(5):F533–F539
Lai L, Feng X, Liu D et al (2012) Dietary salt modulates the sodium chloride cotransporter expression likely through an aldosterone-mediated WNK4-ERK1/2 signaling pathway. Pflugers Arch: Eur J Physiol 463(3):477–485
Shi H, Asher C, Chigaev A et al (2002) Interactions of beta and gamma ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J Biol Chem 277(16):13539–13547
Arroyo JP, Lagnaz D, Ronzaud C et al (2011) Nedd4-2 modulates renal Na+–Cl− cotransporter via the aldosterone-SGK1-Nedd4-2 pathway. J Am Soc Nephrol 22(9):1707–1719
Ronzaud C, Loffing-Cueni D, Hausel P et al (2013) Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest 123(2):657–665
Schlatter E, Greger R, Schafer JA (1990) Principal cells of cortical collecting ducts of the rat are not a route of transepithelial Cl− transport. Pflugers Arch: Eur J Physiol 417(3):317–323
Greger R (1988) Chloride transport in thick ascending limb, distal convolution, and collecting duct. Annu Rev Physiol 50:111–122
Royaux IE, Wall SM, Karniski LP et al (2001) Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 98(7):4221–4226
Wall SM, Kim YH, Stanley L et al (2004) NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl− conservation. Hypertension 44(6):982–987
Jacques T, Picard N, Miller RL et al (2013) Overexpression of pendrin in intercalated cells produces chloride-sensitive hypertension. J Am Soc Nephrol 24(7):1104–1113
Estevez R, Boettger T, Stein V et al (2001) Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 414(6863):558–561
Nissant A, Paulais M, Lachheb S et al (2006) Similar chloride channels in the connecting tubule and cortical collecting duct of the mouse kidney. Am J Physiol Renal Physiol 290(6):F1421–F1429
Boettger T, Hubner CA, Maier H et al (2002) Deafness and renal tubular acidosis in mice lacking the K–Cl co-transporter Kcc4. Nature 416(6883):874–878
Melo Z, Cruz-Rangel S, Bautista R et al (2013) Molecular evidence for a role for K(+)–Cl(−) cotransporters in the kidney. Am J Physiol Renal Physiol 305(10):F1402–F1411
Sansom SC, Weinman EJ, O’Neil RG (1984) Microelectrode assessment of chloride-conductive properties of cortical collecting duct. Am J Physiol 247(2 Pt 2):F291–F302
Leviel F, Hubner CA, Houillier P et al (2010) The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120(5):1627–1635
Terada Y, Knepper MA (1990) Thiazide-sensitive NaCl absorption in rat cortical collecting duct. Am J Physiol 259(3 Pt 2):F519–F528
Pech V, Thumova M, Kim YH et al (2012) ENaC inhibition stimulates Cl− secretion in the mouse cortical collecting duct through an NKCC1-dependent mechanism. Am J Physiol Renal Physiol 303(1):F45–F55
Sansom SC, O’Neil RG (1985) Mineralocorticoid regulation of apical cell membrane Na+ and K+ transport of the cortical collecting duct. Am J Physiol 248(6 Pt 2):F858–F868
Alexandre MD, Lu Q, Chen YH (2005) Overexpression of claudin-7 decreases the paracellular Cl− conductance and increases the paracellular Na+ conductance in LLC-PK1 cells. J Cell Sci 118(Pt 12):2683–2693
Hou J, Gomes AS, Paul DL et al (2006) Study of claudin function by RNA interference. J Biol Chem 281(47):36117–36123
Tatum R, Zhang Y, Salleng K et al (2010) Renal salt wasting and chronic dehydration in claudin-7-deficient mice. Am J Physiol Renal Physiol 298(1):F24–F34
Hou J, Renigunta A, Yang J et al (2010) Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc Natl Acad Sci USA 107(42):18010–18015
Fujita H, Hamazaki Y, Noda Y et al (2012) Claudin-4 deficiency results in urothelial hyperplasia and lethal hydronephrosis. PLoS One 7(12):e52272
Le Moellic C, Boulkroun S, Gonzalez-Nunez D et al (2005) Aldosterone and tight junctions: modulation of claudin-4 phosphorylation in renal collecting duct cells. Am J Physiol Cell Physiol 289(6):C1513–C1521
Amasheh S, Milatz S, Krug SM et al (2009) Na+ absorption defends from paracellular back-leakage by claudin-8 upregulation. Biochem Biophys Res Commun 378(1):45–50
Schambelan M, Sebastian A, Rector FC Jr (1981) Mineralocorticoid-resistant renal hyperkalemia without salt wasting (type II pseudohypoaldosteronism): role of increased renal chloride reabsorption. Kidney Int 19(5):716–727
Take C, Ikeda K, Kurasawa T et al (1991) Increased chloride reabsorption as an inherited renal tubular defect in familial type II pseudohypoaldosteronism. N Engl J Med 324(7):472–476
Yamauchi K, Rai T, Kobayashi K et al (2004) Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc Natl Acad Sci USA 101(13):4690–4694
Tatum R, Zhang Y, Lu Q et al (2007) WNK4 phosphorylates ser(206) of claudin-7 and promotes paracellular Cl(−) permeability. FEBS Lett 581(20):3887–3891
Kahle KT, Macgregor GG, Wilson FH et al (2004) Paracellular Cl− permeability is regulated by WNK4 kinase: insight into normal physiology and hypertension. Proc Natl Acad Sci USA 101(41):14877–14882
Tomita K, Pisano JJ, Burg MB et al (1986) Effects of vasopressin and bradykinin on anion transport by the rat cortical collecting duct. Evidence for an electroneutral sodium chloride transport pathway. J Clin Invest 77(1):136–141
Tomita K, Pisano JJ, Knepper MA (1985) Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J Clin Invest 76(1):132–136
Eladari D, Chambrey R, Peti-Peterdi J (2012) A new look at electrolyte transport in the distal tubule. Annu Rev Physiol 74:325–349
Matsuzaki K, Stokes JB, Schuster VL (1989) Stimulation of Cl− self exchange by intracellular HCO3− in rabbit cortical collecting duct. Am J Physiol 257(1 Pt 1):C94–C101
Chambrey R, Kurth I, Peti-Peterdi J et al (2013) Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci USA 110(19):7928–7933
Hentschke M, Hentschke S, Borgmeyer U et al (2009) The murine AE4 promoter predominantly drives type B intercalated cell specific transcription. Histochem Cell Biol 132(4):405–412
Lipovich L, Lynch ED, Lee MK et al (2001) A novel sodium bicarbonate cotransporter-like gene in an ancient duplicated region: SLC4A9 at 5q31. Genome Biol 2(4):RESEARCH0011
Romero MF, Fulton CM, Boron WF (2004) The SLC4 family of HCO3 − transporters. Pflugers Arch: Eur J Physiol 447(5):495–509
Ko SB, Luo X, Hager H et al (2002) AE4 is a DIDS-sensitive Cl(−)/HCO(−)(3) exchanger in the basolateral membrane of the renal CCD and the SMG duct. Am J Physiol Cell Physiol 283(4):C1206–C1218
Parker MD, Boron WF (2013) The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol Rev 93(2):803–959
Quentin F, Chambrey R, Trinh-Trang-Tan MM et al (2004) The Cl−/HCO3− exchanger pendrin in the rat kidney is regulated in response to chronic alterations in chloride balance. Am J Physiol Renal Physiol 287(6):F1179–F1188
Vallet M, Picard N, Loffing-Cueni D et al (2006) Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol 17(8):2153–2163
Kim YH, Pech V, Spencer KB et al (2007) Reduced ENaC protein abundance contributes to the lower blood pressure observed in pendrin-null mice. Am J Physiol Renal Physiol 293(4):F1314–F1324
Verlander JW, Hassell KA, Royaux IE et al (2003) Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 42(3):356–362
Gueutin V, Vallet M, Jayat M et al (2013) Renal beta-intercalated cells maintain body fluid and electrolyte balance. J Clin Invest 123(10):4219–4231
Pech V, Pham TD, Hong S et al (2010) Pendrin modulates ENaC function by changing luminal HCO3. J Am Soc Nephrol 21(11):1928–1941
Carattino MD, Hughey RP, Kleyman TR (2008) Proteolytic processing of the epithelial sodium channel gamma subunit has a dominant role in channel activation. J Biol Chem 283(37):25290–25295
Soleimani M, Barone S, Xu J et al (2012) Double knockout of pendrin and Na−Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc Natl Acad Sci USA 109(33):13368–13373
Acknowledgments
R. Chambrey, J. Hadchouel, and D. Eladari are funded by Institut National de la Santé et de la Recherche Médicale (INSERM) and N. Picard is funded by Centre National de la Recherche Scientifique (CNRS). This work was also funded by grants from l’Agence Nationale de la Recherche (BLANC-2010-R10164DD to DE, BLANC-2012-R13011KK to RC, and 2012-ISV1-0001-01 to JH), from the Fondation pour la Recherche sur l’Hypertension Arterielle to JH, from the Société de Néphrologie (subvention de recherche 2013 AMGEN) to NP, and the Fondation du rein (Prix Jeune Chercheur 2012) to NP.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eladari, D., Chambrey, R., Picard, N. et al. Electroneutral absorption of NaCl by the aldosterone-sensitive distal nephron: implication for normal electrolytes homeostasis and blood pressure regulation. Cell. Mol. Life Sci. 71, 2879–2895 (2014). https://doi.org/10.1007/s00018-014-1585-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-014-1585-4