
Abstract
Obesity-related insulin resistance is a chronic inflammatory condition that often gives rise to type 2 diabetes (T2D). Much evidence supports a role for pro-inflammatory T cells and macrophages in promoting local inflammation in tissues such as visceral adipose tissue (VAT) leading to insulin resistance. More recently, B cells have emerged as an additional critical player in orchestrating these processes. B cells infiltrate VAT and display functional and phenotypic changes in response to diet-induced obesity. B cells contribute to insulin resistance by presenting antigens to T cells, secreting inflammatory cytokines, and producing pathogenic antibodies. B cell manipulation represents a novel approach to the treatment of obesity-related insulin resistance and potentially to the prevention of T2D. This review summarizes the roles of B cells in governing VAT inflammation and the mechanisms by which these cells contribute to altered glucose homeostasis in insulin resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes (T2D) is a chronic disease characterized by hyperglycemia, pancreatic beta cell insufficiency, and insulin resistance, a suboptimal tissue response to insulin that is largely driven by obesity. By 2030, it is estimated that close to 400 million people worldwide will be affected by diabetes, with 90 % of these being T2D [1]. Obesity-related insulin resistance is thought to be the major precursor driving the development of T2D, and it targets multiple tissues including the brain, liver, skeletal muscle, pancreas, and adipose tissue. There are many causative factors governing obesity-related insulin resistance, but chronic low-grade inflammation is emerging as a dominant overarching theme [2]. Immune cells of the innate and adaptive immune system infiltrate insulin responsive tissues, such as visceral adipose tissue (VAT), during obesity and incite inflammatory responses [3–6]. This inflammation leads to local and systemic increases in pro-inflammatory molecules, including tumor necrosis factor α (TNF-α), interleukin (IL)-1β, IL-6, interferon-γ (IFNγ), resistin, and free fatty acids. Several of these molecules can directly induce insulin resistance, often through c-Jun amino terminal kinase and inhibitor of nuclear factor kappa B kinase subunit β-mediated induction of serine phosphorylation of the insulin receptor and/or its substrates, including insulin receptor substrate-1 (IRS-1) [7, 8]. Accordingly, chronic inflammation of VAT associated with obesity is thought to be a major driver of insulin resistance.
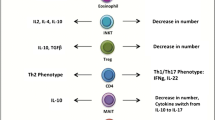
Extensive work has been done on the biology of T cell and macrophage-mediated inflammation in insulin resistance [9]. In obesity, IFNγ-producing CD8+ T cells and Th1 CD4+ T cells infiltrate inflamed VAT and promote pro-inflammatory “classically activated” M1 macrophage formation and function [10–12]. These M1 macrophages produce large amounts of TNF-α, IL-1β, and IL-6, which contribute to local insulin resistance, but can also escape into the circulation and contribute to systemic insulin resistance, through reduction in insulin signaling (for example, via increased serine phosphorylation of IRS-1) in distant organs such as muscle and liver [8]. In lean individuals, it is thought that IL-4-producing eosinophils, IL-4, IL-5, and IL-13-producing Th2 CD4+ T cells, IL-5, and IL-13-producing innate lymphoid cells, as well as IL-10-producing CD4+ Foxp3+ regulatory T cells and iNKT cells dominate in VAT [11, 13–16]. The cytokines secreted by these cells favor the maintenance of “alternatively activated” M2 macrophages, which produce mainly anti-inflammatory cytokines such as IL-10 that maintain insulin sensitivity. Thus, insulin sensitivity in adipose tissue inflammation is profoundly affected by the complex crosstalk between adaptive and innate immune cells and adipocytes.
Like T cells and macrophages, B cells also have been shown to play important roles in many chronic inflammatory and autoimmune diseases, including systemic lupus erythematosus, rheumatoid arthritis, scleroderma, type 1 diabetes and multiple sclerosis [17, 18], but only recently has their role in obesity-related insulin resistance been revealed. B cells infiltrate VAT during diet-induced obesity, and exhibit a variety of functional changes during insulin resistance [19, 20]. B cells regulate systemic and local adipose tissue inflammation during obesity-related insulin resistance through antigen presentation, cytokine secretion, and antibody production [20, 21]. Through these processes, B cells have the unique ability to modulate T cell and macrophage cytokine production and polarization at multiple levels, and thus potentially represent an attractive new target for immune therapy of insulin resistance. Moreover, recent studies of B cells have yielded novel information on distinct antigens targeted by the immune system during insulin resistance [20]. These findings lay the groundwork for novel antibody-based diagnostics or vaccination approaches to insulin resistance. This review will highlight recent insights into the role of B cells in governing adipose tissue inflammation and obesity-related insulin resistance.
B cell subsets and functions
Like T cells, B cells consist of distinct subsets with differing surface phenotypes, functions, and cytokine secretion profiles. B cells can be divided into two broad classes, B-1 or B-2 cells, which display unique development, phenotypes, functions, and cytokine secretion profiles.
B-1 cells are typically enriched in mucosal tissues and body cavities including pleural, peritoneal, and pericardial cavities, and are also enriched in fatty tissues such as the omentum and fat pads near the peritoneal cavity [22]. B-1 cells are further divided into B-1a cells, the major producers of natural IgM antibody in the body, and B-1b cells which give rise to adaptive humoral immune responses to T cell-independent antigens [23].
In the steady state, there is constant B-1 cell trafficking between the peritoneal cavity and abdominal VAT, such as the omentum, where B-1 cells often co-localize with other leukocytes into clusters called “milky spots” [24]. Milky spots in the omentum sample fluids and antigens from the peritoneal cavity, and contribute to an ensuing pro-inflammatory or immune suppressing response [25]. B-1 cells are found in similar lymphoid clusters in mesenteric and gonadal adipose tissue known as fat-associated lymphoid clusters (FALCs), where they interact with both phenotypically mature and immature immune cells [26]. During breach of the intestinal barrier, or in the presence of intraperitoneal bacteria, peritoneal cavity B-1 cells become highly activated and produce T-independent antibodies. Through a CXCL13-dependent process, these cells exit the peritoneal cavity and traffic to omentum, spleen, and mesentery [27]. Indeed, B-1 cells are highly responsive to bacterial products such as lipopolysaccharide (LPS) [27] and thus are considered as one of the first lines of defense against bacterial pathogens. B-1 cells also show prominent responses to some viral infections, including influenza [28]. It is also interesting to speculate that priming of the adaptive immune response from potential VAT antigens may occur at least partially in FALCs [29].
B-1 cells show a less diverse but more polyreactive antibody repertoire compared to B-2 cells [30]. Antibodies produced by B-1 cells are typically IgM, though these cells can also produce natural IgA in the gut mucosa [31]. One of the best-studied natural polyreactive IgM antibodies produced by B-1 cells is the T15 idiotype, which reacts against phosphorylcholine on Streptococcus pneumonia. This antibody cross-reacts with membrane lipids on apoptotic cells and with modified forms of circulating lipids such as oxidized low-density lipoprotein (oxLDL) [32, 33]. Importantly, neutralization of oxLDL by natural IgM has been shown to protect against inflammation associated with atherosclerosis [34, 35]. B-1a cells are also major producers of IL-10, and can exert anti-inflammatory effects through this cytokine [36].
Recently, human B-1 cells have been identified in umbilical cord and adult peripheral blood based on functional criteria that they share with mouse B-1 cells, such as spontaneous secretion of IgM and tonic intracellular signaling [37]. It will be interesting to compare these cells to mouse B-1 cells in terms of their putative roles in pathophysiology across multiple disease states.
B-2 cells produce specific antibodies to T-dependent antigens and are enriched in secondary lymphoid organs [38]. B-2 cells can be subdivided into more conventional B cells such as mature transitional cells (T1, T2, and T3) and mature follicular B cells [39]. In addition, B-2 cells include marginal zone B cells [39]. Conventional B-2 cells are stimulated by a variety of ligands to achieve an activated phenotype. In the naive state, B-2 cells typically require engagement of a combination of surface immunoglobulin, the co-activator CD40, and a third signal, which is often provided by engagement of a Toll-like receptor (TLR) [40]. Antigen-induced B-2 cell activation typically occurs in secondary lymphoid structures, where the hallmark “germinal center reaction” occurs. During germinal center reactions, B-2 cells undergo clonal expansion, class switch recombination (CSR) to another isotype, somatic hypermutation (SHM) of VH genes, and affinity maturation for increased binding of antibody to a unique antigenic epitope [41]. CSR and SHM are mediated by an enzyme called activation-induced cytidine deaminase [42]. Conventional B-2 cells can then differentiate into antibody-secreting plasma cells or can switch into memory B cells that subsequently require less surface engagement for activation [43].
B-2 cells can produce a variety of cytokines, with some populations capable of producing more pro-inflammatory Th1 polarizing cytokines such as IFNγ, IL-12, and TNF-α, and others producing cytokines more associated with Th2 responses such as IL-2, IL-4, and IL-13 [39]. More recently, regulatory B cells (“Breg” cells) with the ability to suppress inflammatory responses have been described [44]. One subset of these cells includes B cells called “B10” cells, which produce large quantities of IL-10 and may co-express CD1d and CD5 [45]. These cells mature in the presence of IL-21 and CD40-dependent cognate interaction with T cells, and exert potent anti-inflammatory effects on T cell activation and can markedly reduce autoimmune disease in vivo [46].
Effects of obesity and insulin resistance on B cell development
In mice and humans, B cell development typically occurs in the fetal or adult bone marrow. Early B cell development from progenitor B cells (pro-B cells) to precursor B cells (pre-B cells) occurs through upregulation of a pre-B cell receptor (BCR) in murine and human pre-B cells [47], followed by expression of the BCR, which promotes survival of mature B cells in the periphery [48]. Many of the processes of B cell development are under the control of the transcription factor, Pax5 [41].
During obesity, there is conflicting evidence as to whether bone marrow B cell development is enhanced or compromised. In one recent study, in which C57BL/6 mice were fed a high-fat diet (HFD) for 180 days, marked increases in pre-B cells, immature and mature B cells were seen starting 90 days after initiation of HFD and persisting throughout the study [49]. Increased B cell lymphopoiesis was at least partially attributed to increases in leptin, which were associated with an up-to-sixfold increase in bone marrow adipocytes [49]. Consistently, leptin-deficient obese ob/ob and leptin receptor-deficient db/db mice show deficits in B lymphocyte progenitors [50]. In another recent study in which mice were maintained on a HFD for ~210 days, C57BL/6 mice showed a 52 % reduction in B cells in bone marrow, associated with a trend to reduced bone marrow Pax5 expression, consistent with reduced B cell lymphopoiesis [51]. Differences between these studies may be due the type and duration of HFD and methods of B cell quantification. It will be interesting to determine if chronic obesity can lead to leptin resistance in B cell precursors as a mechanism for altered B cell lymphopoiesis. Given the emerging importance of B cells in metabolic diseases, further investigation of the effects of obesity and insulin resistance on B cell development is warranted to better understand this fundamentally important biological process.
Pathophysiology of B cells in insulin resistance
Phenotypic changes and temporal response patterns of B cells in obesity
During the course of high-fat feeding, B cell populations infiltrate inflamed tissues such as VAT and undergo functional and phenotypic changes. B cell infiltration into VAT of C57BL/6 mice placed on HFD peaks at around 3–4 weeks after initiation of HFD [19, 20]. B cell infiltration is thought to precede T cell infiltration into VAT, which typically begins with CD8+ T cell influx [10]; however, B cells are not the first immune cell to traffic into VAT. Infiltrating neutrophils have been observed in VAT in as little as 3 days after initiation of HFD [6, 52], while macrophages, including CD11c+ M1 macrophages, have been described in VAT as little as 1 week after the start of a HFD [53].
In addition to localizing with milky spots and FALCs in VAT, B cells, along with macrophages and T cells, can surround stressed or dying adipocytes in so-called “crown-like structures” (CLSs) [8, 20]. Here, B cells likely sample antigen and modulate T cell and macrophage function [20]. In morbidly obese humans, B cells are increased in CLS of subcutaneous adipose tissue, suggesting that they also maintain a presence in multiple fat depots over the long term. More study of B cell infiltration into human VAT is needed to draw comparisons, given the increasing volume of supportive mouse data.
In response to HFD in C57BL/6 mice, total B cell numbers increase in VAT. By 6–12 weeks after initiation of HFD, B cells in VAT show increased frequency of IgM+IgD− cells and increased class switching to IgG+ cells, especially to pro-inflammatory IgG2c [20]. Class switching is a hallmark of an active immune response and provides further evidence for adaptive immune-mediated inflammation occurring in VAT as an active pathophysiological process underlying obesity.
Mechanisms used by B cells to modulate insulin resistance
B cells have a net pathogenic effect on HFD-induced obesity-related insulin resistance. B cell-deficient mice (Bnull) placed on a HFD show reduced glucose levels and less insulin resistance compared to wild-type controls placed on the same diet [20]. Transfer of B-2 cells from diet-induced obese (DIO) mice into DIO Bnull mice worsens metabolic parameters, implicating some B cells as important pathogenic mediators of insulin resistance [20]. As discussed below, B cells have been shown to drive obesity-related insulin resistance through a number of mechanisms, including cytokine production, T cell modulation, and antibody production (Fig. 1).

B cells promote inflammation in insulin resistance through multiple mechanisms. During obesity-related insulin resistance, B cells show altered cytokine production, characterized by decreased secretion of anti-inflammatory IL-10 and increased secretion of chemokines, including IL-8. B cells also modulate T cells in VAT, and systemically in hematolymphoid organs to induce secretion of pro-inflammatory cytokines such as IFNγ and IL-17. Finally, B cells show increased class switching to IgG and production of antibody against distinct antigenic targets linked to insulin resistance. Immune complexes to these targets have the potential to further activate macrophages to produce TNF-α, which further worsens insulin resistance
Cytokine production
B cells are a source of numerous cytokines with important effects in modulating immune-mediated disease. IL-10-producing B cells include B10 and B1 cells in mice, and CD27+ IL-10-producing B cells in humans [54]. B cell-derived IL-10 is mainly an anti-inflammatory cytokine, and has been shown to have beneficial effects in chronic inflammatory diseases such as experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis in mice [44, 55, 56]. In these diseases, IL-10 has been shown to inhibit pro-inflammatory cytokine production, and promote regulatory T cell differentiation. IL-10 also appears to have a dominant protective role in insulin resistance, though this conclusion is less straightforward. IL-10 can prevent HFD-induced insulin resistance in mice by reducing macrophage and cytokine responses in skeletal muscle [57]. In this study, transgenic mice overexpressing IL-10 in skeletal muscle placed on a HFD showed improved whole-body insulin sensitivity, and reduced inflammation as demonstrated by decreased production of TNF-α and IL-6 [57]. Short-term treatment of mice with IL-10 can improve Akt phosphorylation and IRS-1 tyrosine phosphorylation in skeletal muscle, with less effect in white adipose tissue [57]. However, in another study, mice reconstituted with hematopoietic cells lacking IL-10 did not show worsening of inflammation in adipose tissue or liver, or reduced insulin resistance after 12 weeks of HFD [58]. Interpretation of this study, however, was confounded by increased compensatory production of IL-10 from liver and adipose tissues.
In humans, impaired IL-10 production is associated with metabolic syndrome and T2D [59]. Consistently, B cells isolated from the blood of patients with T2D show decreased IL-10 production compared with matched control B cells in response to TLR2, TLR4, or TLR9 agonists [60]. As B cell-derived IL-10 has been shown to reduce chronic inflammation in other diseases, such as EAE, through blocking of Th1 differentiation, it is possible that reduced B cell IL-10 contributes to the increased Th1 cell differentiation seen systemically and in VAT in obesity-related insulin resistance [11, 44, 61]. Thus, B cell-derived IL-10 could potentially act as a critical regulator of T cell polarization and metabolic balance in obesity. Further studies, such as those involving B cell-specific IL-10 knockout mice and dominant IL-10 producers such as B10 and B1 cells, are needed to confirm this possibility and to identify the key cellular sources of this cytokine.
In addition to reduced IL-10 production, B cells isolated from the blood of T2D patients also produce elevated levels of IL-8, a pro-inflammatory chemokine that can recruit neutrophils, but not IL-6 when compared to controls [60]. Serum IL-8 levels correlate with body mass index in obese humans [62], and it is interesting to speculate that B cells may promote neutrophil influx in VAT, although a role for this cytokine in insulin resistance has not yet been confirmed.
Changes in B cell cytokine production during obesity might also be influenced by increasing leptin levels associated with the obese state. The leptin receptor is expressed on B cells, T cells, monocytes, and macrophages [63]. In human B cells, leptin induces phosphorylation of Janus activation kinase 2, signal transducer and activator of transcription 3, p38 mitogen-activated protein kinase, and extracellular signal-regulated kinase (ERK1/2), leading to dose-dependent increases of TNF-α [64]. Leptin signaling also acts to promote B cell survival by inhibiting apoptosis and inducing cell-cycle entry through activation of bcl-2 and cyclin D1 [65]. Leptin has potent Th1 T cell-polarizing properties, and so it is interesting to speculate that some of the pro-inflammatory changes seen in T cells and B cells in VAT inflammation are influenced by leptin. Additional studies analyzing the effects of leptin or leptin receptor deficiency (e.g., in ob/ob and db/db mice) on VAT B cell function are needed to further characterize these mechanisms.
In addition to leptin, B cells in VAT are also exposed to increased saturated fatty acids, which have been shown to stimulate TLR4 in a manner similar to LPS, which is a strong B cell stimulus [66]. Toll-like receptor activation by saturated fatty acids leads to the activation of the NF-κB pathway and production of proinflammatory cytokines IL-6 and TNF-α in adipocytes and macrophages in the context of diet-induced obesity [66]. However, more studies are needed to determine whether the local inflammatory environment, rich in fatty acids, in VAT could also contribute to alterations in B cell activation and cytokine production.
T cell modulation
In addition to producing cytokines, B cells are also important antigen-presenting cells and through this activity they can profoundly influence T cells. B cells regulate pro-inflammatory T cell function in models of infection, cancer [67], autoimmunity [68], and chronic inflammatory disease such as atherosclerosis [34]. In diet-induced obesity, B cells can interact with T cells in an MHC-dependent manner to induce T cell IFNγ expression, which contributes to local and systemic inflammation and associated insulin resistance [20]. Indeed, transfer of B cells lacking MHC class I or II expression from HFD-fed mice into diet-induced obese B cell-deficient mice fails to worsen glucose tolerance. Moreover, transfer of wild-type B cells from HFD-fed mice into diet-induced obese recombination activating gene-deficient mice, which lack T and B cells, has only a minor negative effect on glucose tolerance [20]. These findings indicate that B cell modulation of T cells, likely through cognate interactions, is an important mechanism by which B cells influence the development of insulin resistance. These interactions are known to take place in VAT or FALCs, but they may also occur in other metabolic tissues such as muscle or in hematolymphoid organs, such as the spleen. Very recently, these findings have been reproduced and extended in important work that shows marked reduction of inflammatory T cell signature genes and cytokines, such as IFNγ and IL-17, in VAT of B cell-deficient mice compared to controls, as well as a critical role for contact-dependent B cell regulation of T cell-derived IL-17 from the blood of human subjects with T2D [21].
T cells can also modulate B cells, and it is possible that reciprocal interactions occur, with T cells functioning to alter B cell cytokine production, immunoglobulin class switching, or activation state, potentially through CD40–CD40L interactions [69]. Given the restricted T cell receptor repertoire that develops in obesity-related insulin resistance in VAT, the above findings support a role for antigen-specific immunity in VAT inflammation, with antigen presentation by B cells as a potential driver of this process [11].
Although interactions between CD40L on T cells and CD40 on B cells play an important role in germinal center formation, memory B cell development, and immunoglobulin class switching, the role of these interactions in insulin resistance is controversial. The first study of CD40L-deficient mice fed a high-fat diet showed improved insulin resistance as well as reduced macrophage and T cell accumulation in adipose tissue [69]. Mice treated with anti-CD40L inhibiting antibody recapitulated many of these results and also showed decreased IL-6 and leptin plasma levels [69]. However, other studies of CD40L- or CD40-deficient mice have failed to demonstrate improvement in insulin resistance [70, 71]. In one study, CD40L-deficient mice showed reduced immune cell VAT infiltration, inflammation, and IgG antibody levels against oxidized lipids, but these mice were not protected from insulin resistance [70]. Another report showed that when mice deficient in CD40 were fed a high-fat diet, they had significantly increased VAT inflammation and insulin resistance by comparison to wild-type mice [71]. Further investigations of the role of CD40 and CD40L in insulin resistance are needed.
In addition to interactions with T cells, direct interactions with adipocytes may also function to shape B cell responses in VAT. For example, along with traditional adipokines including the aforementioned leptin, which may shape B cell responses, adipocytes in VAT are also a source of the potent B cell survival factor, B cell-activating factor [BAFF, or B lymphocyte stimulator (BlyS)]. The levels of BAFF increase in VAT during the course of high-fat-diet feeding and associated insulin resistance [72]. BAFF secretion by adipocytes in vitro is modulated by oxidative stress through NF-κB pathway activation; thus, the local hypoxic environment in obese VAT may be one trigger for B cell survival and activity in VAT during obesity-related insulin resistance [73].
Antibody production
Antibody production is a prototypical function of B cells, and antibodies have important roles in multiple inflammatory diseases including rheumatoid arthritis, lupus, and atherosclerosis. In diet-induced obesity in mice, total splenic B cells show reduced production of spontaneous IgM, but increased IgG secretion [20]. Consistently, studies from Akita mice, in which there is aberrant insulin folding leading to hyperglycemia, show that hyperglycemia alone can delay production of IgM from LPS-stimulated splenocytes [74]. These findings indicate that obesity and hyperglycemia directly influence antibody production. In addition, diet-induced obese mice have increases in class-switched pro-inflammatory IgG2c antibody [20]. Oral feeding of ovalbumin (OVA) antigen in obese mice induces production of pro-inflammatory IgG2c antibodies, while feeding the same antigen to lean mice induces predominantly IgG1, suggesting that a high-fat diet and obesity promote an active pro-inflammatory environment that can contribute to immunoglobulin class switching in B cells [75].
While obesity profoundly impacts antibody production and isotype, antibodies, in turn, have dramatic effects on insulin resistance in obesity. Transfer of purified IgG from DIO mice (HFD IgG), but not from lean mice, induces an Fc-dependent worsening of insulin resistance [20]. Although the antigens recognized by these antibodies have not been identified, the pathogenicity of IgG increases with duration of exposure to HFD, and is also dependent on HFD exposure in the recipient mice, possibly due to diet-induced conditioning or induction of antigen [20]. HFD IgG likely affects multiple cell types, but FcγR activation of macrophages with HFD IgG has been shown to promote TNF-α production in vitro and in vivo. In particular, macrophages isolated from the VAT of DIO B cell-deficient mice and stimulated with HFD IgG show an Fc-dependent increase in TNF-α production in vitro. In addition, transfer of HFD IgG in DIO B cell-deficient mice induces increased M1 macrophage polarization and TNF-α production from VAT stromal vascular cells in vivo [20]. Given that levels of IgG are increased in VAT in DIO mice [20], HFD IgG and their proinflammatory FcγRs likely represent important modulators of VAT macrophage function and polarization during obesity-related insulin resistance. Since adipocytes also express FcγRs, the possibility exists that IgG antibody also has direct effects on adipocyte function in VAT [76]. As antibodies can fix complement, it will also be critical to determine how HFD IgG influences production of C3a, which binds C3aR on macrophages, since both C3a and C3aR have been shown to promote insulin resistance [77].
Antibodies can also regulate obesity at the level of lipid absorption from the gut. Mice lacking B cells or IgA exhibit up-regulation of selected inflammatory pathways, including interferon inducible pathways, in their intestinal epithelium, and an associated reduction in lipid absorption [78]. Consistently, B cell-deficient mice show reduced visceral fat pad weights on HFD compared to wild-type mice [20, 78]. Thus, B cells play an important role in shaping mucosal immunity to gut microbiota, and loss or alteration of this function can lead to changes in nutrient absorption and local inflammatory responses.
Consistent with these findings, there is increasing evidence for a role of the intestinal microflora in regulating obesity and insulin resistance [79, 80]. Interestingly, HFD has been associated with increased permeability across the gut [81], and triglycerides present in HFD can promote absorption of intestinal antigens and LPS into VAT in a chylomicron-dependent manner [82–84]. HFD exposure has been shown to lead to systemic endotoxemia due to such LPS absorption from the gut [85]. These findings beg the question of whether some of the pathogenic IgG identified in DIO mice target food or bacterial antigens derived from the gut. Such findings would be consistent with the dependence of IgG pathogenicity on the duration of HFD in both IgG donors and recipients, as described [20]. Indeed, elevated IgG levels against specific bacterial antigens recently have been reported in obese patients and HFD-fed mice [86], and further study is warranted to determine the pathological significances of these findings.
In addition to targeting potential gut-derived or foreign antigenic targets, antibodies have also been shown in multiple studies to be directed against self-antigens during the course of insulin resistance (Table 1). In a cohort of 32 overweight and obese male human subjects, insulin resistance was linked to a relatively distinct autoantibody profile [20]. Antigens targeted during the course of insulin resistance are predominantly intracellular proteins and are expressed in multiple cell types and tissues such as immune cells, pancreas, liver, nervous system, muscle, and fat. In this study, Golgi SNAP receptor complex member 1 (GOSR1), transcript variant 1, was the most prevalent antigenic target, with autoantibodies present in more than 70 % of insulin resistant subjects [20]. GOSR1 is involved in shuttling proteins between the endoplasmic reticulum (ER) and Golgi, and it will be interesting to investigate how the transcription, splicing, or translation of this protein is influenced during ER stress, a hallmark of insulin resistance, and whether these changes can alter antigenicity [87]. Antibodies against phosphogluconate dehydrogenase, which is highly expressed in adipocytes, are found in approximately 40 % of insulin-resistant overweight male subjects [20]. Thus, it is possible that dying or apoptotic adipocytes, commonly seen in obese insulin resistant VAT at the center of CLSs, represents one potential source of autoantigen and contributes to insulin resistance [29, 88]. Indeed, deletion of the apoptotic inducer, Fas (CD95), in adipocytes has been shown to decrease infiltration of CD11b myeloid cells into VAT, and reverse the adverse effects of HFD on glucose homeostasis [89].
Neural tissue represents another source of autoantigens in insulin resistance. Autoantibodies against glial fibrillary acidic protein (GFAP) in insulin resistance or T2D have been seen in 30–50 % of patients in multiple studies [20, 90]. A recent study showed that HFD-induced obesity is associated with IgG antibody deposition in the hypothalamus of rodents, and it will be interesting to investigate how antibody deposition in a critical feeding and metabolism regulatory center of the brain influences systemic glucose metabolism [91]. GFAP is also expressed in peri-islet glial tissue in the pancreas, and while this tissue is targeted in type 1 diabetes, a role for its targeting in type 2 diabetes is a possibility that warrants future investigation [92].
Interestingly, some autoantibody targets may be exposed following viral or bacterial infection. Autoantibodies against protein disulfide isomerase (PDI), elicited by post-streptococcal infection, can contribute to insulin resistance [93]. Use of an integrative personal omics profile approach in a single individual showed that the development and onset of T2D was preceded by two viral infections during the year, including a human rhinovirus infection early in the study and a respiratory syncytial virus infection which directly preceded the rise in glucose. Viral infection in this subject led to the appearance of novel autoantibodies in the circulation [94]. Some of these autoantibodies targeted potential insulin receptor binding molecules such as DOK6, while others targeted self-antigens previously associated with the insulin resistant state, including BTK and GOSR1 [20]. The findings suggest additional roles for environmental cues, outside of diet, in shaping an immune response that may promote insulin resistance or T2D.
Other studies have shown that antibodies that inhibit endothelial cell function are present in 30 % of T2D patients, and correlate with vascular complications [95–97]. Another recent report has shown that B cells and potential pathogenic IgG are linked to the development of insulin resistance in different lupus prone C57BL/6 mouse models [98]. Increased production of IgG autoantibody in obesity is thought to be mechanistically linked to binding of macrophage-derived apoptosis inhibitor macrophage to pentameric IgM complexes, which reduces clearance of these complexes, potentiating IgG autoantibody production [99].
It is important to note that the autoantibodies identified in the aforementioned studies linked to insulin resistance are distinct from those such as anti-glutamic acid decarboxylase and other autoantibodies characteristic of type 1 diabetes, which are often linked to latent autoimmune diabetes in adults [100]. The list of autoantibodies associated with insulin resistance will likely continue to grow as additional studies investigate their frequency and functions. Ultimately, such information will be crucial in developing novel antibody-based diagnostics for insulin resistance and vaccination approaches to T2D.
B cells as a therapeutic target in insulin resistance
The emergence of inflammatory pathways as important mediators of insulin resistance has led to a number of trials investigating the use of anti-inflammatory drugs for diabetes. Treatments directed against IL-1β [101] or the use of anti-inflammatory drugs such as salsalate [102] have shown promising results in clinical trials. Given their role in governing multiple aspects of insulin resistance pathology, B cells represent a promising new therapeutic target. An anti-CD20 B cell depleting antibody (Rituximab) is FDA approved for use in some autoimmune diseases, such as rheumatoid arthritis and lupus, but carries the potential risk for rare but worrisome side effects [103]. Nonetheless, an anti-CD20-depleting antibody has shown marked therapeutic benefit when given early in the course of diet-induced obesity in mice. The therapeutic effect of B cell depletion with anti-CD20 in mice is linked to marked reductions in proinflammatory cytokines such as IFNγ and TNF-α in VAT, suggesting that B cells may be central regulators in orchestrating T cell and macrophage cytokine production in VAT [20]. Drugs targeting BAFF such as Belimumab, which reduce B cell survival, maturation, and activation [104] have been approved for the treatment of lupus. Mice lacking the BAFF-R show a preferential reduction in pathogenic B2 cells, over potentially beneficial B1a cells, in models of atherosclerosis [105, 106]. These mice also show improved glucose tolerance on HFD [107] and future studies are needed to see if anti-BLyS/BAFF therapy shows benefits in models of obesity-related insulin resistance.
Additional agents that target B cells include transmembrane activator, calcium modulator and cyclophilin ligand interactor fusion proteins, or antibodies or inhibitors to CD19, CD22, spleen tyrosine kinase, and a proliferation-inducing ligand, and studies investigating the effects of these agents on insulin resistance would be worthwhile. As antibodies are potential novel modulators of inflammation in insulin resistance, the effect of FcR-regulating agents, including intravenous immunoglobulin and related therapies, may also show therapeutic benefit. In addition to studying the effect of B cell-modulating drugs in insulin resistance, it will also be important to study the effects that surgical approaches to obesity, such as bariatric surgery, have on B cell function. Bariatric surgery has been associated with dramatic improvement in obesity-related diabetes [108], along with reductions in IFNγ in VAT [109], and so the mechanisms responsible for these results should provide new insights into B cell behavior in obesity-related diabetes control. The use of cell-based therapy, involving transfers of potentially protective regulatory B cells, or B10 B cells, also may yield therapeutic results. Finally, the identification of autoantibody signatures linked to both insulin-resistant and insulin-sensitive states in obese patients [20] opens the door to potential novel antigen-tolerizing approaches to antigens linked to insulin resistance, or to boosting immunity against antigens linked to insulin sensitivity. However, more work is first needed to better elucidate the roles of self-antigen targeting in insulin resistance before such experiments can be undertaken.
Conclusions
B cells are an important new player in the promotion of inflammation associated with insulin resistance and T2D. B cells infiltrate inflamed tissues such as VAT, in insulin resistance, where they sample antigen and promote T cell IFNγ production. Presumably, B cell activation occurs as a result of stimulation with antigen, which may be self-derived or potentially derived from exogenous sources including bacterial or viral antigens. Increased T cell IFNγ, along with reduced B cell-derived IL-10, characteristic of insulin resistance, contributes to increased M1 macrophage polarization in inflamed tissue. M1 macrophages produce TNF-α, and in concert with increasing IFNγ, this cytokine contributes to impaired insulin receptor signaling. B cells also contribute to insulin resistance through production of IgG antibodies, which further enhance local macrophage TNF-α production in an Fc-dependent process, and may have additional effects in targeting insulin signaling molecules and other self- or foreign antigens.
Additional work is needed to identify B cell-targeted antigens in insulin resistance, and studies investigating the frequency of specific BCR sequences in VAT and how these change in response to HFD are underway. Furthermore, additional insight into the roles of distinct B cell subsets, and how each individual subset promotes or regulates insulin resistance is needed. Validation of novel antigenic signatures of insulin resistance in larger clinical studies could lead the way to new diagnostics to help manage the disease. Finally, B cells represent an attractive new target for the treatment of insulin resistance and T2D, and well-designed clinical trials are needed to determine the safety and effectiveness of targeting B cells in patients with these disorders.
Abbreviations
- T2D:
-
Type 2 diabetes
- VAT:
-
Visceral adipose tissues
- FALC:
-
Fat-associated lymphoid cluster
- oxLDL:
-
Oxidized low-density lipoprotein
- CSR:
-
Class switch recombination
- SHM:
-
Somatic hypermutation
- HFD:
-
High-fat diet
- BCR:
-
B cell receptor
- CLS:
-
Crown-like structure
- DIO:
-
Diet-induced obese
- GOSR1:
-
Golgi SNAP receptor complex member 1
- GFAP:
-
Glial fibrillary acidic protein
- BAFF:
-
B-cell activating factor
References
Franks PW, Hanson RL, Knowler WC, Sievers ML, Bennett PH, Looker HC (2010) Childhood obesity, other cardiovascular risk factors, and premature death. N Engl J Med 362:485–493
Johnson AM, Olefsky JM (2013) The origins and drivers of insulin resistance. Cell 152:673–684
Xu H, Barnes GT, Yang Q et al (2003) Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112:1821–1830
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112:1796–1808
Winer S, Winer DA (2012) The adaptive immune system as a fundamental regulator of adipose tissue inflammation and insulin resistance. Immunol Cell Biol 90:755–762
Talukdar S, da Oh Y, Bandyopadhyay G et al (2012) Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18:1407–1412
Ozcan U, Cao Q, Yilmaz E et al (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Olefsky JM, Glass CK (2010) Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72:219–246
Odegaard JI, Chawla A (2013) Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 339:172–177
Nishimura S, Manabe I, Nagasaki M et al (2009) CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15:914–920
Winer S, Chan Y, Paltser G et al (2009) Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 15:921–929
Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117:175–184
Wu D, Molofsky AB, Liang HE et al (2011) Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332:243–247
Feuerer M, Herrero L, Cipolletta D et al (2009) Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15:930–939
Molofsky AB, Nussbaum JC, Liang HE et al (2013) Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 210:535–549
Lynch L, Nowak M, Varghese B et al (2012) Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity 37:574–587
Yanaba K, Bouaziz JD, Matsushita T, Magro CM, St Clair EW, Tedder TF (2008) B-lymphocyte contributions to human autoimmune disease. Immunol Rev 223:284–299
Marino E, Grey ST (2012) B cells as effectors and regulators of autoimmunity. Autoimmunity 45:377–387
Duffaut C, Galitzky J, Lafontan M, Bouloumie A (2009) Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun 384:482–485
Winer DA, Winer S, Shen L et al (2011) B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17:610–617
Defuria J, Belkina AC, Jagannathan-Bogdan M et al (2013) B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci USA 110:5133–5138
Kaminski DA, Randall TD (2010) Adaptive immunity and adipose tissue biology. Trends Immunol 31:384–390
Haas KM, Poe JC, Steeber DA, Tedder TF (2005) B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity 23:7–18
Cui L, Johkura K, Liang Y et al (2002) Biodefense function of omental milky spots through cell adhesion molecules and leukocyte proliferation. Cell Tissue Res 310:321–330
Rangel-Moreno J, Moyron-Quiroz JE, Carragher DM et al (2009) Omental milky spots develop in the absence of lymphoid tissue-inducer cells and support B and T cell responses to peritoneal antigens. Immunity 30:731–743
Moro K, Yamada T, Tanabe M et al (2010) Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463:540–544
Ha SA, Tsuji M, Suzuki K et al (2006) Regulation of B1 cell migration by signals through Toll-like receptors. J Exp Med 203:2541–2550
Choi YS, Baumgarth N (2008) Dual role for B-1a cells in immunity to influenza virus infection. J Exp Med 205:3053–3064
Morris DL, Cho KW, Delproposto JL et al (2013) Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes 62:2762–2772
Martin F, Kearney JF (2001) B1 cells: similarities and differences with other B cell subsets. Curr Opin Immunol 13:195–201
Kunisawa J, Kurashima Y, Gohda M et al (2007) Sphingosine 1-phosphate regulates peritoneal B-cell trafficking for subsequent intestinal IgA production. Blood 109:3749–3756
Kearney JF (2000) Immune recognition of OxLDL in atherosclerosis. J Clin Invest 105:1683–1685
Binder CJ, Silverman GJ (2005) Natural antibodies and the autoimmunity of atherosclerosis. Springer Semin Immunopathol 26:385–404
Ait-Oufella H, Herbin O, Bouaziz JD et al (2010) B cell depletion reduces the development of atherosclerosis in mice. J Exp Med 207:1579–1587
Kyaw T, Tay C, Khan A et al (2010) Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol 185:4410–4419
Mauri C, Ehrenstein MR (2008) The ‘short’ history of regulatory B cells. Trends Immunol 29:34–40
Griffin DO, Holodick NE, Rothstein TL (2011) Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+CD43+ CD70. J Exp Med 208:67–80
Allman D, Pillai S (2008) Peripheral B cell subsets. Curr Opin Immunol 20:149–157
Lund FE (2008) Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol 20:332–338
Ruprecht CR, Lanzavecchia A (2006) Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol 36:810–816
LeBien TW, Tedder TF (2008) B lymphocytes: how they develop and function. Blood 112:1570–1580
Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T (2000) Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102:553–563
Yoshida T, Mei H, Dorner T et al (2010) Memory B and memory plasma cells. Immunol Rev 237:117–139
Mauri C, Bosma A (2012) Immune regulatory function of B cells. Annu Rev Immunol 30:221–241
Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF (2008) A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 28:639–650
Yoshizaki A, Miyagaki T, DiLillo DJ et al (2012) Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature 491:264–268
Melchers F (2005) The pre-B-cell receptor: selector of fitting immunoglobulin heavy chains for the B-cell repertoire. Nat Rev Immunol 5:578–584
Lam KP, Kuhn R, Rajewsky K (1997) In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 90:1073–1083
Trottier MD, Naaz A, Li Y, Fraker PJ (2012) Enhancement of hematopoiesis and lymphopoiesis in diet-induced obese mice. Proc Natl Acad Sci USA 109:7622–7629
Claycombe K, King LE, Fraker PJ (2008) A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc Natl Acad Sci USA 105:2017–2021
Chan ME, Adler BJ, Green DE, Rubin CT (2012) Bone structure and B-cell populations, crippled by obesity, are partially rescued by brief daily exposure to low-magnitude mechanical signals. FASEB J: Off Publ Fed Am Soc Exp Biol 26:4855–4863
Elgazar-Carmon V, Rudich A, Hadad N, Levy R (2008) Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res 49:1894–1903
Nguyen MT, Favelyukis S, Nguyen AK et al (2007) A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 282:35279–35292
Duddy M, Niino M, Adatia F et al (2007) Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J Immunol 178:6092–6099
Mauri C, Gray D, Mushtaq N, Londei M (2003) Prevention of arthritis by interleukin 10-producing B cells. J Exp Med 197:489–501
Matsushita T, Yanaba K, Bouaziz JD, Fujimoto M, Tedder TF (2008) Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J Clin Invest 118:3420–3430
Hong EG, Ko HJ, Cho YR et al (2009) Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 58:2525–2535
Kowalski GM, Nicholls HT, Risis S et al (2011) Deficiency of haematopoietic-cell-derived IL-10 does not exacerbate high-fat-diet-induced inflammation or insulin resistance in mice. Diabetologia 54:888–899
van Exel E, Gussekloo J, de Craen AJ, Frolich M, Bootsma-Van Der Wiel A, Westendorp RG (2002) Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: the Leiden 85-plus study. Diabetes 51:1088–1092
Jagannathan M, McDonnell M, Liang Y et al (2010) Toll-like receptors regulate B cell cytokine production in patients with diabetes. Diabetologia 53:1461–1471
Jagannathan-Bogdan M, McDonnell ME, Shin H et al (2011) Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol 186:1162–1172
Azar Sharabiani MT, Vermeulen R, Scoccianti C et al (2011) Immunologic profile of excessive body weight. Biomarkers 16:243–251
Papathanassoglou E, El-Haschimi K, Li XC, Matarese G, Strom T, Mantzoros C (2006) Leptin receptor expression and signaling in lymphocytes: kinetics during lymphocyte activation, role in lymphocyte survival, and response to high-fat diet in mice. J Immunol 176:7745–7752
Agrawal S, Gollapudi S, Su H, Gupta S (2011) Leptin activates human B cells to secrete TNF-alpha, IL-6, and IL-10 via JAK2/STAT3 and p38MAPK/ERK1/2 signaling pathway. J Clin Immunol 31:472–478
Lam QL, Wang S, Ko OK, Kincade PW, Lu L (2010) Leptin signaling maintains B-cell homeostasis via induction of Bcl-2 and Cyclin D1. Proc Natl Acad Sci USA 107:13812–13817
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116:3015–3025
DiLillo DJ, Yanaba K, Tedder TF (2010) B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol 184:4006–4016
Bouaziz JD, Yanaba K, Venturi GM et al (2007) Therapeutic B cell depletion impairs adaptive and autoreactive CD4+ T cell activation in mice. Proc Natl Acad Sci USA 104:20878–20883
Poggi M, Engel D, Christ A et al (2011) CD40L deficiency ameliorates adipose tissue inflammation and metabolic manifestations of obesity in mice. Arterioscler Thromb Vasc Biol 31:2251–2260
Wolf D, Jehle F, Ortiz Rodriguez A et al (2012) CD40L deficiency attenuates diet-induced adipose tissue inflammation by impairing immune cell accumulation and production of pathogenic IgG-antibodies. PLoS ONE 7:e33026
Guo CA, Kogan S, Amano SU et al (2013) CD40 deficiency in mice exacerbates obesity-induced adipose tissue inflammation, hepatic steatosis, and insulin resistance. Am J Physiol Endocrinol Metab 304:E951–E963
Hamada M, Abe M, Miyake T et al (2011) B cell-activating factor controls the production of adipokines and induces insulin resistance. Obesity (Silver Spring) 19:1915–1922
Tada F, Abe M, Kawasaki K et al (2013) B cell activating factor in obesity is regulated by oxidative stress in adipocytes. J Clin Biochem Nutr 52:120–127
Nikolajczyk BS, Jagannathan-Bogdan M, Shin H, Gyurko R (2011) State of the union between metabolism and the immune system in type 2 diabetes. Genes Immun 12:239–250
Mito N, Kaburagi T, Yoshino H, Imai A, Sato K (2006) Oral-tolerance induction in diet-induced obese mice. Life Sci 79:1056–1061
Palming J, Gabrielsson BG, Jennische E et al (2006) Plasma cells and Fc receptors in human adipose tissue–lipogenic and anti-inflammatory effects of immunoglobulins on adipocytes. Biochem Biophys Res Commun 343:43–48
Mamane Y, Chung Chan C, Lavallee G et al (2009) The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes 58:2006–2017
Shulzhenko N, Morgun A, Hsiao W et al (2011) Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med 17:1585–1593
Tilg H, Kaser A (2011) Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest 121:2126–2132
Qin J, Li Y, Cai Z et al (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490:55–60
Lam YY, Ha CW, Campbell CR et al (2012) Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS ONE 7:e34233
Wang Y, Li J, Tang L, Charnigo R, de Villiers W, Eckhardt E (2010) T-lymphocyte responses to intestinally absorbed antigens can contribute to adipose tissue inflammation and glucose intolerance during high-fat feeding. PLoS ONE 5:e13951
Wang Y, Ghoshal S, Ward M, de Villiers W, Woodward J, Eckhardt E (2009) Chylomicrons promote intestinal absorption and systemic dissemination of dietary antigen (ovalbumin) in mice. PLoS ONE 4:e8442
Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E (2009) Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res 50:90–97
Cani PD, Amar J, Iglesias MA et al (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772
Mohammed N, Tang L, Jahangiri A, de Villiers W, Eckhardt E (2012) Elevated IgG levels against specific bacterial antigens in obese patients with diabetes and in mice with diet-induced obesity and glucose intolerance. Metabolism 61:1211–1214
Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140:900–917
Alkhouri N, Gornicka A, Berk MP et al (2010) Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem 285:3428–3438
Wueest S, Rapold RA, Schumann DM et al (2010) Deletion of Fas in adipocytes relieves adipose tissue inflammation and hepatic manifestations of obesity in mice. J Clin Invest 120:191–202
Gomez-Tourino I, Camina-Darriba F, Otero-Romero I et al (2010) Autoantibodies to glial fibrillary acid protein and S100beta in diabetic patients. Diabet Med 27:246–248
Yi CX, Tschop MH, Woods SC, Hofmann SM (2012) High-fat-diet exposure induces IgG accumulation in hypothalamic microglia. Dis Model Mech 5:686–690
Winer S, Tsui H, Lau A et al (2003) Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat Med 9:198–205
Aran A, Weiner K, Lin L et al (2010) Post-streptococcal auto-antibodies inhibit protein disulfide isomerase and are associated with insulin resistance. PLoS ONE 5:e12875
Chen R, Mias GI, Li-Pook-Than J et al (2012) Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148:1293–1307
Fredrikson GN, Anand DV, Hopkins D et al (2009) Associations between autoantibodies against apolipoprotein B-100 peptides and vascular complications in patients with type 2 diabetes. Diabetologia 52:1426–1433
Zimering MB, Pan Z (2009) Autoantibodies in type 2 diabetes induce stress fiber formation and apoptosis in endothelial cells. J Clin Endocrinol Metab 94:2171–2177
Hempel P, Karczewski P, Kohnert KD et al (2009) Sera from patients with type 2 diabetes contain agonistic autoantibodies against G protein-coupled receptors. Scand J Immunol 70:159–160
Gabriel CL, Smith PB, Mendez-Fernandez YV, Wilhelm AJ, Ye AM, Major AS (2012) Autoimmune-mediated glucose intolerance in a mouse model of systemic lupus erythematosus. Am J Physiol Endocrinol Metab 303:E1313–E1324
Arai S, Maehara N, Iwamura Y et al (2013) Obesity-associated autoantibody production requires AIM to retain the immunoglobulin m immune complex on follicular dendritic cells. Cell Rep 3:1187–1198
Tuomi T, Groop LC, Zimmet PZ, Rowley MJ, Knowles W, Mackay IR (1993) Antibodies to glutamic acid decarboxylase reveal latent autoimmune diabetes mellitus in adults with a non-insulin-dependent onset of disease. Diabetes 42:359–362
Larsen CM, Faulenbach M, Vaag A et al (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356:1517–1526
Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE (2010) The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med 152:346–357
Tesfa D, Ajeganova S, Hagglund H et al (2011) Late-onset neutropenia following rituximab therapy in rheumatic diseases: association with B lymphocyte depletion and infections. Arthritis Rheum 63:2209–2214
Rauch M, Tussiwand R, Bosco N, Rolink AG (2009) Crucial role for BAFF–BAFF-R signaling in the survival and maintenance of mature B cells. PLoS ONE 4:e5456
Kyaw T, Tay C, Hosseini H et al (2012) Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS ONE 7:e29371
Sage AP, Tsiantoulas D, Baker L et al (2012) BAFF receptor deficiency reduces the development of atherosclerosis in mice: brief report. Arterioscler Thromb Vasc Biol 32:1573–1576
Kawasaki K, Abe M, Tada F et al (2013) Blockade of B-cell-activating factor signaling enhances hepatic steatosis induced by a high-fat diet and improves insulin sensitivity. Lab Invest 93:311–321
Mingrone G, Panunzi S, De Gaetano A et al (2012) Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med 366:1577–1585
Zhang H, Wang Y, Zhang J, Potter BJ, Sowers JR, Zhang C (2011) Bariatric surgery reduces visceral adipose inflammation and improves endothelial function in type 2 diabetic mice. Arterioscler Thromb Vasc Biol 31:2063–2069
Acknowledgments
This work was supported in part by NIH grant 1R01DK096038 (EE), CIHR Grant 119414 (DW), and CDA grants, OG-3-12-3844 (DW) and CS-5-12-3886 (DW).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Winer, D.A., Winer, S., Chng, M.H.Y. et al. B Lymphocytes in obesity-related adipose tissue inflammation and insulin resistance. Cell. Mol. Life Sci. 71, 1033–1043 (2014). https://doi.org/10.1007/s00018-013-1486-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-013-1486-y