
Abstract
Endonuclease V (endo V) was first discovered as the fifth endonuclease in Escherichia coli in 1977 and later rediscovered as a deoxyinosine 3′ endonuclease. Decades of biochemical and genetic investigations have accumulated rich information on its role as a DNA repair enzyme for the removal of deaminated bases. Structural and biochemical analyses have offered invaluable insights on its recognition capacity, catalytic mechanism, and multitude of enzymatic activities. The roles of endo V in genome maintenance have been validated in both prokaryotic and eukaryotic organisms. The ubiquitous nature of endo V in the three domains of life: Bacteria, Archaea, and Eukaryotes, indicates its existence in the early evolutionary stage of cellular life. The application of endo V in mutation detection and DNA manipulation underscores its value beyond cellular DNA repair. This review is intended to provide a comprehensive account of the historic aspects, biochemical, structural biological, genetic and biotechnological studies of this unusual DNA repair enzyme.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
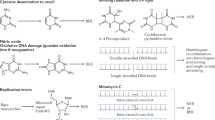
Deamination of DNA bases is a common lesion caused by endogenous and environmental agents [1–8]. By hydrolytic or nitrosative deamination, cytidine (C), adenosine (A), and guanosine (G) are converted to uridine (U), inosine (I, the corresponding base is hypoxanthine), and xanthosine (X) and oxanosine (O) (Fig. 1). The amino-to-keto conversion alters the hydrogen bonding properties of the damaged bases from a hydrogen bond donor to a hydrogen bond acceptor, which may result in mutation during DNA synthesis. As a small chemical modification occurs in DNA bases, deaminated lesions are removed from DNA by two repair pathways. The base excision repair (BER) pathway initiated by DNA glycosylase is well known for its ability to remove deaminated base damage. Enzymes in the uracil DNA glycosylase (UDG) superfamily can remove uracil, hypoxanthine, xanthine, and oxanine from DNA [9–15]. Deaminated base repair activities have been reported from E. coli AlkA and endo VIII, and mammalian AAG and NEIL1 [14, 16–18]. For additional information on the BER pathway related to repair of deaminated base damage, excellent reviews are available [19–24]. The second pathway of deaminated base repair is initiated by endonuclease V (endo V), which makes a hydrolytic nick at the 3′ side one nucleotide downstream of a lesion. This review attempts to provide a comprehensive account of the history of discovery, structure, catalytic mechanism and function, role in repair, repair pathway, and application of endonuclease V.

Chemical structures of deaminated DNA nucleosides. A adenosine, C cytidine, G guanosine, I inosine, U uridine, X xanthosine, O oxanosine
Historic aspects
The discovery of endonuclease V dates back to 1977 in Stuart Linn’s laboratory at the University of California, Berkeley [25, 26]. At the time, an endonuclease that nicks DNA but not RNA or RNA/DNA hybrid was purified from an Escherichia coli (E. coli) K12 strain deficient in endonuclease I. The 2.3 S small protein was found active on single-stranded DNA at pH 9.5, DNA treated with osmium tetroxide, DNA irradiated with ultraviolet light, DNA exposed to pH 5, and DNA isolated from Bacillus subtilis phage PBS2, which contained uracil instead of thymine in its genome. The enzyme was designated as endonuclease V, after endonuclease I, endonuclease II/exonuclease III, endonuclease III, and endonuclease IV. By convention in E. coli genetics, the corresponding gene was designated as nfi. Both endo II/exo III and endo IV are now known as AP endonucleases nicking abasic sites. Endo III is in fact a bifunctional DNA glycosylase/endolyase. In a follow-up work, additional characterization indicated that endo V also acted on AP sites and adducts of 7-bromobenzanthracene [27]. It was found that endo V acted on lesions in DNA processively [27].
In 1988, a hypoxanthine DNA glycosylase was partially purified from E. coli with a molecular weight about 56 kD and a sedimentation coefficient of 4.0 S [28]. Its enzymatic activity required Mg2+ and was completely inhibited by the presence of EDTA. This special feature is particularly interesting because another hypoxanthine DNA glycosylase purified earlier was independent of metal requirement [29]. In the early 1990s, in search for genes encoding for hypoxanthine DNA glycosylases in E. coli, Yoke Wah Kow at Emory University undertook a traditional biochemical approach to identify the molecular nature of the hypoxanthine repair activities. After 24,800-fold purification from E. coli extracts, instead of finding a Mg2+-dependent hypoxanthine DNA glycosylase, he and his coworkers found a deoxyinosine endonuclease that hydrolyzed the second phosphodiester bond 3′ to an deoxyinosine in DNA [30]. A subsequent series of biochemical studies defined the DNA repair properties of the E. coli deoxyinosine 3′ endonuclease [31–34]. In the mid 1990s, Bernard Weiss, working independently at the University of Michigan, was interested in isolating an nfi mutant of E. coli in order to study its repair properties. Based on the high level of single-stranded endonuclease activity in high pH previously detected in endo V [25, 35], he and his coworkers successfully purified E. coli endo V protein from a strain deficient in endonuclease I (encoded by endA) and UDG (encoded by ung), and identified the nfi gene from an N-terminal protein sequence revealed by Edman degradation [36]. At the same time, Yoke Wah Kow at Emory also deduced the gene sequence of the deoxyinosine 3′ endonuclease protein [33, 34]. Facilitated by Susan Wallace at the University of Vermont, when Weiss and Kow compared the genes they independently discovered, they found that the gene responsible for encoding deoxyinosine 3′ endonuclease discovered in the Kow laboratory was identical to the encoding gene for endonuclease V found in the Weiss laboratory. In a later account, the previously reported Mg2+-dependent hypoxanthine DNA glycosylase appeared to be the outcome of a combination of Mg2+-dependent phosphodiesterase and nucleotidase and nucleosidase [37]. As a note of caution, cellular endonuclease V is not to be confused with another repair enzyme of the same name from E. coli T4 phage [38, 39]. T4 endonuclease V (encoded by the denV gene) is a bifunctional DNA glycosylase that initiates repair of UV-induced pyrimidine dimer (PD) by its glycosylase and lyase activity. By coincidence, it was named endonuclease V after the endonuclease I–IV discovered in E. coli [40].
Domains, sequences, and structures
With the breakthrough made in E. coli and an increasingly larger amount of sequencing information, it soon became clear that endo V was not limited to bacteria. It is ubiquitously distributed in many species in Bacteria, Archaea, and Eukaryotes. Human endo V was initially found through EST sequencing and later located in chromosome 17q25.3. In most species, including mammals, endo V enzymes exist as small proteins of 200–300 amino acids (Fig. 2). However, fusion of the endo V domain with other domains has been detected, as summarized in Pfam [41]. In some Archaea, another DNA repair enzyme, O6-alkylguanine-DNA alkyltransferase, is fused upstream of the endo V domain (Fig. 2a). In the nematode Caenorhabditis, an Alg6_Alg8 glycosyltransferase domain is linked to the N-terminus of the endo V domain. In Entamoeba histolytica, a protein kinase domain is inserted in front of the endo V domain. The homologous sequences maintain highly conserved catalytic residues as seen in Thermotoga maritima (Tma) endo V. However, the enzymatic activity of endo V in Caenorhabditis and Entamoeba have not been demonstrated to my knowledge. The functional significance of domain fusion with endo V remains to be determined. In a PHI-BLAST search, endo V was found to share weak sequence homology with UvrC, a dual endonuclease involved in nucleotide excision repair (NER) [42]. Endo V and UvrC are classified as a superfamily in Pfam database (clan 0189). Unlike endo V, which cleaves at the 3′ side of a lesion, UvrC nicks at the 5′ side [43]. The incision at the 3′ side during NER is mediated by the GIY-YIG endonuclease domain in UvrC (Fig. 2b).

Endonuclease V and endo V domains. a Endo V domain-containing proteins. Methyltransferase: O6-alkylguanine-DNA alkyltransferase; Alg6_Alg8: Alg6_Alg8 glycosyltransferase; PKinase: protein kinase. b UvrC. GIY-YIG: GIY-YIG endonuclease; UVR: domain interacting with UvrB; Endo V-like: endonuclease domain that shares similarity with endo V; HhH: helix-hairpin-helix motif
Extensive sequence alignment of endo V family proteins uncovered several conserved motifs (Fig. 3). The DED triad (D43, E89, D110 in thermophilic bacterium Thermotoga maritima (Tma) endo V) is well established as a metal-binding site based on biochemical and structural studies, as will be detailed later. By comparison, UvrC family proteins utilize a DDH triad to coordinate a metal ion. The endo V-UvrC superfamily and the RNase H-integrase superfamily also show conservation of both the sequence and structure of the metal-binding site (Fig. 3). More information on the RNase H-integrase superfamily can be found in several review articles [44, 45]. Of particular interest is the conservation of DEDD tetrad in E. coli RNase HI with the DEDH tetrad in Tma endo V, which will be discussed in more detail under catalytic mechanism. The PIWI domain of the argonaute protein involved in RNAi shows a similar conservation as to metal coordination [46, 47]. Proteins in both superfamilies are folded as α/β proteins. They all show two highly conserved Asp residues located in the middle or at the end of a β-strand (D43 and D110 in Tma endo V, Figs. 3, 4). In endo V and RNase H1, a Glu residue sandwiched by the two Asp residues provides the third ligand from the middle of an α-helix (Figs. 3, 4). On the other hand, a His residue downstream of the two Asp residues provides the third ligand for metal binding in UvrC and Argonaute (Figs. 3, 4).

Sequence alignment of endo V and related endonucleases. GenBank accession numbers are shown after the species names. Endo V: Tma: Thermotoga maritima, NP_229661; Eco: Escherichia coli, NP_418426; Sty: Salmonella typhimurium, NP_463037; Ype: Yersinia pestis, NP_667835; Sco: Streptomyces coelicolor, CAB40676; Bsu: Bacillus subtilis, BSUB0019; Spo: Schizosaccharomyces pombe, 1723511; Cel: Caenorhabditis elegans, 1731299; Ath: Arabidopsis thaliana, T10669; Mmu: Mus musculus, XP_203558; Hsa: Homo sapiens, BAC04765; Afu: Archaeoglobus fulgidus, NP_068968; Tac: Thermoplasma acidophilum, CAC11602; Fac: Ferroplasma acidarmanus, ZP_00001774; Sso: Sulfolobus solfataricus, NP_343804; Pfu: Pyrococcus furiosus, NP_578716. UvrC: Tma: Thermotoga maritima MSB8, NP_228078.1; Tvi: Treponema vincentii ATCC 35580, ZP_05623437.1; Eco: Escherichia coli STEC_S1191, EGX18131.1; Bsu: Bacillus subtilis subsp. subtilis str. SC-8, EHA30951.1; Mtu: Mycobacterium tuberculosis H37Rv, NP_215936.1; Ngo: Neisseria gonorrhoeae F62, ZP_06643037.1. RNase H1: Eco: Escherichia coli O157:H7 str. EDL933, NP_285902.1. Argonaute: Pfu: Pyrococcus furiosus DSM 3638, NP_578266.1

Structural comparison of endo V-UvrC superfamily and RNase H superfamily. a Endo V from Thermotoga maritima (pdb, 2w35). D43, red; E89, green; D110, purple. b Endo V-like domain (residues 340–495) in UvrC from Thermotoga maritima (pdb, 2nrz). D367, red; D429, purple; H488, green. c RNase H1 from Escherichia coli (pdb 1G15). D10, red; E48, green; D70, purple. d Argonaute PIWI domain from Pyrococcus furiosus (pdb, 1U04). D558, red; D628, purple; H745, green
An E. coli endo V-DNA model was constructed using a comparative modeling approach in 2009 [48]. A few months later, Tma endo V-DNA cocrystal structures became available, which offered valuable information on endo V [49]. Currently, both endo V protein and endo V–DNA complex structures are available in the Protein Data Bank. The overall interactions between Tma endo V and DNA are shown in Fig. 5a. The cocrystal structures of endo V–DNA complex present several interesting features that explain the base recognition mechanism and several enzymatic properties. Similar to what has been observed in other repair enzymes and methylases [50–52], the damaged base, in this case hypoxanthine, is flipped out of the helix by a 90° rotation (Fig. 5b). To lock the flipped base in the base recognition pocket, endo V inserts a highly conserved PYIP wedge into the space vacated by the hypoxanthine base (Fig. 5b). An invariant Tyr residue (Y80 in Tma endo V) in motif III acts like a surrogate base in the DNA helix (Fig. 5b). Substitutions of the Tyr residue in Tma endo V exert a significant effect on the enzymatic behavior. The wild-type Tma endo V is a single-turnover enzyme, as the product turnover is limited by very slow product dissociation after the nicking event [53]. When the Tyr residue in the wedge was substituted by Phe, which still maintained the phenyl ring, the Y80F mutant enzyme showed modest reduction in binding affinity to hypoxanthine and still retained the single-turnover property [54]. However, when the Tyr residue was substituted by Ala, the Y80A mutant enzyme bound more weakly to both the deoxyinosine-containing substrate and the nicked deoxyinosine-containing product and behaved as a multiple turnover enzyme [54, 55]. Therefore, the wedge centered around the invariant Tyr residue is required to maintain a high affinity to the hypoxanthine-containing DNA. Without the Tyr residue inserted into the helix, the hypoxanthine base may flip back to the helix and cause dissociation of the DNA.

Protein–DNA interactions in Tma endonuclease V. a Overall structure of endo V-inosine-containing DNA complex. b Flipping of hypoxanthine base and insertion of PYIP wedge into DNA. Y80 is shown in red and inosine is shown in element style. c Hydrophobic packing of hypoxanthine base. L85, yellow; L142, red. Inosine is shown in element style. d Base recognition pocket in Tma endo V. G83, green; L85, pink; Q112, orange; H116, silver; I122, lime. Inosine is shown in element style. e Tautomerization of hypoxanthine and base recognition by mainchain interactions. f Interactions with 5′-phosphate at the scissile bond in the post cleavage (PC) complex. K139 and H214 are shown in green
The cocrystal structures also reveal two interesting features in the base recognition pocket [49]. First, the hypoxanthine base is packed between two hydrophobic residues, L85 in motif III and L142 in motif IV (Fig. 5c). Second, the recognition of the hypoxanthine base is mediated completely by mainchain interactions (Fig. 5d). Based on the structural information, it was proposed that the hypoxanthine may experience tautomerization when complexed with endo V (Fig. 5e). The deprotonation at the N1 position allows hydrogen bonding with the mainchain-NH from I122. The resulting OH at the C6 position is stabilized by interactions with G83 and L85. Recognition of N3 and N7 is mediated by the mainchain-NH from G83 and Q112 (Fig. 5e). A similar tautomerization model was proposed for xanthine recognition [49]. The recognition pocket defined by mainchain interactions may underlie the ability of endo V to accommodate a variety of damaged bases and mismatched base pairs. In the post-cleavage complex, the 5′-phosphate generated by the nicking at the second phosphodiester bond downstream of the inosine position is coordinated by the sidechains of K139 and H214 residues (Fig. 5f). The phosphate backbone interactions, wedging effect, hydrophobic packing and mainchain interactions in the recognition pocket enable endo V to maintain tight binding to the nicked inosine-containing DNA. Loss of this tight binding is accompanied by conversion of endo V from a single-turnover enzyme to a multiple turnover enzyme [54].
Catalytic mechanism
As an authentic endonuclease, endo V is a metal-dependent enzyme. In addition to Mg2+, Mn2+, Co2+ or Ni2+ can serve as the metal cofactor for its enzymatic activity [25, 30, 56, 57]. The metal coordination is well defined in the crystal structures [49]. The D43 in motif II and the D110 in motif IV directly coordinate the Mg2+ ion through the sidechains, while the E89 in motif III interacts with the Mg2+ ion through water molecules (Fig. 6). This Mg2+ ion coordinated by the DED triad is responsible for the cleavage of the phosphodiester bond [49]. As mentioned earlier, of particular interest is the high-level resemblance of metal coordination between endo V and RNase H1 both sequentially and structurally (Figs. 3, 6). In both cases, the catalytic metal ion, which activates a water molecule for the hydrolysis reaction, is coordinated by a DED triad, which is D43–E89–D110 in endo V and D10–E48–D70 in RNase H1 [49, 58]. In the crystal structure, a single Mg2+ ion was observed in the active site [49]. Indeed, endo V appeared to follow single-metal kinetics in the presence of Mg2+ [59]. However, a complicated behavior was observed when Mn2+ was used as a metal cofactor [59]. The endonuclease activity was initially inhibited with increasing Mn2+ concentrations, but later with additional increases of Mn2+ concentrations, the activity was enhanced [59]. Such a peculiar behavior led to the speculation that endo V may possess two-metal-binding sites for certain metal ions such as Mn2+ [59]. A subsequent study provided more experimental support of the catalytic and regulatory two-metal model [56]. This study took advantage of an experimental approach of using dual metal ions in the assay system to discern two-metal mechanisms, which was previously applied in the study of restriction endonuclease EcoRV and human AP endonuclease Ape1 [60–62]. In the Mn2+–Ca2+ combination, in which Ca2+ was known as a catalytically inactive metal ion for endo V, DNA cleavage activity was stimulated [56]. In the Mg2+–Mn2+ combination, the DNA cleavage activity was stimulated at low Mn2+ concentrations and then inhibited as the Mn2+ concentration increased [56]. Data from the metal combination experiments have led to a two-metal catalytic and regulatory model, in which the catalytic high affinity metal-binding site (M1) possesses affinity to metal ions in the order of Mg2+ > Mn2+ > Ca2+ and the regulatory low affinity metal-binding site (M2) binds metal ions in the order of Ca2+ > Mn2+ > Mg2+ (Fig. 6). In the crystal structure of E. coli RNase H1, two Mn2+ ions were observed, with the activating Mn2+ coordinated by D10–E48–D70 and the attenuating Mn2+ coordinated by D10–D134 (Fig. 6b) [58]. In the RNA/DNA complex structure of Bacillus halodurans RNase H, two Mg2+ ions are found in the active site [63]. In the endo V–DNA–Mg2+ structure, the observation of a single Mg2+ ion is probably due to the rather low affinity of the M2 site to Mg2+. Two-metal models have been proposed for a variety of other endonucleases as well [64–66], indicating that the use of multiple metal ions in catalysis and regulation of catalysis is a common theme in hydrolysis of phosphodiester bonds in nucleic acids. The readers are referred to recent reviews for further information on the roles of metal ions in nucleases [67, 68].

Metal coordination in Tma endo V and E. coli RNase HI. a Coordination of Mg2+ ion in the active site of Tma endo V by D43–E89–D110. D43, red; E89, green; D110, purple. Mg2+, black. b Coordination of two Mn2+ ions in the active site of E. coli RNase HI by D10–E48–D70–D134. D10, red; E48, green, D70, purple; D134, blue. Mn2+, silver. c Position of H214 of Tma endo V relative to the DED metal-binding triad shown in (a) H214, blue. e Catalytic and regulatory two-metal model for endonuclease V. The catalytic metal is shown as a solid black circle. The regulatory metal is shown as a solid black circle followed by a question mark
Endo V family enzymes in Bacteria, Archaea, and Eukaryotes
Endo V is ubiquitously distributed in nature. In bacteria, endo V can be found in Gram-negative and Gram-positive organisms, in mesophiles and thermophiles, and in nonpathogens and pathogens. In Archaea, endo V can be found in euryarchaeotes, crenarchaeotes, thaumarchaeota, candidatus, and nanoarchaeum. In eukaryotes, endo V is found in both single-cell organisms such as fission yeast Schizosaccharomyces pombe (but not in budding yeast Saccharomyces cerevisiae) and multi-cellular organisms, in algae and plants, and in animals including mammals.
Bacterial endo V enzymes have been studied the most. As stated above, endo V was discovered and rediscovered in E. coli. E. coli endo V was the first enzyme to be extensively characterized biochemically. E. coli endo V was initially discovered as an enzyme that degraded deoxyuridine-containing DNA in Bacillus subtilis phage PBS2 [25, 27]. After the rediscovery of endo V as a deoxyinosine endonuclease [30], E. coli endo V was found as a deoxyxanthosine endonuclease and a deoxyuridine endonuclease using lesion-containing deoxyoligonucleotide substrates [34, 69]. The enzyme also recognizes urea and AP sites but not 8-oxoguanine or 8-oxoadenine [30]. E. coli endo V can recognize mismatched base pairs except for C/A, C/T, and C/C, which are recognized poorly [31]. In the presence of Mn2+, E. coli endo V can even cleave insertions/deletions, flap, and pseudo-Y DNA structures [33]. Gel mobility shift analysis revealed that E. coli endo V bound to both deoxyinosine-containing DNA and nicked products tightly, but bound to deoxyuridine-containing DNA weakly [32, 34]. At high enzyme concentrations, E. coli endo V formed two complexes with deoxyinosine-containing DNA. While the lower molecular weight complex protected 4–5 nucleotides 5′ to the deoxyinosine, formation of the second complex extended the protection to at least 13 nucleotides 3′ to deoxyinosine, suggesting the possibility that a second E. coli endo V molecule bound to the primary complex through protein–protein interactions [32]. A close homolog of E. coli endo V, Salmonella typhimurium endo V was also characterized [70]. Interestingly, S. typhimurium endo V cleaved mismatched base pairs more evenly and was the only endo V that showed a retarded band in binding assays using deoxyoxanosine-containing DNA [59].
In the late 1990s, the release of raw sequencing data by The Institute of Genome Research (TIGR) facilitated manual assembly of an endo V homolog found in the thermophilic bacterium Thermotoga maritima before the whole-genome assembly became available. Tma endo V offers an excellent model for biochemical and structural investigations. The thermostable nature of the Tma endo V allows for purification of site-directed mutant enzymes with ease. The wild-type Tma endo V shows similarly broad substrate specificity as the E. coli enzyme [53, 59]. Like many other nucleases, the specificity of Tma endo V is relaxed with Mn2+ as a metal cofactor. Its endonuclease activity on mismatched base pairs and its nonspecific nuclease activity on undamaged DNA are enhanced in the presence of Mn2+ [53]. Its ability to recognize and nick mismatched base pairs has been used in mutation detection, as described later; however, there is no indication that such activity plays a role in mismatch repair in vivo. The exonucleases in Tma endo V described below are also Mn2+-dependent. The single-turnover nature of Tma endo V on the T/I substrate was demonstrated by kinetics analysis [53]. As a result, the rate of cleavage appeared higher with the T/U substrate than with the T/I substrate. This is due to the high affinity of endo V to inosine-containing substrates [53]. The caveat is that a substrate with a higher rate due to multiple turnover in vitro may not be the physiological substrate in vivo. This has been demonstrated in endo V by in vivo experiments [71]. Through a series of mutational analysis, amino acid residues important for interactions with DNA and catalysis were identified [54–56]. Using a fluorescence resonance energy transfer (FRET)-based system, the single-molecule behavior of Tma endo V on single-stranded deoxyinosine-containing DNA was studied [72]. The oligonucleotide substrate was labeled at the 3′ side with a TAMRA fluorescence group. The inosine site was four nucleotides away from the 3′ end. The single-molecule catalytic cycle of approximately 30 s on this substrate can be characterized by an association phase (5.9 s), recognition/cleavage phase (14.5 s) and a cleavage/dissociation phase (9.1 s).
A particularly interesting observation made in Tma endo V is that of multiple exonuclease activities. Previous studies raised the prospect that E. coli endo V contains weak 5′ exonuclease activity [33], but definitive proof was difficult to find due to multiple exonuclease activities in the E. coli host. The study of exonuclease activities is facilitated by the thermostable nature of Tma endo V, which allows for removal of the host nucleases by heat treatment. The availability of a variety of active site mutant proteins offers additional means to rule out nuclease contamination from the host. A study using 5′, 3′ and internally labeled oligonucleotide substrates in the presence of Mn2+ establishes that Tma endo V is not only a lesion-specific endonuclease, but also an unusual lesion-dependent 3′ exonuclease and nonspecific 5′ exonuclease [73]. The implication of these activities in DNA repair pathway will be discussed later.
Knowledge of archaeal endo V enzymes is limited. Studies on endo V from Archaeoglobus fulgidus suggest that it only recognizes deoxyinosine [74]. However, endo V from Ferroplasma acidarmanus, in which the endo V is fused downstream of an O6-alkylguanine-DNA alkyltransferase, is active on deoxyinosine-, deoxanthosine- and deoxyuridine-containing DNA [75]. Deoxyinosine endonuclease activity is also reported in endo V from Pyrococcus furiosus [76], but its substrate specificity remains to be determined.
Mammalian endo V proteins are larger than their prokaryotic homologs. Mouse and human endo V contain 338 and 282 amino acids, respectively. An earlier study indicates that mouse endo V is a deoxyinosine endonuclease with the highest activity on single-stranded deoxyinosine-containing DNA [77]. Human endo V gene, located in chromosome 17q25.3, shares about 30 % of its sequence with bacterial endo V. A recent study revealed that human endo V is primarily a deoxyinosine endonuclease but with minor deoxyxanthosine endonuclease activity [57]. The endonuclease activity on deoxyinosine-containing DNA follows the order of single-stranded I > G/I > T/I > A/I > C/I. Like bacterial endo V enzymes, human endo V is most active with Mg2+ ion, but can also use Mn2+, Ni2+, and Co2+ as a metal cofactor. Unlike bacterial endo V enzymes, human endo V shows no detectable endonuclease activity on deoxyoxanosine and deoxyuridine. The human enzyme is not as robust as bacterial enzymes. For example, the apparent rate constant measure under single turnover condition for single-stranded deoxyinosine-containing DNA was 0.1/min for S. typhimurium endo V versus 0.025/min for human endo V [57, 70].
Roles of endo V in DNA repair
When it was first discovered, endo V was known as a DNA repair enzyme. However, it was not understood until after its rediscovery which of its broad specificities were related to DNA repair in vivo. Genetic analysis using nfi deletion strains shows that E. coli endo V prevents mutations due to nitrosative damage but does not have significant effects on hydrogen peroxide exposure, and γ-ray and UV irradiation [71]. Because nitrous acid is known for its ability to deaminate DNA bases, the genetic effects E. coli endo V exhibited correlate well with the rediscovery of endo V as a deamination repair enzyme. The initial genetic indication that endo V is involved in repair of adenine deamination comes from increased frequency of nitrite-induced mutations to streptomycin resistance in an nfi mutant [71]. Using trp and lac as indicators, a more detailed study showed a markedly increased frequency of nitrous acid-induced A/T to G/C and G/C to A/T transition mutations in E. coli nfi mutants [78]. These results indicate that the nfi gene is an antimutator under nitrosative stress enabling the repair of adenine and guanine base damage. By the same mechanism, endo V also plays a role in repair of nitrosative deamination during nitrate/nitrite respiration in E. coli [79]. In addition to repair of adenine and guanine deamination, genetic analysis also indicates E. coli endo V in the repair of AP sites and N6-hydroxylaminopurine (HAP) [71, 80]. The single-stranded endonuclease activity occurring at high pH as initially reported, however, does not seem to be relevant in vivo [25, 71].
Endo V was initially found active on uracil-containing Bacillus phage PBS2 DNA and deoxyuridine-containing deoxyoligonucleotides [25, 34]. Through a careful analysis using a combination of E. coli ung and nfi mutants [71], endo V did not appear to play any significant role in the repair of cytosine deamination in vivo. However, a recent study found that the deletion of the nfi gene alone or in combination with the ung gene in B. subtilis greatly increased mutation frequency induced by bisulfite treatment, which deaminated cytosine to uracil [81]. This observation raises the possibility that the nfi gene in B. subtilis is involved in repair of uracil.
Endo V enzymes from eukaryotic sources appear to play a similar antimutator role in vivo. Lack of nfi gene in fission yeast Schizosaccharomyces pombe elicits a mutator phenotype [49]. Both mouse and human nfi genes complement an nfi mutant in E. coli [57, 77]. Most profoundly, nfi −/− knockout mice showed a cancer-prone phenotype, suggesting that endo V-initiated repair is an important mechanism in maintaining genome integrity [49].
Another interesting feature is the ability of endo V to cause double-strand breaks. The E. coli nfi mutants showed a slightly increased survival than the wild-type strain after nitrous acid treatment [71]. It is conceivable that if two deaminated bases in the opposing strands are close to each other, nicking at both strands by endo V will result in a double strand break, which is more detrimental to cell survival than base damage. Interestingly, Tma endo V is able to cleave DNA with two inosine opposing each other as an I/I base pair and two molecules of endo V can bind to the I/I base pair [53]. The ability of endo V to cleave single-stranded deoxyinosine-containing DNA also facilitates the generation of double-strand breaks. Indeed, the nfi gene was found to suppress the lethality caused by rdgB recA in E. coli [82]. The rdgB mutation, which affects dITPase activity, results in incorporation of deoxyinosine into DNA.
Endo V-initiated repair pathway
A typical DNA repair pathway for removal of base lesions requires damage recognition, strand nicking, damage removal, repair synthesis and nick sealing. Obviously, endo V plays the initiating role in base recognition and strand nicking (Fig. 7). Because endo V nicks at the 3′ side of the lesion, the base damage is not removed from DNA. Several proposals have been put forward to explain the removal of the damaged base by 3′ exonuclease or endonuclease action [20, 54, 69, 77, 80, 83]. In one proposal, using a circular plasmid-based in vitro system, E. coli DNA polymerase I is implicated as the enzyme to remove an inosine lesion from DNA due to its 3′ exonuclease activity [83]. Subsequently, DNA pol I and DNA ligase can carry out repair synthesis and nick sealing. However, because endo V remains bound to the nicked DNA after the initial endonucleolytic cleavage, it remains to be determined whether endo V can be displaced by DNA pol I.

Proposed pathway for endo V-initiated repair. I. inosine. Endo V is shown as an oval in yellow. Pol I, E. coli DNA polymerase I
Another proposal is based on the observation that Tma endo V not only possesses lesion-specific endonuclease activity, but also displays 3′ exonuclease activity in vitro [54]. A more detailed biochemical study more clearly revealed that Tma endo V can act as a lesion-specific 3′ exonuclease and a nonspecific 5′ exonuclease in the presence of Mn2+ [73]. These unusual combination of enzymatic activities have led to the suggestion that in the cellular environment and with the assistance of a partner protein, Tma endo V may switch from an endonuclease mode to exonuclease mode upon making the initial nick [54, 73]. The lesion-dependent 3′ exonuclease activity will allow the enzyme to remove the inosine lesion from DNA and the nonspecific and weak 5′ exonuclease activity will expand the gap. The inosine site will be reverted back to an adenosine site through repair synthesis by a DNA polymerase and the nick sealed by a DNA ligase.
In an elegant study using an nfi deletion strain and deoxyinosine-containing oligonucleotides, endo V-initiated repair was studied in vivo [84]. In E. coli cells, it appears that the vast majority of deoxyinosine repair is carried out by endo V. More importantly, this study provides valuable information on the repair patch. Using a clever heteroduplex strategy, it was found that the repair patch encompassed three nucleotides 3′ and two nucleotides 5′ from the initial cleavage site [84]. A salient but interesting inference from this study is that it appears that the creation of the gap involves both 3′ and 5′ nuclease activities.
Application of endo V in biotechnology
The unusual enzymatic properties not only made endo V an interesting repair enzyme, but it also attracted interest in exploiting its use in biotechnology. Endo V’s ability to recognize and nick mismatched base pairs has led to its use in mutation detection. In one case, E. coli endo V and T/G DNA glycosylase are used to detect mutations in the BRCA1 gene [85]. In another approach, Tma endo V is combined with thermostable DNA ligase for mutation scanning [86, 87]. Because Tma endo V has a weak nonspecific endonuclease activity [53], the addition of high-fidelity DNA ligase serves to reduce spurious nicks. The native Tma endo V enzyme is weak in cleavage of C-containing mismatches. As described earlier, an Ala substitution at Y80 position renders the enzyme capable of multiple turnovers on inosine-containing DNA [55]. A further analysis found that the Y80A mutant enzyme also altered the base preference of Tma endo V so that it now cleaves C-containing mismatches [88]. It was shown that the Y80A mutant enzyme enhanced the ability of Tma endo V to cleave an A/C mismatch in the K-ras G13D sequence [88]. In addition to mutation detection, the nicking activity of Tma endo V has been used in combination with DNA polymerase from Bacillus stearothermophilus to amplify DNA by strand displacement [89].
DNA shuffling is a technique to recombine homologous sequences in vitro. The ability of endo V to nick uracil-containing DNA is explored for use in DNA shuffling as well [90]. Previous DNA shuffling techniques for protein evolution relied on DNase I to generate nicks on DNA [91, 92]. An alternative method was developed based on incorporation of dUMP into DNA by PCR and subsequent fragmentation near the uracil sites by E. coli endo V [90]. The success of this method was demonstrated by regeneration of a full-length green fluorescence protein. A more recent method added a random mutagenesis step mediated by dITP incorporation prior to DNA fragmentation by endo V [93]. Random mutations were generated by the pairing of dITP with different bases during PCR. The transition mutations were still the predominant type in the mutation pool. The mutation frequency was adjusted by dNTP/dITP ratio. DNA shuffling was carried out by endo V and the gene assembled by PCR.
Concluding remarks
Endonuclease V was first discovered as the fifth endonuclease in E. coli in 1977 and rediscovered 17 years later as a deoxyinosine 3′ endonuclease. Since then, tremendous progress has been made on understanding its structure and function relationship and its role as a DNA repair enzyme. Its ability to recognize a variety of deaminated lesions enables endo V to repair base damage in vivo. Its ability to act on mismatched base pairs offers a useful tool for detection of single base changes. Its ability to initiate DNA amplification and DNA shuffling takes it from deaminated lesion removal to genetic manipulations. The unusual properties exhibited by endo V for its repair functions and application value depend on its unique structural arrangement in the active site. The importance of endo V-initiated repair should not be underestimated given its role in reducing mutation load and preventing cancer development in mammals. Looking ahead, there are still fundamental questions to be answered. The most outstanding one is the downstream process after the endonucleolytic cleavage at the DNA lesion. Even though several candidate enzymes have been proposed, the real player involved in the removal of damaged bases in vivo has yet to be identified or validated. Given the role endo V plays in cancer etiology, it is essential to understand the physiological and pathological role of the nfi gene in mammalian systems. The effect the lack of an nfi gene has on other tumor suppressor genes and oncogenes needs to be investigated. The two-metal model needs additional structural details to validate Mn2+ coordination. Given the history of endo V’s discovery, it would not be surprising if more surprises are on the horizon.
References
Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR (1999) The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res 424:37–49
Dedon PC, Tannenbaum SR (2004) Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys 423:12–22
Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362:709–715
Nguyen T, Brunson D, Crespi CL, Penman BW, Wishnok JS, Tannenbaum SR (1992) DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc Natl Acad Sci USA 89:3030–3034
Shapiro R (1981) Damage to DNA caused by hydrolysis. In: Seeberg E, Kleppe K (eds) Chromosome damage and repair. Plenum Press, New York, pp 3–18
Spencer JP, Whiteman M, Jenner A, Halliwell B (2000) Nitrite-induced deamination and hypochlorite-induced oxidation of DNA in intact human respiratory tract epithelial cells. Free Radic Biol Med 28:1039–1050
Wink DA, Kasprzak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WH, Andrews AW, Allen JS et al (1991) DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 254:1001–1003
Suzuki T, Yamaoka R, Nishi M, Ide H, Makino K (1996) Isolation and characterization of a novel product, 2′-deoxyoxanosine, from 2′-deoxyguanosine, oligodeoxynucleotide and calf thymus DNA treated by nitrous-acid and nitric-oxide. J Am Chem Soc 118:2515–2516
Parikh SS, Putnam CD, Tainer JA (2000) Lessons learned from structural results on uracil-DNA glycosylase. Mutat Res 460:183–199
Pearl LH (2000) Structure and function in the uracil-DNA glycosylase superfamily. Mutat Res 460:165–181
Mi R, Dong L, Kaulgud T, Hackett KW, Dominy BN, Cao W (2009) Insights from xanthine and uracil DNA glycosylase activities of bacterial and human SMUG1: switching SMUG1 to UDG. J Mol Biol 385:761–778
Lee HW, Brice AR, Wright CB, Dominy BN, Cao W (2010) Identification of Escherichia coli mismatch-specific uracil DNA glycosylase as a robust xanthine DNA glycosylase. J Biol Chem 285:41483–41490
Lee HW, Dominy BN, Cao W (2011) New family of deamination repair enzymes in uracil-DNA glycosylase superfamily. J Biol Chem 286:31282–31287
Dong L, Meira LB, Hazra TK, Samson LD, Cao W (2008) Oxanine DNA glycosylase activities in mammalian systems. DNA Repair 7:128–134
Dong L, Mi R, Glass RA, Barry JN, Cao W (2008) Repair of deaminated base damage by Schizosaccharomyces pombe thymine DNA glycosylase. DNA Repair 7:1962–1972
Hitchcock TM, Dong L, Connor EE, Meira LB, Samson LD, Wyatt MD, Cao W (2004) Oxanine DNA glycosylase activity from mammalian alkyladenine glycosylase. J Biol Chem 279:38177–38183
Saparbaev M, Laval J (1994) Excision of hypoxanthine from DNA containing dIMP residues by the Escherichia coli, yeast, rat, and human alkylpurine DNA glycosylases. Proc Natl Acad Sci USA 91:5873–5877
Terato H, Masaoka A, Asagoshi K, Honsho A, Ohyama Y, Suzuki T, Yamada M, Makino K, Yamamoto K, Ide H (2002) Novel repair activities of AlkA (3-methyladenine DNA glycosylase II) and endonuclease VIII for xanthine and oxanine, guanine lesions induced by nitric oxide and nitrous acid. Nucleic Acids Res 30:4975–4984
Cortazar D, Kunz C, Saito Y, Steinacher R, Schar P (2007) The enigmatic thymine DNA glycosylase. DNA Repair 6:489–504
Kow YW (2002) Repair of deaminated bases in DNA. Free Radic Biol Med 33:886–893
Krokan HE, Drablos F, Slupphaug G (2002) Uracil in DNA—occurrence, consequences and repair. Oncogene 21:8935–8948
Dalhus B, Laerdahl JK, Backe PH, Bjoras M (2009) DNA base repair–recognition and initiation of catalysis. FEMS Microbiol Rev 33:1044–1078
Barnes DE, Lindahl T (2004) Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet 38:445–476
Stivers JT, Jiang YL (2003) A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem Rev 103:2729–2759
Gates FT 3rd, Linn S (1977) Endonuclease V of Escherichia coli. J Biol Chem 252:1647–1653
Linn S (2012) Life in the serendipitous lane: excitement and gratification in studying DNA repair. DNA Repair 11:595–605
Demple B, Linn S (1982) On the recognition and cleavage mechanism of Escherichia coli endodeoxyribonuclease V, a possible DNA repair enzyme. J Biol Chem 257:2848–2855
Harosh I, Sperling J (1988) Hypoxanthine-DNA glycosylase from Escherichia coli. Partial purification and properties. J Biol Chem 263:3328–3334
Karran P, Lindahl T (1978) Enzymatic excision of free hypoxanthine from polydeoxynucleotides and DNA containing deoxyinosine monophosphate residues. J Biol Chem 253:5877–5879
Yao M, Hatahet Z, Melamede RJ, Kow YW (1994) Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3′ endonuclease, from Escherichia coli. J Biol Chem 269:16260–16268
Yao M, Kow YW (1994) Strand-specific cleavage of mismatch-containing DNA by deoxyinosine 3′-endonuclease from Escherichia coli. J Biol Chem 269:31390–31396
Yao M, Kow YW (1995) Interaction of deoxyinosine 3′-endonuclease from Escherichia coli with DNA containing deoxyinosine. J Biol Chem 270:28609–28616
Yao M, Kow YW (1996) Cleavage of insertion/deletion mismatches, flap and pseudo-Y DNA structures by deoxyinosine 3′-endonuclease from Escherichia coli. J Biol Chem 271:30672–30676
Yao M, Kow YW (1997) Further characterization of Escherichia coli endonuclease V. J Biol Chem 272:30774–30779
Demple B, Gates FT, 3rd, Linn S (1980) Purification and properties of Escherichia coli endodeoxyribonuclease V. Methods Enzymol 65:224–231
Guo G, Ding Y, Weiss B (1997) nfi, the gene for endonuclease V in Escherichia coli K-12. J Bacteriol 179:310–316
Dianov G, Lindahl T (1991) Preferential recognition of I.T base-pairs in the initiation of excision-repair by hypoxanthine-DNA glycosylase. Nucleic Acids Res 19:3829–3833
Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T (2006) DNA repair and mutagenesis, 2nd edn. ASM Press, Washington, DC
Sekiguchi M (2012) My path toward DNA repair. DNA Repair 11:606–615
Yasuda S, Sekiguchi M (1970) T4 endonuclease involved in repair of DNA. Proc Natl Acad Sci USA 67:1839–1845
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD (2012) The Pfam protein families database. Nucleic Acids Res 40:D290–301
Aravind L, Walker DR, Koonin EV (1999) Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res 27:1223–1242
Van Houten B, Croteau DL, DellaVecchia MJ, Wang H, Kisker C (2005) ‘Close-fitting sleeves’: DNA damage recognition by the UvrABC nuclease system. Mutat Res 577:92–117
Nowotny M (2009) Retroviral integrase superfamily: the structural perspective. EMBO Rep 10:144–151
Haren L, Ton-Hoang B, Chandler M (1999) Integrating DNA: transposases and retroviral integrases. Annu Rev Microbiol 53:245–281
Rand TA, Ginalski K, Grishin NV, Wang X (2004) Biochemical identification of Argonaute 2 as the sole protein required for RNA-induced silencing complex activity. Proc Natl Acad Sci USA 101:14385–14389
Song JJ, Smith SK, Hannon GJ, Joshua-Tor L (2004) Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305:1434–1437
Majorek KA, Bujnicki JM (2009) Modeling of Escherichia coli endonuclease V structure in complex with DNA. J Mol Model 15:173–182
Dalhus B, Arvai AS, Rosnes I, Olsen OE, Backe PH, Alseth I, Gao H, Cao W, Tainer JA, Bjoras M (2009) Structures of endonuclease V with DNA reveal initiation of deaminated adenine repair. Nat Struct Mol Biol 16:138–143
Huffman JL, Sundheim O, Tainer JA (2005) DNA base damage recognition and removal: new twists and grooves. Mutat Res 577:55–76
Roberts RJ, Cheng X (1998) Base flipping. Annu Rev Biochem 67:181–198
Scharer OD, Campbell AJ (2009) Wedging out DNA damage. Nat Struct Mol Biol 16:102–104
Huang J, Lu J, Barany F, Cao W (2001) Multiple cleavage activities of endonuclease V from Thermotoga maritima: recognition and strand nicking mechanism. Biochemistry 40:8738–8748
Feng H, Dong L, Klutz AM, Aghaebrahim N, Cao W (2005) Defining amino acid residues involved in DNA–protein interactions and revelation of 3′-exonuclease activity in endonuclease V. Biochemistry 44:11486–11495
Huang J, Lu J, Barany F, Cao W (2002) Mutational analysis of endonuclease V from Thermotoga maritima. Biochemistry 41:8342–8350
Feng H, Dong L, Cao W (2006) Catalytic mechanism of endonuclease v: a catalytic and regulatory two-metal model. Biochemistry 45:10251–10259
Mi R, Alford-Zappala M, Kow YW, Cunningham RP, Cao W (2012) Human endonuclease V as a repair enzyme for DNA deamination. Mutat Res 735:12–18
Goedken ER, Marqusee S (2001) Co-crystal of Escherichia coli RNase HI with Mn2+ ions reveals two divalent metals bound in the active site. J Biol Chem 276:7266–7271
Hitchcock TM, Gao H, Cao W (2004) Cleavage of deoxyoxanosine-containing oligodeoxyribonucleotides by bacterial endonuclease V. Nucleic Acids Res 32:4071–4080
Horton NC, Perona JJ (2004) DNA cleavage by EcoRV endonuclease: two metal ions in three metal ion binding sites. Biochemistry 43:6841–6857
Kostrewa D, Winkler FK (1995) Mg2+ binding to the active site of EcoRV endonuclease: a crystallographic study of complexes with substrate and product DNA at 2 A resolution. Biochemistry 34:683–696
Vipond IB, Baldwin GS, Halford SE (1995) Divalent metal ions at the active sites of the EcoRV and EcoRI restriction endonucleases. Biochemistry 34:697–704
Nowotny M, Gaidamakov SA, Crouch RJ, Yang W (2005) Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 121:1005–1016
Pingoud V, Wende W, Friedhoff P, Reuter M, Alves J, Jeltsch A, Mones L, Fuxreiter M, Pingoud A (2009) On the divalent metal ion dependence of DNA cleavage by restriction endonucleases of the EcoRI family. J Mol Biol 393:140–160
Noble CG, Maxwell A (2002) The role of GyrB in the DNA cleavage-religation reaction of DNA gyrase: a proposed two metal-ion mechanism. J Mol Biol 318:361–371
Zheng L, Li M, Shan J, Krishnamoorthi R, Shen B (2002) Distinct roles of two Mg2+ binding sites in regulation of murine flap endonuclease-1 activities. Biochemistry 41:10323–10331
Dupureur CM (2010) One is enough: insights into the two-metal ion nuclease mechanism from global analysis and computational studies. Metallomics: integrated biometal science 2:609–620
Yang W (2011) Nucleases: diversity of structure, function and mechanism. Q Rev Biophys 44:1–93
He B, Qing H, Kow YW (2000) Deoxyxanthosine in DNA is repaired by Escherichia coli endonuclease V. Mutat Res 459:109–114
Feng H, Klutz AM, Cao W (2005) Active site plasticity of endonuclease V from Salmonella typhimurium. Biochemistry 44:675–683
Guo G, Weiss B (1998) Endonuclease V (nfi) mutant of Escherichia coli K-12. J Bacteriol 180:46–51
Lin J, Gao H, Schallhorn KA, Harris RM, Cao W, Ke PC (2007) Lesion recognition and cleavage by endonuclease V: a single-molecule study. Biochemistry 46:7132–7137
Mi R, Abole AK, Cao W (2011) Dissecting endonuclease and exonuclease activities in endonuclease V from Thermotoga maritima. Nucleic Acids Res 39:536–544
Liu J, He B, Qing H, Kow YW (2000) A deoxyinosine specific endonuclease from hyperthermophile. Archaeoglobus fulgidus: a homolog of Escherichia coli endonuclease V. Mutat Res 461:169–177
Kanugula S, Pauly GT, Moschel RC, Pegg AE (2005) A bifunctional DNA repair protein from Ferroplasma acidarmanus exhibits O6-alkylguanine-DNA alkyltransferase and endonuclease V activities. Proc Natl Acad Sci USA 102:3617–3622
Emptage K, O’Neill R, Solovyova A, Connolly BA (2008) Interplay between DNA polymerase and proliferating cell nuclear antigen switches off base excision repair of uracil and hypoxanthine during replication in archaea. J Mol Biol 383:762–771
Moe A, Ringvoll J, Nordstrand LM, Eide L, Bjoras M, Seeberg E, Rognes T, Klungland A (2003) Incision at hypoxanthine residues in DNA by a mammalian homologue of the Escherichia coli antimutator enzyme endonuclease V. Nucleic Acids Res 31:3893–3900
Schouten KA, Weiss B (1999) Endonuclease V protects Escherichia coli against specific mutations caused by nitrous acid. Mutat Res 435:245–254
Weiss B (2001) Endonuclease V of Escherichia coli prevents mutations from nitrosative deamination during nitrate/nitrite respiration. Mutat Res 461:301–309
Burgis NE, Brucker JJ, Cunningham RP (2003) Repair system for noncanonical purines in Escherichia coli. J Bacteriol 185:3101–3110
Lopez-Olmos K, Hernandez MP, Contreras-Garduno JA, Robleto EA, Setlow P, Yasbin RE, Pedraza-Reyes M (2012) Roles of endonuclease V, uracil-DNA glycosylase, and mismatch repair in Bacillus subtilis DNA base-deamination-induced mutagenesis. J Bacteriol 194:243–252
Bradshaw JS, Kuzminov A (2003) RdgB acts to avoid chromosome fragmentation in Escherichia coli. Mol Microbiol 48:1711–1725
Lee CC, Yang YC, Goodman SD, Yu YH, Lin SB, Kao JT, Tsai KS, Fang WH (2010) Endonuclease V-mediated deoxyinosine excision repair in vitro. DNA Repair 9:1073–1079
Weiss B (2008) Removal of deoxyinosine from the Escherichia coli chromosome as studied by oligonucleotide transformation. DNA Repair 7:205–212
Bazar L, Collier G, Vanek P, Siles B, Kow Y, Doetsch P, Cunningham R, Chirikjian J (1999) Mutation identification DNA analysis system (MIDAS) for detection of known mutations. Electrophoresis 20:1141–1148
Huang J, Kirk B, Favis R, Soussi T, Paty P, Cao W, Barany F (2002) An endonuclease/ligase based mutation scanning method especially suited for analysis of neoplastic tissue. Oncogene 21:1909–1921
Pincas H, Pingle MR, Huang J, Lao K, Paty PB, Friedman AM, Barany F (2004) High sensitivity EndoV mutation scanning through real-time ligase proofreading. Nucleic Acids Res 32:e148
Gao H, Huang J, Barany F, Cao W (2007) Switching base preferences of mismatch cleavage in endonuclease V: an improved method for scanning point mutations. Nucleic Acids Res 35:e2
Turner DJ, Pingle MR, Barany F (2006) Harnessing asymmetrical substrate recognition by thermostable EndoV to achieve balanced linear amplification in multiplexed SNP typing. Biochem Cell Biol (Biochimie et biologie cellulaire) 84:232–242
Miyazaki K (2002) Random DNA fragmentation with endonuclease V: application to DNA shuffling. Nucleic Acids Res 30:e139
Stemmer WP (1994) DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc Natl Acad Sci USA 91:10747–10751
Stemmer WP (1994) Rapid evolution of a protein in vitro by DNA shuffling. Nature 370:389–391
Wang Z, Wang HY, Feng H (2012) A simple and reproducible method for directed evolution: combination of random mutation with dITP and DNA fragmentation with endonuclease V. Mol Biotechnol. doi:10.1007/s12033-012-9516-9
Acknowledgments
This project was supported in part by CSREES/USDA (SC-1700274, technical contribution No. 6059), the Department of Defense (W81XWH-10-1-0385), and the National Institutes of Health (GM090141). I thank Drs. Bernard Weiss, Yoke Wah Kow, Richard Cunningham, Francis Barany, Brian Dominy and Rongjuan Mi for discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cao, W. Endonuclease V: an unusual enzyme for repair of DNA deamination. Cell. Mol. Life Sci. 70, 3145–3156 (2013). https://doi.org/10.1007/s00018-012-1222-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-1222-z